
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Backbone conformation affects duplex initiation and duplex propagation in hybridisation of synthetic H-bonding oligomers†
Giulia
Iadevaia
 ,
Diego
Núñez-Villanueva
,
Alexander E.
Stross
and
Christopher A.
Hunter
*
,
Diego
Núñez-Villanueva
,
Alexander E.
Stross
and
Christopher A.
Hunter
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB21EW, UK. E-mail: herchelsmith.orgchem@ch.cam.ac.uk
First published on 23rd May 2018
Abstract
Synthetic oligomers equipped with complementary H-bond donor and acceptor side chains form multiply H-bonded duplexes in organic solvents. Comparison of the duplex forming properties of four families of oligomers with different backbones shows that formation of an extended duplex with three or four inter-strand H-bonds is more challenging than formation of complexes that make only two H-bonds. The stabilities of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes formed between length complementary homo-oligomers equipped with either phosphine oxide or phenol recognition modules were measured in toluene. When the backbone is very flexible (pentane-1,5-diyl thioether), the stability increases uniformly by an order of magnitude for each additional base-pair added to the duplex: the effective molarities for formation of the first intramolecular H-bond (duplex initiation) and subsequent intramolecular H-bonds (duplex propagation) are similar. This flexible system is compared with three more rigid backbones that are isomeric combinations of an aromatic ring and methylene groups. One of the rigid systems behaves in exactly the same way as the flexible backbone, but the other two do not. For these systems, the effective molarity for formation of the first intramolecular H-bond is the same as that found for the other two backbones, but additional H-bonds are not formed between the longer oligomers. The effective molarities are too low for duplex propagation in these systems, because the oligomer backbones cannot adopt conformations compatible with formation of an extended duplex.
:1 complexes formed between length complementary homo-oligomers equipped with either phosphine oxide or phenol recognition modules were measured in toluene. When the backbone is very flexible (pentane-1,5-diyl thioether), the stability increases uniformly by an order of magnitude for each additional base-pair added to the duplex: the effective molarities for formation of the first intramolecular H-bond (duplex initiation) and subsequent intramolecular H-bonds (duplex propagation) are similar. This flexible system is compared with three more rigid backbones that are isomeric combinations of an aromatic ring and methylene groups. One of the rigid systems behaves in exactly the same way as the flexible backbone, but the other two do not. For these systems, the effective molarity for formation of the first intramolecular H-bond is the same as that found for the other two backbones, but additional H-bonds are not formed between the longer oligomers. The effective molarities are too low for duplex propagation in these systems, because the oligomer backbones cannot adopt conformations compatible with formation of an extended duplex.
Introduction
Nucleic acids store and express genetic information via sequence selective duplex formation and template directed synthesis.1 Variation in the chemical structure of the backbone,2 the base pairing system,3 the phosphate diester linker4 and the sugar5 have been explored, and the ability to form a stable duplex is maintained despite these modifications. This observation suggests that oligomers with very different chemical structures may also be able to form duplexes in a sequence selective manner. A number of synthetic oligomers have been reported that form duplexes through different types of non-covalent interaction, including metal–ligand coordination,6 aromatic stacking,7 salt bridges8 and H-bonding.9Fig. 1 shows the structures of four different H-bonded duplexes formed by oligomers equipped with phenol and phosphine oxide recognition units.10 All combinations of 2-mers that have the N7, N8 and C8 backbones shown in Fig. 1 form duplexes with similar stabilities.10b,d In fact, the effective molarities for formation of the intramolecular H-bonds that lead to duplex formation in all of the systems shown in Fig. 1 are remarkably similar (7–20 mM).11 These results suggest that conformational properties of the backbone are not critical for duplex formation, so oligomers with the highly flexible C9 backbone form duplexes that have similar stability to the duplexes formed by oligomers with the more rigid C8 backbone. Here we describe a new family of oligomers where the conformational properties of the backbone compromise duplex formation between longer oligomers.
 | ||
| Fig. 1 H-bonded duplexes. Different backbones lead to equally stable 2-mer duplexes. The C8 backbone is highlighted in purple, the N8 backbone in orange, the N7 backbone in green, and the C9 in blue. R is 2-ethylhexyloxy and R′ is n-hexyl. | ||
The stepwise equilibria for the assembly of two complementary oligomers into a duplex are shown in Fig. 2.12 After the first intermolecular interaction, that has an association constant of K, the subsequent intramolecular interactions have an equilibrium constant of KEMN, where EMN is the stepwise effective molarity, and N is the number of intramolecular interactions formed. Fully bound duplexes are formed, if the values of KEMN are all significantly greater than one. By comparing the stabilities of duplexes of increasing chain length, it is possible to experimentally determine values of EMN. In this paper, we show that the effective molarity for the first intramolecular interaction responsible for duplex initiation (EM1) can be very different from the values of EM for the subsequent intramolecular interactions that lead to duplex propagation.
 | ||
| Fig. 2 Stepwise assembly of a duplex from two complementary oligomers. K is the association constant for formation of an intermolecular interaction between two complementary H-bonding sites, and EMN are the stepwise effective molarities for formation of the intramolecular interactions that lead to zipping up of the duplex. | ||
The synthesis of the homo-oligomers up to four recognition units in length was previously reported for the C8 and C9 backbones.10a Here we describe the synthesis of the corresponding homo-oligomers with the N7 and N8 backbones. Measurement of the stabilities of the duplexes formed by all complementary pairs of oligomers allows characterisation of the stepwise effective molarities for duplex initiation and propagation.
Results and discussion
Synthesis
A convergent approach was taken to obtain the oligomers from a set of common building blocks. Secondary anilines 1a and 1b, which were used as chain end caps, were prepared via reductive amination of the corresponding primary anilines (Scheme 1). The H-bond donor primary aniline, 4-aminophenol, is commercially available, and synthesis of the H-bond acceptor primary aniline bearing a phosphine oxide was reported previously.10b | ||
| Scheme 1 | ||
Scheme 2 shows the synthetic route to the 3-mers and 4-mers for the N8 backbone. Monoprotected aldehyde 3 was prepared by refluxing 2 with one equivalent of ethylene glycol in toluene (Scheme 2).13 Aldehydes 4a and 4b were synthesized by reductive amination of 3 with 1a or 1b, followed by acetal deprotection (Scheme 2). 3-mers 5a and 5b were obtained by reductive amination of 4a or 4b with the corresponding primary aniline. The bisanilines 6a and 6b were prepared from 2 as described previously.10b Reductive amination of aldehyde 4a with aniline 6a gave the all donor 4-mer 7a, and reductive amination of aldehyde 4b with aniline 6b gave the all acceptor 4-mer 7b.
 | ||
| Scheme 2 | ||
A similar synthetic route was used for the synthesis of the N7 backbone oligomers (Scheme 3). Reductive amination of monoprotected aldehyde 9 with 1a or 1b gave aldehydes 10a and 10b respectively. The 3-mers 11a and 11b were obtained by reductive amination of 10a or 10b with the corresponding primary aniline. The bisanilines 12a and 12b were prepared from 8 as described previously.10b Reductive amination of aldehyde 10a with 12a the all donor 4-mer 13a, and reductive amination of aldehyde 10b with 12b gave the all acceptor 4-mers 13b (Scheme 3).
 | ||
| Scheme 3 | ||
NMR binding studies
Association constants between complementary H-bond donor (D) and H-bond acceptor (A) oligomers were determined via31P NMR titrations in toluene-d8. In all cases, the H-bond acceptor phosphine oxide oligomer was used as the host, and the data fit well to 1:1 binding isotherms. The results are summarized in Table 1, together with data for the corresponding 1-mer and 2-mer complexes previously reported.10a,b,dTable 1 also reports the complexation-induced changes in chemical shift of the 31P signals. The large increases in 31P chemical shift are characteristic of H-bond formation.14 The association constants for the 3-mer complexes (AAA·DDD) and for the 4-mer complexes (AAAA·DDDD) are of the same order of magnitude as the association constant for the corresponding 2-mer complexes (AA·DD) (Table 1).
:1 complexes in toluene-d8 at 298 K
| Backbone | Complex | logKN/M−1 |
Δδ31P/ppm |
|---|---|---|---|
| — | A·D | 2.4 ± 0.1 | 5.0 |
| N8 | AA·DD | 3.4 ± 0.1 | 3.9 |
| N8 | AAA·DDD | 3.5 ± 0.2 | 3.8, 4.7 |
| N8 | AAAA·DDDD | 3.8 ± 0.1 | 4.3, 4.3 |
| N7 | AA·DD | 3.1 ± 0.2 | 6.9 |
| N7 | AAA·DDD | 3.4 ± 0.1 | 7.6, 7.6 |
| N7 | AAAA·DDDD | 3.3 ± 0.1 | 8.5, 8.6 |
Fig. 3 shows the relationship between the association constant for duplex formation (logKN) and the number of potential H-bonding interactions (N). The association constants for the C8 and C9 backbones increase uniformly by an order of magnitude for each H-bonding site added to the chain. For the N8 and N7 backbones, the 2-mer duplex is an order of magnitude more stable than the singly H-bonded 1-mer complex, but there is no significant increase in association constant for the 3-mer or 4-mer complexes. These results suggest that in the longer N8 and N7 oligomers only two H-bonds are formed and that the fully assembled duplexes are not stable. One would expect a corresponding difference in the magnitudes of the complexation-induced changes in 31P NMR chemical shift for the systems that are not fully H-bonded, but the changes in chemical shift are actually larger for the longer N8 and N7 oligomers. This discrepancy must reflect some difference in three-dimensional structure for these complexes or the formation of higher order aggregates that do not significantly affect the shape of the binding isotherm.
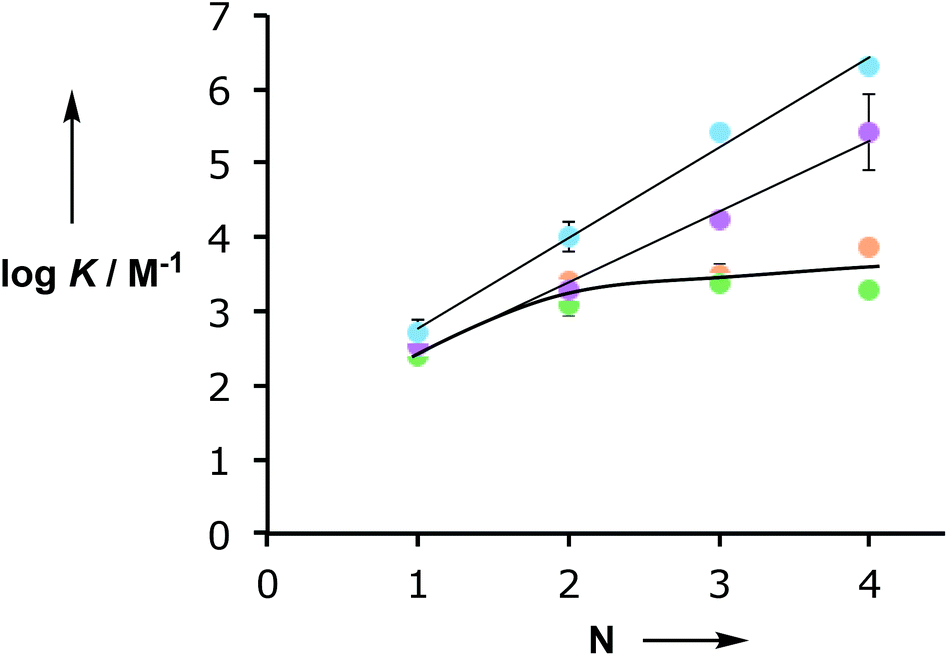 | ||
| Fig. 3 Relationship between the association constant for duplex formation (logKN) and the number of recognition units in an oligomer (N) for the C8 (purple),10a C9 (blue),10d N8 (orange) and N7 (green) backbones. | ||
Conformational analysis
The difference in behaviour between the four different systems is likely to be due to geometrical incompatibility of the N8 and N7 backbones with the fully bound duplex. The geometrical properties of the backbones were therefore investigated using molecular mechanic calculations.15 By using a distance constraint to make sure that at least one intermolecular H-bond is formed in the duplex, it is possible to restrict the conformational search space to a tractable size. Fig. 4 shows the lowest energy structures obtained from conformational searches for 2-mer AA·DD duplexes with the four different backbones. In all cases, the fully assembled duplex with two A·D H-bonds was found. Fig. 5 shows the lowest energy structures obtained from conformational searches for 3-mer AAA·DDD duplexes with the four different backbones. In this case, the fully assembled duplex with three A·D H-bonds was found for the C8 and C9 backbones, but not for the N8 and N7 backbones. In the N8 and N7 backbone structures, only two of the recognition units are properly paired. Moreover, the fully H-bonded duplex with all three A·D H-bonds was not located at all in the conformational search for either of these systems, which suggests that this structure is considerably higher in energy than the global energy minimum. | ||
| Fig. 4 Lowest energy conformations of the 2-mer AA·DD duplexes from conformational searches (MMFFs force-field and CHCl3 solvation implemented in Macromodel)15 for the (a) N8, (b) N7, (c) C8 and (d) C9 backbones. The H-bond donor oligomer is shown in red and the H-bond acceptor oligomer in blue. The recognition modules are shown as atomic spheres, H-bonds are shown in yellow, and hydrogen atoms on carbon are not shown for clarity. The chemical structures highlight the key interactions made in the calculated structures. | ||
 | ||
| Fig. 5 Lowest energy conformations of the 3-mer AAA·DDD duplexes from conformational searches (MMFFs force-field and CHCl3 solvation implemented in Macromodel)15 for the (a) N8, (b) N7, (c) C8 and (d) C9 backbones. The H-bond donor oligomer is shown in red and the H-bond acceptor oligomer in blue. The recognition modules are shown as atomic spheres, H-bonds are shown in yellow, and hydrogen atoms on carbons are not shown for clarity. The chemical structures highlight the key interactions made in the calculated structures. | ||
The computational results are consistent with the experimental observations. All of the backbones are compatible with formation of the first and second H-bond of the duplex, but only the C8 and C9 backbones can make the third H-bond. For the two complexes that do not form the fully assembled duplex, the unsatisfied H-bond donor makes an additional interaction with an alternative site. For the N8 backbone, the unpaired terminal phenol interacts with the central phosphine oxide that is already engaged in a H-bond with the complementary central phenol. For the N7 backbone, the unpaired terminal phenol interacts with the central phenol that is already H-bonded to the complementary central phosphine oxide. The experimental association constants indicate that if these additional interactions are present, then they do not contribute significantly to the stabilities of the complexes.
The molecular mechanics calculations should provide some insight into the reasons for the failure of the N8 and N7 backbones, but the structures in Fig. 5 are rather complicated. Further experimental information was obtained from X-ray crystallography. Single crystals suitable for X-ray analysis were obtained for the phosphine oxide 2-mer with the N8 backbone. Fig. 6 compares the backbone conformation in the solid state with the conformations of the 2-mer and 3-mer backbones from the energy minimum molecular mechanics structures of the duplexes shown in Fig. 4 and 5.
 | ||
| Fig. 6 Conformation of the N8 backbone. (a) Chemical structure of the AA 2-mer; (b) X-ray structure of the AA 2-mer; (c) conformation of the AA 2-mer in the lowest energy molecular mechanics structure of the AA·DD duplex; (d) conformation of the AAA 3-mer in the lowest energy molecular mechanics structure of the AAA·DDD duplex. The first two recognition modules that are H-bonded in the duplex are highlighted in blue for comparison with (b) and (c), and the third recognition module that is not involved in H-bonding in the duplex is shown in grey. | ||
The X-ray and the molecular mechanics structures of the 2-mer are very similar: the two recognition modules are oriented in an anti arrangement, and this motif is repeated in the 3-mer molecular mechanics structure. The X-ray structure indicates that the molecular mechanics calculations provide a reasonable description of the conformational properties of these molecules. Moreover, the results suggest a reason why the N8 backbone does not make an extended multiply H-bonded duplex. Duplex formation requires a syn arrangement of the recognition modules with all of the interaction sites pointing in the same direction, but the structures in Fig. 6 imply that the N8 backbone prefers to adopt an alternating anti arrangement of recognition modules along the chain.
Conclusions
The conformational flexibility of the backbone plays an important role in determining the ability of complementary H-bonding oligomers to form a fully bound duplex. For a relatively rigid backbone that has conformational properties compatible with duplex formation (e.g. C8), the stability of the duplex increases uniformly with the number of recognition sites present in the oligomers. For the C8 systems, the effective molarity for formation of the first intramolecular H-bond, i.e. duplex initiation (EM1), and the effective molarities for subsequent intramolecular interactions that lead to zipping up of the duplex, i.e. duplex propagation (EM2, EM3etc.), have similar values (Fig. 7a). The same is true for highly flexible backbones (e.g. C9) that will always be able to find conformations compatible with a fully bound duplex (Fig. 7b). | ||
| Fig. 7 Relationship between the conformational properties of the backbone and stepwise effective molarities in the formation of non-covalent duplexes. (a) A rigid backbone will form a duplex if it is geometrically compatible. (b) A very flexible backbone will always be able find a conformational state that is geometrically compatible with the duplex. (c) If a rigid backbone is not geometrically compatible with the duplex, the first effective molarity for duplex initiation (EM1) and the subsequent effective molarities for duplex propagation (EM2) will be different, so only the 2-mer duplex will assemble. | ||
The other two backbones described in this paper (N8 and N7) have quite different properties. These oligomers have relatively rigid backbones that appear to be incompatible with a fully assembled duplex. The effective molarities for duplex initiation (EM1) for both of these systems are similar to the values found for the C8 and C9 backbones, but the effective molarities for duplex propagation (EM2) are much lower. The reason is that formation of a doubly H-bonded complex imposes little conformational constraint on the backbone, whereas formation of a triply H-bonded complex can only be achieved within a much more restricted conformational window. As a result, the N7 and N8 backbones are not compatible with the formation of an extended multiply H-bonded duplex, even though the 2-mers behave in exactly the same way as the C8 and C9 2-mers.
The results described here, together with our previous observations on the folding of AD 2-mers, indicate that the choice of backbone is critical to the successful development of synthetic information molecules designed to form H-bonded duplexes between complementary recognition sites. If the backbone is very flexible (C9), duplex formation will always be favourable for homo-sequence oligomers, but intramolecular interactions will complete with duplex formation in mixed sequence oligomers. The solution is to use a more rigid backbone, but as the results here show, this approach carries the risk that the conformational states accessible to the backbone may not be compatible with duplex formation. We have studied three isomeric backbones (C8, N7 and N8) that are a semi-flexible combination of an aromatic ring and two methylene groups. One of these systems forms stable duplexes and the other two do not. Conformational analysis using molecular modelling is consistent with the experimental properties of these systems and should provide a useful tool for future backbone design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC and ERC for funding. We thank Mr Harry Adams (University of Sheffield) for help with X-ray crystallographic data collection and analysis.Notes and references
- (a) J. D. Watson and F. H. Crick, Nature, 1953, 171, 964 CrossRef PubMed; (b) R. R. Sinden, DNA Structure and Function, Academic Press, 1994 Search PubMed.
- (a) A. Eschenmoser, Science, 1999, 284, 2118 CrossRef PubMed; (b) S. A. Benner, Acc. Chem. Res., 2004, 37, 784 CrossRef PubMed; (c) S. A. Benner, F. Chen, Z. Yang, P. L. Luisi and C. Chiarabelli, in Chemical Synthetic Biology, John Wiley & Sons, Ltd, Chichester, UK, 2011, p. 69 Search PubMed; (d) S. A. Benner, Biol. Theory, 2013, 8, 357 CrossRef; (e) C. Wilson and A. D. Keef, Curr. Opin. Chem. Biol., 2006, 10, 607 CrossRef PubMed; (f) D. H. Appella, Curr. Opin. Chem. Biol., 2009, 13, 687 CrossRef PubMed; (g) E. T. Kool, Curr. Opin. Chem. Biol., 2000, 4, 602 CrossRef PubMed.
- Base modification of nucleic acids: (a) J. A. Piccirilli, T. Krauch, S. E. Moroney and S. A. Benner, Nature, 1990, 343, 33 CrossRef PubMed; (b) Z. Yang, D. Hutter, P. Sheng, A. M. Sismour and S. A. Benner, Nucleic Acids Res., 2006, 34, 6095 CrossRef PubMed; (c) F. Wojciechowski and C. J. Leumann, Chem. Soc. Rev., 2011, 40, 5669 RSC; (d) S. A. Benner, Curr. Opin. Chem. Biol., 2012, 16, 581 CrossRef PubMed; (e) H. Liu, J. Gao, S. R. Lynch, Y. D. Saito, L. Maynard and E. T. Kool, Science, 2003, 302, 868 CrossRef PubMed; (f) E. T. Kool, Acc. Chem. Res., 2002, 35, 936 CrossRef PubMed; (g) E. T. Kool, H. Lu, S. J. Kim, S. Tan, J. N. Wilson, J. Gao and H. Liu, Nucleic Acids Symp. Ser., 2006, 50, 15 CrossRef PubMed; (h) J. N. Wilson and E. T. Kool, Org. Biomol. Chem., 2006, 4, 4265 RSC.
- Phosphate modifications of nucleic acids: (a) S. A. Benner and D. Hutter, Bioorg. Chem., 2002, 30, 62 CrossRef PubMed; (b) Z. Huang, K. C. Schneider and S. A. Benner, J. Org. Chem., 1991, 56, 3869 CrossRef; (c) Z. Huang and S. A. Benner, J. Org. Chem., 2002, 67, 3996 CrossRef PubMed; (d) C. Richert, A. L. Roughton and S. A. Benner, J. Am. Chem. Soc., 1996, 118, 4518 CrossRef; (e) P. Li, Z. A. Sergueeva, M. Dobrikov and B. R. Shaw, Chem. Rev., 2007, 107, 4746 CrossRef PubMed; (f) H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Org. Lett., 2008, 10, 3729 CrossRef PubMed; (g) M. Eriksson and P. E. Nielsen, Rev. Biophys., 1996, 29, 369 CrossRef; (h) P. E. Nielsen, Chem. Biodiversity, 2010, 7, 786 CrossRef PubMed; (i) P. E. Nielsen and M. Egholm, Curr. Issues Mol. Biol., 1999, 1, 89 Search PubMed; (j) P. E. Nielsen and G. Haaima, Chem. Soc. Rev., 1997, 26, 73 RSC; (k) Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73 CrossRef PubMed.
- Sugar modifications of nucleic acids: (a) K. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347 CrossRef; (b) A. van Aerschot, I. Verheggen, C. Hendrix and P. Herdewijn, Angew. Chem., Int. Ed. Engl., 1995, 34, 1338 CrossRef; (c) D. Renneberg and C. J. Leumann, J. Am. Chem. Soc., 2002, 124, 5993 CrossRef PubMed; (d) D. A. Braasch and D. R. Corey, Chem. Biol., 2001, 8, 1 CrossRef PubMed; (e) S. K. Singh, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455 RSC; (f) H. V. Nguyen, Z. Y. Zhao, A. Sallustrau, S. L. Horswell, L. Male, A. Mulas and J. H. R. Tucker, Chem. Commun., 2012, 48, 12165 RSC; (g) L. Zhang, A. Peritz and E. Meggers, J. Am. Chem. Soc., 2005, 127, 4174 CrossRef PubMed; (h) M. K. Schlegel, A. E. Peritz, K. Kittigowittana, L. Zhang and E. Meggers, ChemBioChem, 2007, 8, 927 CrossRef PubMed; (i) P. Karri, V. Punna, K. Kim and R. Krishnamurthy, Angew. Chem., Int. Ed., 2013, 52, 5840 CrossRef PubMed.
- (a) R. Kramer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CrossRef PubMed; (b) A. Marquis, V. Smith, J. Harrowfield, J.-M. Lehn, H. Herschbach, R. Sanvito, E. Leize-Wagner and A. van Dorsselaer, Chem. – Eur. J., 2006, 12, 5632 CrossRef PubMed; (c) H. L. Anderson, Inorg. Chem., 1994, 33, 972 CrossRef; (d) P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538 CrossRef.
- (a) V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720 CrossRef PubMed; (b) V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem. – Eur. J., 2001, 7, 2810 CrossRef PubMed.
- (a) Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2005, 117, 3935 CrossRef; (b) H. Ito, Y. Furusho, T. Hasegawa and E. Yashima, J. Am. Chem. Soc., 2008, 130, 14008 CrossRef PubMed; (c) Y. Furusho and E. Yashima, Macromol. Rapid Commun., 2011, 32, 136 CrossRef PubMed; (d) H. Yamada, Y. Furusho, H. Ito and E. Yashima, Chem. Commun., 2010, 46, 3487 RSC.
- (a) J. Sánchez-Quesada, C. Seel, P. Prados, J. de Mendoza, I. Dalcol and E. Giralt, J. Am. Chem. Soc., 1996, 118, 277 CrossRef; (b) A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856 CrossRef; (c) A. P. Bisson and C. A. Hunter, Chem. Commun., 1996, 1723 RSC; (d) B. Gong, Y. Yan, H. Zeng, E. Skrzypczak-Jankunn, Y. W. Kim, J. Zhu and H. Ickes, J. Am. Chem. Soc., 1999, 121, 5607 CrossRef; (e) B. Gong, Synlett, 2001, 582 CrossRef; (f) H. Zeng, R. S. Miller, R. A. Flowers and B. Gong, J. Am. Chem. Soc., 2000, 122, 2635 CrossRef; (g) H. Zeng, H. Ickes, R. A. Flowers and B. Gong, J. Org. Chem., 2001, 66, 3574 CrossRef PubMed; (h) B. Gong, Polym. Int., 2007, 56, 436 CrossRef; (i) B. Gong, Acc. Chem. Res., 2012, 45, 2077 CrossRef PubMed; (j) Y. Yang, Z.-Y. Yang, Y.-P. Yi, J.-F. Xiang, C.-F. Chen, L.-J. Wan and Z.-G. Shuai, J. Org. Chem., 2007, 72, 4936 CrossRef PubMed; (k) W.-J. Chu, Y. Yang and C.-F. Chen, Org. Lett., 2010, 12, 3156 CrossRef PubMed; (l) W.-J. Chu, J. Chen, C.-F. Chen, Y. Yang and Z. Shuai, J. Org. Chem., 2012, 77, 7815 CrossRef PubMed; (m) E. A. Archer and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 5074 CrossRef PubMed; (n) H. Gong and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 1719 CrossRef PubMed.
- (a) A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 94 RSC; (b) G. Iadevaia, A. E. Stross, A. Neumann and C. A. Hunter, Chem. Sci., 2016, 7, 1760 RSC; (c) A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 5686 RSC; (d) D. Nűñez-Villanueva and C. A. Hunter, Chem. Sci., 2017, 8, 206 RSC; (e) D. Núñez-Villanueva, G. Iadevaia, A. E. Stross, M. A. Jinks, J. A. Swain and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 6654 CrossRef PubMed; (f) E. Stross, G. Iadevaia, D. Núñez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 12655 CrossRef PubMed.
- Although all of the interactions in these systems are strictly intermolecular interactions, we will use the term intramolecular to describe H-bonds that are formed after the first intermolecular H-bond has brought the two molecules together to form a complex.
- (a) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef PubMed; (b) H. J. Hogben, J. K. Sprae, M. Hoffmann, M. Pawlicki and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 20962 CrossRef PubMed.
- C. Xue and F.-T. Luo, J. Org. Chem., 2003, 68, 4417 CrossRef PubMed.
- (a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235 CrossRef; (b) U. Mayer, W. Gerger and V. Gutmann, Monatsh. Chem., 1977, 108, 489 CrossRef; (c) V. Gutmann, Pure Appl. Chem., 1979, 51, 2197 CrossRef.
- MacroModel, version 9.8, Schrödinger, LLC, New York, NY, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1835177. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob00819a |
| This journal is © The Royal Society of Chemistry 2018 |