
Synthesis of plasmodione metabolites and 13C-enriched plasmodione as chemical tools for drug metabolism investigation†
Liwen
Feng,  a
Katharina
Ehrhardt,
b
Herbert
Zimmermann,
c
François
Fenaille,
d
Elisabeth
Davioud-Charvet
*a
a
Katharina
Ehrhardt,
b
Herbert
Zimmermann,
c
François
Fenaille,
d
Elisabeth
Davioud-Charvet
*a
a
Katharina
Ehrhardt,
b
Herbert
Zimmermann,
c
François
Fenaille,
d
Elisabeth
Davioud-Charvet
*a
Abstract
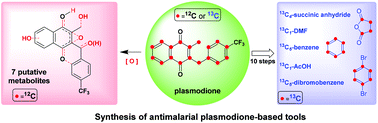
Malaria is a tropical parasitic disease threatening populations in tropical and sub-tropical areas. Resistance to antimalarial drugs has spread all over the world in the past 50 years, thus new drugs are urgently needed. Plasmodione (benzylmenadione series) has been identified as a potent antimalarial early lead drug, acting through a redox bioactivation on asexual and young sexual blood stages. To investigate its metabolism, a series of plasmodione-based tools, including a fully 13C-labelled lead drug and putative metabolites, have been designed and synthesized for drug metabolism investigation. Furthermore, with the help of UHPLC-MS/MS, two of the drug metabolites have been identified from urine of drug-treated mice.
- This article is part of the themed collection: Celebrating excellence in research: women of organic chemistry
 Please wait while we load your content...
Please wait while we load your content...

