
Asymmetric synthesis of trans-4,5-disubstituted γ-butyrolactones involving a key allylboration step. First access to (−)-nicotlactone B and (−)-galbacin†
F.
Carreaux  *a
*a
*a
Abstract
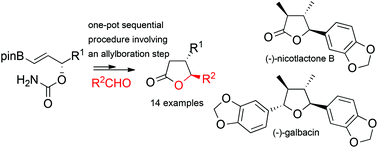
An efficient asymmetric synthesis of trans-4,5-disubstituted γ-butyrolactones from aldehydes and enantioenriched γ-carbamate alkenylboronates is reported. The cornerstone of this strategy is the implementation of sequential [3,3]-allyl cyanate rearrangement/allylboration/nucleophilic addition/cyclisation reactions. Diverse γ-butyrolactones such as the flavouring compounds, (+)-trans-whiskey lactone and (+)-trans-cognac lactone, as well as an advanced intermediate towards the first synthesis of natural products, (−)-nicotlactone B and (−)-galbacin, have thus been obtained.
 Please wait while we load your content...
Please wait while we load your content...

