
Small-molecule anticancer agents kill cancer cells by harnessing reactive oxygen species in an iron-dependent manner†

Abstract
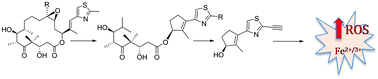
In the course of generating a library of open-chain epothilones, we discovered a new class of small molecule anticancer agents that has no effect on tubulin but instead kills selected cancer cell lines by harnessing reactive oxygen species in an iron-dependent manner. Results of the preliminary studies are consistent with the recently described cell death mechanism ferroptosis. Studies are in progress to confirm ferroptosis as the cell death mechanism and to identify the specific molecular targets of these small molecule anticancer agents.
 Please wait while we load your content...
Please wait while we load your content...

