
Kinetic basis for the activation of human cyclooxygenase-2 rather than cyclooxygenase-1 by nitric oxide†
 *a
*a
Abstract
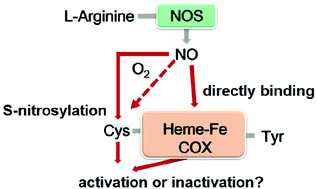
Numerous studies have shown that nitric oxide (NO) interacts with human cyclooxygenase (COX); however, conflicting results exist with respect to their interactions. Herein, recombinant human COX-1 and COX-2 were prepared and treated with NO donors individually under anaerobic and aerobic conditions. The S-nitrosylation detection and subsequent kinetic investigations into the arachidonic acid (AA) oxidation of COX enzymes indicate that NO S-nitrosylates both COX-1 and COX-2 in an oxygen-dependent manner, but enhances only the dioxygenase activity of COX-2. The solution viscosity, deuterium kinetic isotope effect (KIE), and oxygen-18 KIE experiments further demonstrate that NO activates COX-2 by altering the protein conformation to stimulate substrate association/product release and by accelerating the rate of hydrogen abstraction from AA by catalytic tyrosine radicals. These novel findings provide useful information for designing new drugs with less cardiotoxic effects that can block the interaction between NO and COX.
 Please wait while we load your content...
Please wait while we load your content...

