
A semi-rigid isoindoline-derived nitroxide spin label for RNA†

Abstract
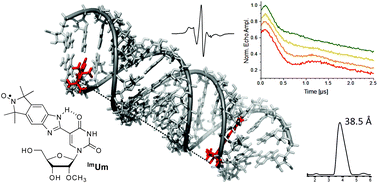
A new isoindoline-derived benzimidazole nitroxide spin label, ImUm, was synthesized and incorporated into RNA oligoribonucleotides. ImUm is the first example of a conformationally unambiguous spin label for RNA, in which the nitroxide N–O bond lies on the same axis as the single bond used to attach the rigid isoindoline-based spin label to a uridine base. This results in minimal displacement of the nitroxide upon rotation of this single bond, which is a useful property for a label to be used for distance measurements. Continuous-wave (CW) EPR measurements of RNA duplexes containing ImUm indicate a restricted rotation around this single bond, presumably due to an intramolecular hydrogen bond between the benzimidazole N–H and O4 of the uracil. Orientation-selective pulsed electron–electron double resonance (PELDOR, also called double electron–electron resonance, or DEER) distance measurements between two spin labels in two RNA duplexes showed in one case a strong orientation dependence, further confirming the restricted motion of the spin labels in RNA duplexes.
- This article is part of the themed collection: Nucleic Acid Modifications
 Please wait while we load your content...
Please wait while we load your content...

