
Design and synthesis of a hybrid framework of indanone and chromane: total synthesis of a homoisoflavanoid, brazilane†
Jinwoo
Kim
and
Ikyon
Kim
 *
*
College of Pharmacy and Yonsei Institute of Pharmaceutical Sciences, Yonsei University, 85 Songdogwahak-ro, Yeonsu-gu, Incheon, 21983, Republic of Korea. E-mail: ikyonkim@yonsei.ac.kr; Fax: +82 32 749 4105; Tel: +82 32 749 4515
First published on 27th November 2017
Abstract
A chemical backbone of tetracyclic homoisoflavanoid natural products such as brazilin inspired us to design a new chemical scaffold, 6a,11b-dihydroindeno[2,1-c]chromen-7(6H)-one, which is a hybrid structure of indanone and chromane. Pd-catalyzed Suzuki–Miyaura cross-coupling of 4-chloro-2H-chromene-3-carbaldehydes with (hetero)aryl boronic acids was employed as a means to introduce a wide variety of (hetero)aryl groups as the D ring and intramolecular Friedel–Crafts acylation was utilized to construct the C ring of this skeleton. Total synthesis of the natural product, brazilane, was also demonstrated via this new chemical framework.
Introduction
Biologically active natural products have been a rich source for new drug development for the last several decades.1 At an early stage of the research, it is critical to evaluate which functional groups in natural products are essential for desired pharmacological effects and to modulate the potency and/or toxicity. For this purpose, strategic design and construction to rapidly realize a number of synthetic analogues in a divergent manner are highly important for structure–activity relationship (SAR) studies in small-molecule drug discovery programs.2In connection with our interest on homoisoflavanoid natural products (Fig. 1),3 we have developed several synthetic approaches to enable diverse analogues to be synthesized in a highly efficient manner.4 Structurally, tetracyclic homoisoflavanoids (1 in Scheme 1) can be viewed as a hybrid structure5 of indane and chromane, both of which are bicyclic frameworks embedded in many natural and synthetic products and are known to exhibit a wide variety of interesting biological functions depending on the substitution patterns of these basic cores. Inspired by this scaffold, we came up with a new structural analogue, 6a,11b-dihydroindeno[2,1-c]chromen-7(6H)-one (2), which has not been reported in the literature.6 Since it may have some intriguing biological properties as a hybrid molecule of indanone7 and chromane,8 we decided to develop a flexible synthetic route to this skeleton by which access to a number of analogues is feasible. Retrosynthetically, it was envisioned that the C ring of 2 could be formed via the intramolecular Friedel–Crafts acylation9 of 3. We chose the Suzuki–Miyaura cross-coupling reaction10 of 4 with (hetero)arylboronic acids as a means to introduce diverse D rings. The requisite β-halo-α,β-unsaturated aldehyde 4 could be easily derived from chromanone 5 by the known Vilsmeier–Haack reaction.11
 | ||
| Fig. 1 Some representative homoisoflavanoids. | ||
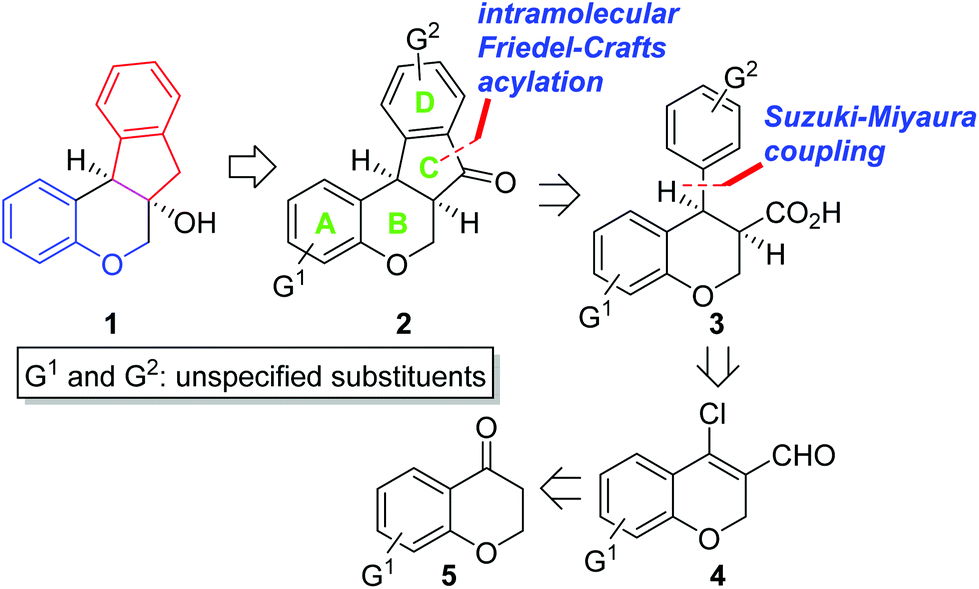 | ||
| Scheme 1 Synthetic plan. | ||
Results and discussion
Our approach commenced with the preparation of 4 by using the known procedure (Scheme 2). The Vilsmeier–Haack reaction of 5a–c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 provided the corresponding β-chloroenals (4a–c) in good to excellent yields. Although several Suzuki–Miyaura coupling reactions of β-halo-α,β-unsaturated enal systems have been known in the literature,13 the use of β-chloro-2H-chromene-3-carbaldehydes (4) as substrates for Suzuki–Miyaura cross-coupling reactions is rare.14
12 provided the corresponding β-chloroenals (4a–c) in good to excellent yields. Although several Suzuki–Miyaura coupling reactions of β-halo-α,β-unsaturated enal systems have been known in the literature,13 the use of β-chloro-2H-chromene-3-carbaldehydes (4) as substrates for Suzuki–Miyaura cross-coupling reactions is rare.14
 | ||
| Scheme 2 Synthesis of β-chloro-2H-chromene-3-carbaldehydes (4). | ||
Initially, we found that the Suzuki–Miyaura coupling of 4 with arylboronic acid in the presence of Pd(PPh3)4 and K3PO4 in EtOH at room temperature provided the desired β-aryl-α,β-unsaturated aldehyde 6 under mild conditions (Table 1). In some cases, however, yields were poor so coupling reactions were conducted in other catalytic systems where Pd2dba3 and SPhos were used as a Pd source and a ligand, respectively. Generally, electron-rich aromatic groups as well as electron-poor aromatic groups were installed in good yields under these conditions. The reactions with heteroarylboronic acids afforded the corresponding adducts in 72–81% yields (entries 15, 16, and 19).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 4 | Conditions | ArB(OH)2 | 6 | |
| a Conditions A: A mixture of 4, arylboronic acid (1.1 equiv.), Pd(PPh3)4 (0.1 equiv.), and K3PO4 (2 equiv.) in EtOH (1 mL) was stirred at rt. Conditions B: A mixture of 4, arylboronic acid (1.1 equiv.), Pd2dba3 (0.05 equiv.), SPhos (0.15 equiv.), and K3PO4 (2 equiv.) in THF (2 mL) was stirred at 100 °C. b Isolated yield (%). | |||||
| 1 | 4a | A |
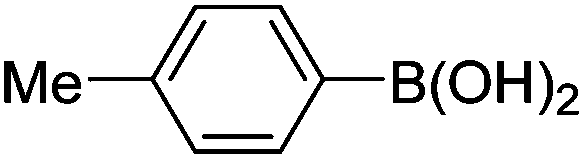
|

|
6a (52) |
| 2 | 4a | A |
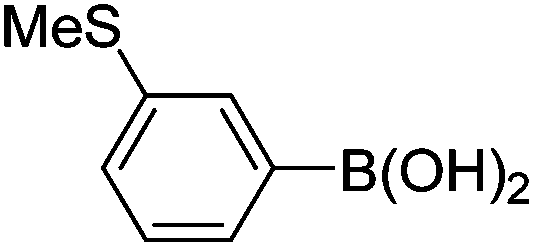
|

|
6b (47) |
| 3 | 4a | A |

|

|
6c (69) |
| 4 | 4a | A |

|

|
6d (68) |
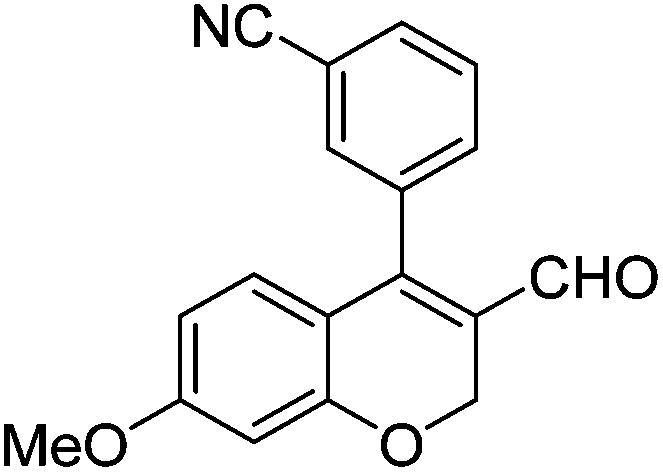
| 5 | 4a | A |

|

|
6e (66) |
| 6 | 4a | A |

|

|
6f (61) |
| 7 | 4a | B |

|

|
6g (85) |
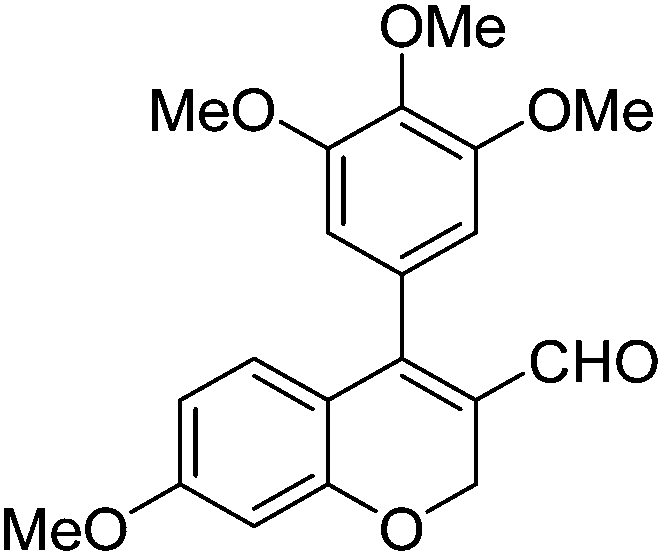
| 8 | 4b | B |
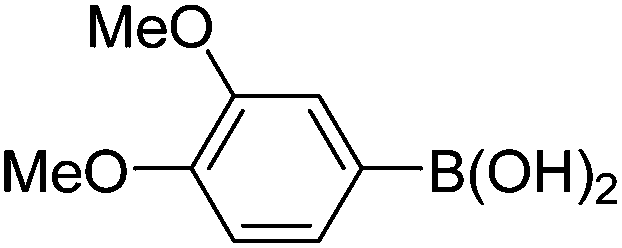
|

|
6h (88) |
| 9 | 4b | B |

|

|
6i (88) |
| 10 | 4b | B |
|

|
6j (90) |
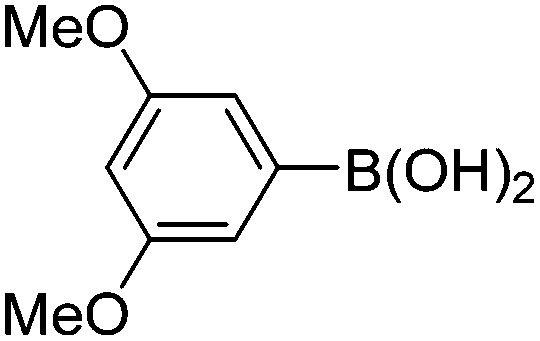
| 11 | 4b | B |

|
|
6k (92) |
| 12 | 4b | B |

|
|
6l (38) |
| 13 | 4b | B |

|

|
6m (88) |
| 14 | 4b | B |

|

|
6n (80) |
| 15 | 4b | B |

|

|
6o (72) |
| 16 | 4b | B |

|

|
6p (73) |
| 17 | 4c | B |

|

|
6q (80) |
| 18 | 4c | B |

|

|
6r (39) |
| 19 | 4c | B |

|

|
6s (81) |

Having secured a wide variety of 4-(hetero)aryl-2H-chromene-3-carbaldehydes (6) in hand, we next directed our attention to the elaboration of 6 to 2. Initial attempts including the intramolecular Friedel–Crafts alkylation with 6h under acidic conditions15 did not yield the desired cyclized product (Scheme 3). After some experimentation, we decided to reduce the olefin bond in 6h first by using catalytic hydrogenation conditions (H2, 10% Pd/C), which reduced not only the double bond but also aldehyde in 6h to afford alcohol 7 in an 85% yield. The Dess–Martin periodinane oxidation16 of 7 proceeded smoothly to give 8, with which cyclization efforts in the presence of various acids did not lead to success. Finally, the Kraus–Pinnick oxidation17 of 8 delivered carboxylic acid which was exposed to POCl3 at 80 °C (ref. 18) to furnish indanone (2h) in a 55% yield (for two steps).
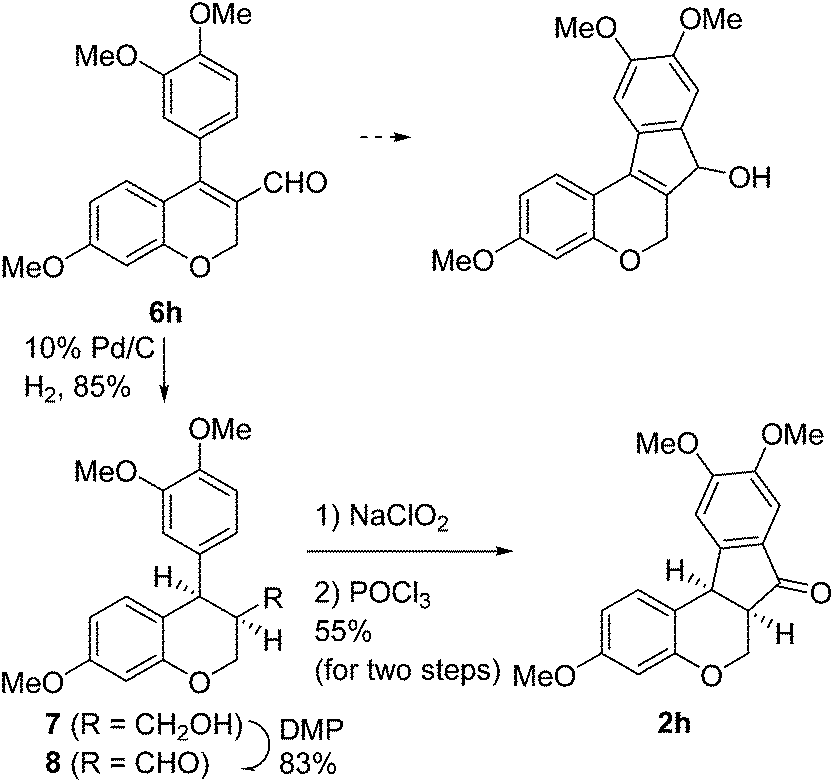 | ||
| Scheme 3 Synthesis of 2h. | ||
In the meantime, the conversion of 2h to naturally occurring brazilane was attempted (Scheme 4). The diastereoselective reduction of 2h provided benzylic alcohol 9 which was dehydrated under acidic media to give alkene 10 instead of 10′ as a result of the facile acid-promoted isomerization of 10′ (trisubstituted) to 10 with the more stable double bond (tetrasubstituted and double conjugation with two aromatics). The catalytic hydrogenation of the olefin bond in 10 furnished the trimethyl ether analogue (11) of brazilane.19,20 Alternatively, the carbonyl group in 2h was removed by the catalytic hydrogenation of 2h to directly deliver 11. Global deprotection with excess BBr3 provided brazilane in a 64% yield. The spectral data of the synthetic brazilane matched the literature values.
 | ||
| Scheme 4 Total synthesis of brazilane. | ||
In conclusion, a new chemical scaffold based on a hybrid structure of indanone and chromane, a tetracyclic isoflavanoid natural product analogue, was designed and synthesized. Suzuki–Miyaura cross-coupling and intramolecular Friedel–Crafts acylation were employed to construct the D and C rings of this motif, respectively. Further functionalization of this basic skeleton and biological tests of the synthesized compounds are currently in progress.
Experimental section
General methods
Unless specified, all reagents and starting materials were purchased from commercial sources and used as received without purification. “Concentrated” refers to the removal of volatile solvents via distillation using a rotary evaporator. “Dried” refers to pouring onto, or passing through, anhydrous magnesium sulfate followed by filtration. Flash chromatography was performed using silica gel (230–400 mesh) with hexanes, ethyl acetate, and dichloromethane as the eluent. All reactions were monitored by thin-layer chromatography on 0.25 mm silica plates (F-254) visualized with UV light. Melting points were measured using a capillary melting point apparatus. 1H and 13C NMR spectra were recorded on a 400 MHz NMR spectrometer and were described as chemical shifts, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), coupling constant in hertz (Hz), and number of protons. HRMS were measured with an electrospray ionization (ESI) and Q-TOF mass analyzer.General procedure for the synthesis of 4
To a stirred solution of 5 (3.91 mmol) in CH2Cl2 (13 mL) was dropwise added a pre-mixed solution of POCl3 (2.19 mL, 23.47 mmol) and DMF (2.26 mL, 29.34 mmol) at 0 °C. After being stirred at 0 °C for 17 h, the reaction mixture was quenched with H2O (5 mL) at 0 °C and concentrated in vacuo to give the residue. The resulting residue was basified with aq. NaOH solution (pH ∼ 8) and the precipitated solid was filtered and dried to afford 4.Yellow solid, mp: 78.3–79.9 °C; 1H NMR (400 MHz, CDCl3) δ 10.10 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.41–7.34 (m, 5H), 6.68 (dd, J = 2.0, 8.8 Hz, 1H), 6.51 (d, J = 2.0 Hz, 1H), 5.09 (s, 2H), 5.00 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 187.6, 164.0, 158.6, 143.8, 136.0, 128.9, 128.5, 128.3, 127.6, 122.3, 114.0, 110.2, 102.4, 70.5, 64.8; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C17H14ClO3 301.0626, found 301.0621.
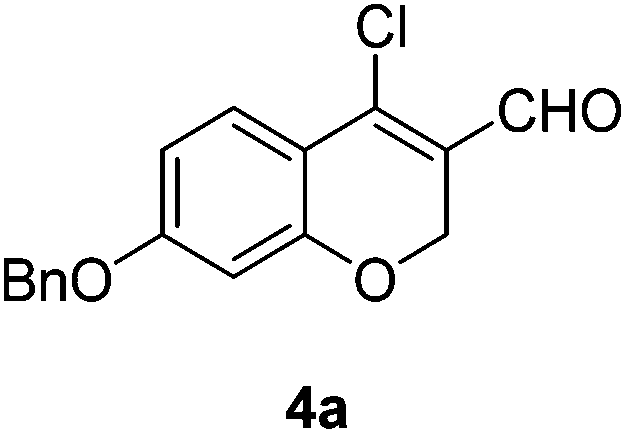
Yellow solid, mp: 83.2–85.8 °C; 1H NMR (400 MHz, CDCl3) δ 10.11 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 6.61 (dd, J = 2.4, 8.8 Hz, 1H), 6.44 (d, J = 2.4 Hz, 1H), 5.01 (s, 2H), 3.84 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.7, 164.9, 158.7, 143.9, 128.3, 122.2, 113.8, 109.6, 101.5, 64.9, 55.8; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C11H10ClO3 225.0313, found 225.0314.

Yellow solid, mp: 102.1–103.2 °C; 1H NMR (400 MHz, CDCl3) δ 10.10 (s, 1H), 7.57 (s, 1H), 6.43 (s, 1H), 4.99 (s, 2H), 3.87 (s, 3H), 1.37 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 187.6, 163.9, 157.2, 144.8, 133.2, 124.9, 121.7, 112.3, 99.8, 64.8, 55.6, 34.8, 29.8; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C15H18ClO3 281.0939, found 281.0932.

General procedure for the synthesis of 6
Conditions A: To a stirred solution of 4 (0.10 mmol) in EtOH (1 mL) were added arylboronic acid (1.1 equiv.), K3PO4 (2 equiv.), and Pd(PPh3)4 (0.1 equiv.) at rt. After being stirred at rt for 16 h, the reaction mixture was suction-filtered through a pad of Celite and the filtrate was concentrated under reduced pressure to give the residue which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 20:1:2) to give 6.
Conditions B: To a stirred solution of 4 (0.10 mmol) in THF (2 mL) were added arylboronic acid (1.1 equiv.), K3PO4 (2 equiv.), Pd2dba3 (0.05 equiv.), and SPhos (0.15 equiv.) at rt. After being stirred at 100 °C for 16 h, the reaction mixture was suction-filtered through a pad of Celite and the filtrate was concentrated under reduced pressure to give the residue which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 10:1:2) to give 6.
Brown solid, mp: 114.5–116.1 °C; 1H NMR (400 MHz, CDCl3) δ 9.41 (s, 1H), 7.41–7.35 (m, 5H), 7.28–7.26 (m, 2H), 7.20 (d, J = 7.2 Hz, 2H), 6.84 (d, J = 8.4 Hz, 1H), 6.57 (s, 1H), 6.51 (d, J = 8.4 Hz, 1H), 5.07 (s, 2H), 5.06 (s, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 190.4, 163.0, 158.3, 152.3, 139.4, 136.3, 130.4, 130.2, 130.1, 129.2, 128.8, 128.4, 127.6, 124.0, 117.0, 109.6, 102.5, 70.3, 63.8, 21.5; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C24H21O3 357.1485, found 357.1485.

Brown solid, mp: 80.9–81.3 °C; 1H NMR (400 MHz, CDCl3) δ 9.40 (s, 1H), 7.41–7.33 (m, 7H), 7.15 (s, 1H), 7.07 (d, J = 6.8 Hz, 1H), 6.81 (d, J = 8.8 Hz, 1H), 6.57 (s, 1H) 6.52 (d, J = 8.4 Hz, 1H), 5.07 (s, 4H), 2.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 190.0, 163.1, 158.2, 151.5, 139.6, 136.2, 133.9, 130.1, 128.9, 128.8, 128.4, 127.6, 127.4, 126.9, 126.8, 124.2, 116.6, 109.7, 102.6, 70.4, 63.7, 15.6; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C24H21O3S 389.1206, found 389.1201.

Yellow solid, mp: 121.3–123.7 °C; 1H NMR (400 MHz, CDCl3) δ 9.36 (s, 1H), 8.15 (d, J = 7.2 Hz, 2H), 7.42–7.35 (m, 7H), 6.71 (d, J = 8.8 Hz, 1H), 6.58 (s, 1H), 6.51 (d, J = 8.8 Hz, 1H), 5.08 (s, 4H), 3.97 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.6, 166.6, 163.2, 158.2, 150.9, 138.0, 136.1, 131.0, 130.4, 129.9, 129.8, 128.8, 128.4, 127.6, 124.3, 116.3, 109.8, 102.7, 70.4, 63.6, 52.6; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C25H21O5 401.1384, found 401.1386.

Yellow solid, mp: 172.6–174.3 °C; 1H NMR (400 MHz, CDCl3) δ 9.32 (s, 1H), 7.79 (d, J = 7.6 Hz, 1H), 7.63 (t, J = 7.2 Hz, 2H), 7.57 (d, J = 7.6 Hz, 1H), 7.41–7.36 (m, 5H), 6.65 (d, J = 8.8 Hz, 1H), 6.59 (s, 1H), 6.54 (d, J = 8.8 Hz, 1H), 5.08 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 188.9, 163.5, 158.3, 149.3, 136.0, 134.8, 134.6, 133.5, 132.9, 129.7, 129.6, 128.9, 128.5, 127.6, 124.7, 118.1, 116.0, 113.3, 110.0, 102.9, 70.5, 63.6; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C24H18NO3 368.1281, found 368.1283.

Brown solid, mp: 110.1–113.3 °C; 1H NMR (400 MHz, CDCl3) δ 9.34 (s, 1H), 8.36 (d, J = 6.8 Hz, 2H), 7.53 (d, J = 6.8 Hz, 2H), 7.40–7.36 (m, 5H), 6.64 (d, J = 8.4 Hz, 1H), 6.59 (s, 1H), 6.52 (d, J = 8.8 Hz, 1H), 5.09 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 188.8, 163.5, 158.2, 149.5, 148.5, 140.2, 136.0, 131.3, 129.6, 128.9, 128.5, 127.6, 124.6, 123.9, 115.9, 110.1, 102.9, 70.5, 63.5; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C23H18NO5 388.1179, found 388.1180.

Yellow solid, mp: 89.2–92.1 °C; 1H NMR (400 MHz, CDCl3) δ 9.39 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.42–7.33 (m, 5H), 7.27–7.25 (m, 2H), 6.76 (d, J = 8.8 Hz, 1H), 6.57 (d, J = 2.4 Hz, 1H), 6.52 (dd, J = 2.4, 8.8 Hz, 1H), 5.07 (s, 2H), 5.06 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 189.7, 163.2, 158.3, 150.8, 136.2, 135.6, 131.7, 131.6, 129.9, 129.0, 128.8, 128.4, 127.6, 124.3, 116.5, 109.8, 102.7, 70.4, 63.7; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C23H18ClO3 377.0939, found 377.0940.

Yellow solid, mp: 91.2–93.7 °C; 1H NMR (400 MHz, CDCl3) δ 9.46 (s, 1H), 7.41–7.34 (m, 5H), 6.96 (d, J = 8.0 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 6.79 (s, 1H), 6.57 (s, 1H), 6.53 (d, J = 8.8 Hz, 1H), 5.08–5.02 (m, 4H), 3.95 (s, 3H), 3.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 190.4, 163.0, 158.4, 152.0, 150.0, 149.0, 136.3, 130.2, 128.8, 128.4, 127.6, 125.5, 124.0, 123.5, 117.0, 113.3, 111.0, 109.6, 102.5, 70.4, 63.9, 56.2, 56.1; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C25H23O5 403.1540, found 403.1544.

Yellow solid, mp: 102.5–103.1 °C; 1H NMR (400 MHz, CDCl3) δ 9.46 (s, 1H), 6.97–6.88 (m, 3H), 6.80 (s, 1H), 6.50 (s, 1H), 6.47 (d, J = 8.8 Hz, 1H), 5.06 (d, J = 30.8 Hz, 2H), 3.96 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 190.4, 163.9, 158.5, 152.1, 150.0, 148.9, 130.2, 125.6, 123.9, 123.5, 116.8, 113.3, 110.9, 108.9, 101.6, 63.9, 56.2, 56.1, 55.7; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C19H18NaO5 349.1046, found 349.1049.

Yellow solid, mp: 93.9–95.7 °C; 1H NMR (400 MHz, CDCl3) δ 9.42 (s, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.01 (dd, J = 2.4, 8.4 Hz, 1H), 6.90 (d, J = 7.6 Hz, 1H), 6.85–6.82 (m, 2H), 6.49 (d, J = 2.8 Hz, 1H), 6.44 (dd, J = 2.4, 8.8 Hz, 1H), 5.07 (d, J = 6.4 Hz, 2H), 3.83 (s, 3H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 190.3, 164.0, 159.6, 158.3, 151.9, 134.6, 130.2, 129.7, 123.9, 122.8, 116.5, 115.8, 114.8, 109.0, 101.6, 63.7, 55.7, 55.5; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C18H16NaO4 319.0948, found 319.0945.

Orange solid, mp: 100.3–101.8 °C; 1H NMR (400 MHz, CDCl3) δ 9.46 (s, 1H), 6.88 (d, J = 8.4 Hz, 1H), 6.55 (t, J = 2.2 Hz, 1H), 6.48 (d, J = 2.8 Hz, 1H), 6.46–6.43 (m, 3H), 5.06 (s, 2H), 3.81 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 190.2, 163.9, 160.8, 158.2, 151.9, 135.2, 130.1, 123.8, 116.3, 108.9, 108.4, 101.6, 101.1, 63.7, 55.7, 55.6; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C19H18NaO5 349.1067, found 349.1064.

Yellow solid, mp: 130.2–132.5 °C; 1H NMR (400 MHz, CDCl3) δ 9.48 (s, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.53 (s, 2H), 6.50–6.46 (m, 2H), 5.06 (s, 2H), 3.93 (s, 3H), 3.85 (s, 6H), 3.83 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 190.2, 164.0, 158.3, 153.3, 152.0, 138.6, 130.1, 128.7, 123.8, 116.4, 109.0, 107.6, 101.6, 63.7, 61.1, 56.4, 55.7; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C20H20NaO6 379.1152, found 379.1148.

Orange solid, mp: 135.6–137.4 °C; 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 7.80 (d, J = 7.6 Hz, 1H), 7.65–7.61 (m, 2H), 7.59–7.57 (m, 1H), 6.65 (d, J = 8.8 Hz, 1H), 6.52 (d, J = 2.4 Hz, 1H), 6.47 (dd, J = 2.4, 8.8 Hz, 1H), 5.08 (d, J = 2.8 Hz, 2H), 3.83 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.9, 164.4, 158.4, 149.5, 134.8, 134.6, 133.5, 132.9, 129.7, 129.6, 124.6, 118.1, 115.8, 113.3, 109.4, 101.9, 63.6, 55.8; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C18H13NNaO3 314.0788, found 378.0786.
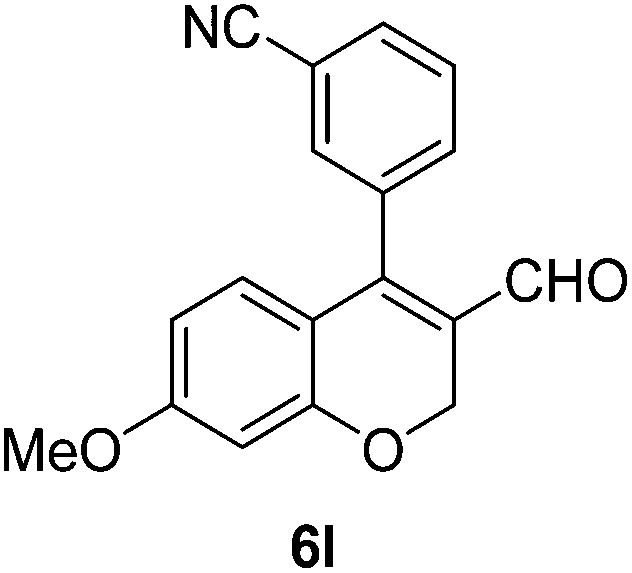
Yellow solid, mp: 99.3–102.2 °C; 1H NMR (400 MHz, CDCl3) δ 9.35 (s, 1H), 8.17 (d, J = 7.6 Hz, 1H), 8.01 (s, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 6.72 (d, J = 8.8 Hz, 1H), 6.51 (d, J = 2.4 Hz, 1H), 6.45 (dd, J = 2.4, 8.4 Hz, 1H), 5.09 (d, J = 1.6 Hz, 2H), 3.94 (s, 3H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.6, 166.5, 164.2, 158.4, 151.0, 134.6, 133.7, 131.3, 130.8, 130.4, 129.9, 128.8, 124.4, 116.3, 109.1, 101.7, 63.7, 55.8, 52.6; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C19H16NaO5 347.0898, found 347.0897.

Yellow solid, mp: 119.6–121.4 °C; 1H NMR (400 MHz, CDCl3) δ 9.36 (s, 1H), 8.07 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 6.71 (d, J = 8.4 Hz, 1H), 6.51 (d, J = 2.4 Hz, 1H), 6.44 (dd, J = 2.6, 8.6 Hz, 1H), 5.09 (s, 2H), 3.82 (s, 3H), 2.68 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.5, 189.5, 164.2, 158.3, 150.9, 138.2, 137.6, 130.7, 129.8, 128.5, 124.2, 116.1, 109.2, 101.8, 63.6, 55.8, 26.9; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C19H16NaO4 331.0945, found 331.0941.

Yellow solid, mp: 146.4–148.8 °C; 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1H), 7.60 (d, J = 7.2 Hz, 2H), 7.12 (d, J = 8.4 Hz, 1H), 6.53–6.49 (m, 3H), 5.02 (s, 2H), 3.83 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.6, 164.0, 158.5, 143.9, 143.5, 143.2, 129.7, 125.1, 117.5, 115.9, 112.7, 109.1, 101.7, 63.7, 55.7; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C15H12NaO4 279.0630, found 279.0628.

Yellow solid, mp: 129.2–130.4 °C; 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1H), 7.73 (d, J = 3.6 Hz, 1H), 7.20 (d, J = 3.6 Hz, 1H), 7.02–7.00 (m, 1H), 6.51–6.48 (m, 2H), 5.04 (s, 2H), 3.83 (s, 3H), 2.62 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 190.5, 188.9, 164.4, 158.2, 146.8, 143.5, 141.4, 132.1, 131.6, 129.6, 126.1, 115.9, 109.4, 101.8, 63.6, 55.8, 27.0; HRMS (ESI-QTOF) m/z [M + Na]+ calcd for C17H14NaO4S 337.0503, found 337.0505.

Yellow solid, mp: 122.4–123.8 °C; 1H NMR (400 MHz, CDCl3) δ 9.35 (s, 1H), 8.16 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 6.68 (s, 1H), 6.50 (s, 1H), 5.08 (s, 2H), 3.98 (s, 3H), 3.87 (s, 3H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 189.5, 166.6, 163.1, 156.7, 151.7, 138.2, 132.6, 130.9, 130.5, 129.7, 126.6, 123.7, 114.7, 100.1, 63.6, 55.5, 52.5, 34.6, 29.7; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C23H25O5 381.1697, found 381.1696.

Brown solid, mp: 81.3–83.8 °C; 1H NMR (400 MHz, CDCl3) δ 9.35 (s, 1H), 8.08 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 8.4 Hz, 2H), 6.68 (s, 1H), 6.50 (s, 1H), 5.08 (s, 2H), 3.87 (s, 3H), 2.69 (s, 3H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 197.6, 189.5, 163.1, 156.8, 151.6, 138.4, 137.6, 132.7, 130.7, 128.4, 126.6, 123.8, 114.6, 100.1, 63.6, 55.5, 34.6, 29.7, 26.9; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C23H25O4 365.1747, found 365.1752.

Brown solid, mp: 63.2–65.6 °C; 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1H), 7.74 (d, J = 3.6 Hz, 1H), 7.21 (d, J = 4.0 Hz, 1H), 6.99 (s, 1H), 6.49 (s, 1H), 5.03 (s, 2H), 3.87 (s, 3H), 2.63 (s, 3H), 1.24 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 190.6, 188.9, 163.4, 156.7, 146.8, 144.2, 141.7, 132.9, 132.1, 131.7, 126.3, 125.5, 114.4, 100.1, 63.6, 55.5, 34.7, 29.8, 27.0; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C21H23O4S 371.1312, found 371.1317.

A solution of 6h (1091 mg, 3.34 mmol) in ethyl acetate (11 mL) was hydrogenated under H2 balloon pressure in the presence of Pd–C (10% by weight, 35.58 mg) for 16 h. The reaction mixture was suction-filtered through a pad of Celite and the filtrate was concentrated to give the crude product which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 10
 :1:2 to 5:1:2) to give 7. White solid, mp: 79.6–80.8 °C; 1H NMR (400 MHz, CDCl3) δ 6.85 (d, J = 8.0 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.68 (s, 1H), 6.63 (d, J = 8.0 Hz, 1H), 6.45–6.43 (m, 2H), 4.23–4.19 (m, 2H), 4.08 (t, J = 10.4 Hz, 1H), 3.85 (s, 3H), 3.81 (s, 3H), 3.78 (s, 3H), 3.53–3.48 (m, 1H), 3.40–3.34 (m, 1H), 2.50–2.42 (m, 1H), 1.22 (t, J = 4.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 159.5, 155.6, 148.7, 148.0, 134.7, 131.4, 122.0, 116.5, 113.1, 111.0, 108.0, 101.1, 64.4, 61.7, 56.1, 56.0, 55.4, 42.2, 40.1; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H23O5 331.1540, found 331.1541.
:1:2 to 5:1:2) to give 7. White solid, mp: 79.6–80.8 °C; 1H NMR (400 MHz, CDCl3) δ 6.85 (d, J = 8.0 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.68 (s, 1H), 6.63 (d, J = 8.0 Hz, 1H), 6.45–6.43 (m, 2H), 4.23–4.19 (m, 2H), 4.08 (t, J = 10.4 Hz, 1H), 3.85 (s, 3H), 3.81 (s, 3H), 3.78 (s, 3H), 3.53–3.48 (m, 1H), 3.40–3.34 (m, 1H), 2.50–2.42 (m, 1H), 1.22 (t, J = 4.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 159.5, 155.6, 148.7, 148.0, 134.7, 131.4, 122.0, 116.5, 113.1, 111.0, 108.0, 101.1, 64.4, 61.7, 56.1, 56.0, 55.4, 42.2, 40.1; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H23O5 331.1540, found 331.1541.
To a stirred solution of 7 (920 mg, 2.80 mmol) in dry CH2Cl2 (10 mL) was added Dess–Martin periodinane (1426 mg, 3.36 mmol) at 0 °C. After being stirred at rt for 4 h, the reaction mixture was suction-filtered through a pad of Celite and the filtrate was concentrated under reduced pressure to give the residue which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 10
 :1:2 to 5:1:2) to give compound 8. Yellow solid, mp: 54.9–56.2 °C; 1H NMR (400 MHz, CDCl3) δ 9.75 (s, 1H), 6.90 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 6.63 (d, J = 8.0 Hz, 1H), 6.58 (s, 1H), 6.49–6.47 (m, 2H), 4.65 (d, J = 5.2 Hz, 1H), 4.43 (dd, J = 2.6, 11.4 Hz, 1H), 4.30 (t, J = 10.6 Hz, 1H), 3.83 (s, 3H), 3.79 (s, 6H), 3.19–3.16 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 200.9, 159.9, 155.4, 149.0, 148.4, 133.7, 131.2, 121.8, 115.1, 112.7, 111.2, 108.5, 101.3, 62.1, 56.0, 56.0, 55.5, 50.2, 41.3; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H21O5 329.1384, found 329.1386.
:1:2 to 5:1:2) to give compound 8. Yellow solid, mp: 54.9–56.2 °C; 1H NMR (400 MHz, CDCl3) δ 9.75 (s, 1H), 6.90 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 6.63 (d, J = 8.0 Hz, 1H), 6.58 (s, 1H), 6.49–6.47 (m, 2H), 4.65 (d, J = 5.2 Hz, 1H), 4.43 (dd, J = 2.6, 11.4 Hz, 1H), 4.30 (t, J = 10.6 Hz, 1H), 3.83 (s, 3H), 3.79 (s, 6H), 3.19–3.16 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 200.9, 159.9, 155.4, 149.0, 148.4, 133.7, 131.2, 121.8, 115.1, 112.7, 111.2, 108.5, 101.3, 62.1, 56.0, 56.0, 55.5, 50.2, 41.3; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H21O5 329.1384, found 329.1386.
To a stirred solution of 8 (743 mg, 2.26 mmol) in THF (20 mL) were added NaClO2 (2047 mg, 22.63 mmol), NaH2PO4 (3123 mg, 22.63 mmol) in H2O (10 mL), and 2-methyl-2-butene (5 mL) at rt. After being stirred at rt for 4 h, the reaction mixture was concentrated under reduced pressure and diluted with CH2Cl2 (20 mL) and H2O (20 mL). The water layer was extracted with CH2Cl2 (10 mL × 2) two more times. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a crude acid. To a solution of the crude acid (30 mg, 0.087 mmol) in CHCl3 (2 mL) was dropwise added POCl3 (0.041 mL, 0.44 mmol) at 0 °C. After being stirred at 80 °C for 7 h, the reaction mixture was cooled down to rt, and diluted with CH2Cl2 (3 mL) and H2O (3 mL). The water layer was extracted with CH2Cl2 (3 mL × 2) two more times. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a crude mixture which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 10
 :1:2 to 5:1:2) to give 2h. White solid, mp: 147.2–148.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.4 Hz, 1H), 7.16 (s, 1H), 6.98 (s, 1H), 6.63 (dd, J = 2.4, 8.4 Hz, 1H), 6.41 (d, J = 2.4 Hz, 1H), 4.61 (dd, J = 2.8, 11.2 Hz, 1H), 4.55 (d, J = 7.6 Hz, 1H), 4.07 (dd, J = 4.6, 11.0 Hz, 1H), 3.96 (s, 3H), 3.89 (s, 3H), 3.74 (s, 3H), 3.23–3.19 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 215.8, 168.3, 159.5, 156.8, 156.1, 152.2, 129.4, 128.3, 117.5, 109.5, 107.0, 104.2, 103.0, 67.3, 56.4, 56.3, 55.5, 49.6, 37.9; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H19O5 327.1227, found 327.1225.
:1:2 to 5:1:2) to give 2h. White solid, mp: 147.2–148.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.4 Hz, 1H), 7.16 (s, 1H), 6.98 (s, 1H), 6.63 (dd, J = 2.4, 8.4 Hz, 1H), 6.41 (d, J = 2.4 Hz, 1H), 4.61 (dd, J = 2.8, 11.2 Hz, 1H), 4.55 (d, J = 7.6 Hz, 1H), 4.07 (dd, J = 4.6, 11.0 Hz, 1H), 3.96 (s, 3H), 3.89 (s, 3H), 3.74 (s, 3H), 3.23–3.19 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 215.8, 168.3, 159.5, 156.8, 156.1, 152.2, 129.4, 128.3, 117.5, 109.5, 107.0, 104.2, 103.0, 67.3, 56.4, 56.3, 55.5, 49.6, 37.9; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H19O5 327.1227, found 327.1225.
To a solution of 2h (34 mg, 0.104 mmol) in THF/MeOH (1/1, 2 mL) was added NaBH4 (5.91 mg, 0.156 mmol) at 0 °C. After being stirred at rt for 1 h, the reaction mixture was quenched with sat. NH4Cl (1 mL) and concentrated under reduced pressure. The residue was diluted with CH2Cl2 (3 mL) and H2O (3 mL). The water layer was extracted with CH2Cl2 (3 mL × 2) two more times. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give 9. White solid, mp: 157.3–158.9 °C; 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 8.4 Hz, 1H), 6.94 (s, 1H), 6.85 (s, 1H), 6.62 (dd, J = 2.0, 8.4 Hz, 1H), 6.45 (d, J = 2.4 Hz, 1H), 5.51 (t, J = 7.0 Hz, 1H), 4.37 (dd, J = 4.4, 11.2 Hz, 1H), 4.10 (d, J = 6.8 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.86–3.80 (m, 1H), 3.77 (s, 3H), 3.14–3.07 (m, 1H), 2.08 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 159.4, 155.8, 149.8, 149.2, 136.4, 134.1, 130.4, 116.0, 108.3, 107.3, 107.0, 102.1, 76.6, 64.5, 56.2, 55.5, 42.5, 40.6; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H21O5 329.1384, found 329.1386.

To a solution of 9 (10 mg, 0.031 mmol) in acetone (1 mL) was added a drop of 15% HCl solution at rt. After being stirred at 50 °C for 1 h, the reaction mixture was quenched with Et3N (0.5 mL) at 0 °C and concentrated in vacuo to give a residue which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 10
 :1:2) to furnish 10. White solid, mp: 137.9–139.4 °C; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.4 Hz, 1H), 7.35 (s, 1H), 7.10 (s, 1H), 6.60 (dd, J = 2.4, 8.4 Hz, 1H), 6.56 (d, J = 2.4 Hz, 1H), 5.17 (s, 2H), 3.99 (s, 3H), 3.93 (s, 3H), 3.82 (s, 3H), 3.38 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 160.1, 155.2, 148.2, 147.3, 136.2, 134.4, 133.0, 131.8, 123.5, 114.8, 108.5, 107.0, 104.9, 102.7, 67.4, 56.6, 56.4, 55.5, 37.6; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H19O4 311.1278, found 311.1273.
:1:2) to furnish 10. White solid, mp: 137.9–139.4 °C; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.4 Hz, 1H), 7.35 (s, 1H), 7.10 (s, 1H), 6.60 (dd, J = 2.4, 8.4 Hz, 1H), 6.56 (d, J = 2.4 Hz, 1H), 5.17 (s, 2H), 3.99 (s, 3H), 3.93 (s, 3H), 3.82 (s, 3H), 3.38 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 160.1, 155.2, 148.2, 147.3, 136.2, 134.4, 133.0, 131.8, 123.5, 114.8, 108.5, 107.0, 104.9, 102.7, 67.4, 56.6, 56.4, 55.5, 37.6; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H19O4 311.1278, found 311.1273.
From 10: A solution of 10 (13 mg, 0.04 mmol) in ethyl acetate/acetone (2 mL, 1/1) was hydrogenated under H2 balloon pressure in the presence of Pd–C (10% by weight, 1.3 mg) at rt for 2 h. The reaction mixture was suction-filtered through a pad of Celite and the filtrate was concentrated to give the crude product which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 10
 :1:2) to afford 11.
:1:2) to afford 11.
From
2h: A solution of 2h (66 mg, 0.20 mmol) in ethyl acetate (5 mL) was hydrogenated under H2 balloon pressure in the presence of Pd–C (10% by weight, 2.13 mg) at rt for 16 h. The reaction mixture was suction-filtered through a pad of Celite and the filtrate was concentrated to give the crude product which was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 10:1:2) to afford 11. White solid, mp: 55.1–56.5 °C; 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8.4 Hz, 1H), 6.87 (s, 1H), 6.76 (s, 1H), 6.60 (dd, J = 2.6, 8.6 Hz, 1H), 6.43 (d, J = 2.4 Hz, 1H), 4.22 (d, J = 6.8 Hz, 1H), 4.12 (dd, J = 4.6, 11.0 Hz, 1H), 3.84 (s, 6H), 3.77 (s, 3H), 3.62 (t, J = 10.6 Hz, 1H), 3.17 (dd, J = 7.2, 15.6 Hz, 1H), 2.95–2.88 (m, 1H), 2.60 (dd, J = 1.8, 15.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 159.2, 155.6, 148.5, 148.3, 137.4, 132.7, 130.8, 116.0, 108.5, 108.1, 108.0, 101.9, 67.0, 56.2, 56.2, 55.4, 43.3, 37.0, 34.1; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C19H21O4 313.1434, found 313.1430.
To a stirred solution of 11 (18 mg, 0.058 mmol) in CH2Cl2 (3 mL) was dropwise added BBr3 (1 M in CH2Cl2, 0.29 mL, 0.29 mmol) at −78 °C. After being stirred at −78 °C for 1 h, the reaction mixture was stirred at rt for an additional 1 h. After being quenched with MeOH (3 mL), the mixture was concentrated under reduced pressure and diluted with ethyl acetate (3 mL) and H2O (3 mL). The water layer was extracted with ethyl acetate (3 mL × 2) two more times. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate/dichloromethane = 3
 :1:1 to 1:1:1) to give brazilane. Red solid, mp: 152.1–153.7 °C; 1H NMR (400 MHz, acetone-d6) δ 8.16 (s, 1H), 7.63 (s, 1H), 7.56 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 6.82 (s, 1H), 6.70 (s, 1H), 6.49 (dd, J = 2.6, 8.2 Hz, 1H), 6.28 (d, J = 2.4 Hz, 1H), 4.10 (d, J = 6.8 Hz, 1H), 4.06 (dd, J = 4.4, 10.8 Hz, 1H), 3.50 (t, J = 10.6 Hz, 1H), 3.05 (dd, J = 7.2, 15.6 Hz, 1H), 2.84–2.79 (m, 1H), 2.52 (dd, J = 2.0, 15.6 Hz, 1H); 13C NMR (100 MHz, acetone-d6) δ 157.5, 156.4, 145.0, 144.7, 137.9, 132.8, 131.8, 116.2, 112.6, 112.3, 109.4, 104.0, 67.2, 43.6, 37.8, 34.2; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C16H15O4 271.0965, found 271.0971.
:1:1 to 1:1:1) to give brazilane. Red solid, mp: 152.1–153.7 °C; 1H NMR (400 MHz, acetone-d6) δ 8.16 (s, 1H), 7.63 (s, 1H), 7.56 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 6.82 (s, 1H), 6.70 (s, 1H), 6.49 (dd, J = 2.6, 8.2 Hz, 1H), 6.28 (d, J = 2.4 Hz, 1H), 4.10 (d, J = 6.8 Hz, 1H), 4.06 (dd, J = 4.4, 10.8 Hz, 1H), 3.50 (t, J = 10.6 Hz, 1H), 3.05 (dd, J = 7.2, 15.6 Hz, 1H), 2.84–2.79 (m, 1H), 2.52 (dd, J = 2.0, 15.6 Hz, 1H); 13C NMR (100 MHz, acetone-d6) δ 157.5, 156.4, 145.0, 144.7, 137.9, 132.8, 131.8, 116.2, 112.6, 112.3, 109.4, 104.0, 67.2, 43.6, 37.8, 34.2; HRMS (ESI-QTOF) m/z [M + H]+ calcd for C16H15O4 271.0965, found 271.0971.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2017R1A2A2A05069364).References
- (a) J. Clardy and C. Walsh, Nature, 2004, 432, 829 CrossRef CAS PubMed; (b) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2014, 79, 629 CrossRef PubMed; (c) M. S. Butler, A. A. B. Robertson and M. A. Cooper, Nat. Prod. Rep., 2014, 31, 1612 RSC; (d) T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531 CrossRef CAS PubMed.
- (a) M. Pascolutti and R. J. Quinn, Drug Discovery Today, 2014, 19, 215 CrossRef CAS PubMed; (b) S. Rizzo and H. Waldmann, Chem. Rev., 2014, 114, 4621 CrossRef CAS PubMed; (c) J. Szychowski, J.-F. Truchon and Y. L. Bennani, J. Med. Chem., 2014, 57, 9292 CrossRef CAS PubMed; (d) M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302 RSC; (e) B. L. DeCorte, J. Med. Chem., 2016, 59, 9295 CrossRef CAS PubMed; (f) M. W. P. Bebbington, Chem. Soc. Rev., 2017, 46, 5059 RSC.
- (a) B. M. Abegaz, J. Mutanyatta-Comar and M. Nindi, Nat. Prod. Commun., 2007, 2, 475 CAS; (b) L.-G. Lin, Q.-Y. Liu and Y. Ye, Planta Med., 2014, 80, 1053 CrossRef CAS PubMed.
- (a) Y. Jung and I. Kim, J. Org. Chem., 2015, 80, 2001 CrossRef CAS PubMed; (b) Y. Jung and I. Kim, Org. Biomol. Chem., 2015, 13, 4331 RSC; (c) D. K. Singh, Y. Jung and I. Kim, Synthesis, 2017, 49, 1531 CAS.
- For reviews, see: (a) L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996–4028 CrossRef CAS PubMed; (b) V. V. Kouznetsov and A. Gómez-Barrio, Eur. J. Med. Chem., 2009, 44, 3091–3113 CrossRef CAS PubMed; (c) K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422 CrossRef CAS PubMed; (d) S. M. Shaveta and S. Palwinder, Eur. J. Med. Chem., 2016, 124, 500 CrossRef CAS PubMed.
- For the synthesis of other tetracyclic homoisoflavanoid analogues, see: (a) M. Fies and K. Fredrich, J. Prakt. Chem./Chem.-Ztg., 1995, 337, 596 CrossRef CAS; (b) C. Pan, X. Zeng, Y. Guan, X. Jiang, L. Li and H. Zhang, Synlett, 2011, 425 CAS; (c) H. Ishii, H. Koyama, K. Hagiwara, T. Miura, G. Xue, Y. Hashimoto, G. Kitahara, Y. Aida and M. Suzuki, Bioorg. Med. Chem. Lett., 2012, 22, 1469 CrossRef CAS PubMed.
- (a) S. A. Patil, R. Patil and S. A. Patil, Eur. J. Med. Chem., 2017, 138, 182 CrossRef CAS PubMed; (b) M. Turek, D. Szczȩsna, M. Koprowski and P. Baɫczewski, Beilstein J. Org. Chem., 2017, 13, 451 CrossRef CAS PubMed.
- (a) H. C. Shen, Tetrahedron, 2009, 65, 3931 CrossRef CAS; (b) S. Emami and Z. Ghanbarimasir, Eur. J. Med. Chem., 2015, 93, 539 CrossRef CAS PubMed; (c) J. Reis, A. Gaspar, N. Milhazes and F. Borges, J. Med. Chem., 2017, 60, 7941 CrossRef CAS PubMed.
- (a) E. Fillion and D. Fishlock, Tetrahedron, 2009, 65, 6682 CrossRef CAS; (b) K. Kim and I. Kim, Org. Lett., 2010, 12, 5314 CrossRef CAS PubMed; (c) H. F. Motiwala, R. H. Verkariya and J. Aube, Org. Lett., 2015, 17, 5484 CrossRef CAS PubMed; (d) D. Sun, B. Li, J. Lan, Q. Huang and J. You, Chem. Commun., 2016, 52, 3635 RSC.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- G. Jones and S. P. Stanforth, Org. React, Hoboken, NJ, US, 2000, vol. 56, p. 355 Search PubMed.
- 5a and 5b are commercially available. For the syntheses of 5a and 5b, see: (a) K. Koch and M. S. Biggers, J. Org. Chem., 1994, 59, 1216 CrossRef CAS; (b) C.-T. Yen, K. Nakagawa-Goto, T.-L. Hwang, P.-C. Wu, S. Morris-Natschke, W.-C. Lai, K. F. Bastow, F.-R. Chang, Y.-C. Wu and K.-H. Lee, Bioorg. Med. Chem. Lett., 2010, 20, 1037 CrossRef CAS PubMed; (c) Y. Pourshojaei, A. Gouranourimi, S. Hekmat, A. Asadipour, S. Rahmani-Nezad, A. Moradi, H. Nadri, F. H. Moghadam, S. Emami, A. Fouroumadi and A. Shafiee, Eur. J. Med. Chem., 2015, 97, 181 CrossRef CAS PubMed. For the synthesis of 5c, see: (d) P. Cozzi, U. Branzoli, P. P. Lovisolo, G. Orsini, G. Carganico, A. Pillan and A. Chiari, J. Med. Chem., 1986, 29, 404 CrossRef CAS PubMed.
- (a) V. V. Mezhnev and R. C. Geivandov, Russ. Chem. Bull., 2011, 60, 2114 CrossRef CAS; (b) P. Gogoi, P. Bezboruah and R. Boruah, Eur. J. Org. Chem., 2013, 5032 CrossRef CAS; (c) B. Yadagiri, U. D. Hoagunda, R. Bantu, L. Nagarapu, C. G. Kumar, S. Pombala and B. Sridhar, Eur. J. Med. Chem., 2014, 79, 260 CrossRef CAS PubMed; (d) K. S. Hettie and T. E. Glass, Chem. – Eur. J., 2014, 20, 17488 CrossRef CAS PubMed.
- (a) D. Ashok, B. V. Lakshmi, S. Ravi, A. Ganesh and S. Adam, Chem. Heterocycl. Compd., 2015, 51, 462 CrossRef CAS.
- (a) P. B. Reddy, P. P. Singh, S. D. Sawant, S. Koul, S. C. Taneja and H. M. Sampath Kumar, ARKIVOC, 2006,(xiii), 142 CAS; (b) G. B. Womack, J. G. Angeles, V. E. Fanelli, B. Indradas, R. L. Snowden and P. Sonnay, J. Org. Chem., 2009, 74, 5738 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- (a) B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888 CrossRef CAS; (b) G. A. Kraus and M. J. Taschner, J. Org. Chem., 1980, 45, 1175 CrossRef CAS; (c) B. S. Bal, W. E. Childers Jr. and H. W. Pinnick, Tetrahedron, 1981, 37, 2091 CrossRef CAS.
- S. Lee, K.-H. Lim and C.-H. Cheon, Org. Lett., 2017, 19, 2785 CrossRef CAS PubMed.
- B. S. Min, T. D. Cuong, T. M. Hung, B. K. Min, B. S. Shin and M. H. Woo, Bioorg. Med. Chem. Lett., 2012, 22, 7436 CrossRef CAS PubMed.
- For synthesis, see: (a) F. Morsingh and R. Robinson, Tetrahedron, 1970, 26, 281 CrossRef CAS; (b) J. Xu and J. C. Yadan, Tetrahedron Lett., 1996, 37, 2421 CrossRef CAS; (c) C.-C. Lin, T.-M. Teng, C.-C. Tsai, H.-Y. Liao and R.-S. Liu, J. Am. Chem. Soc., 2008, 130, 16417 CrossRef CAS PubMed; (d) J. S. Yadav, A. K. Mishra and S. Das, Tetrahedron, 2014, 70, 7560 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of synthesized compounds. See DOI: 10.1039/c7ob02758c |
| This journal is © The Royal Society of Chemistry 2018 |