
The direct and highly diastereoselective synthesis of 3,4-epoxy-2-piperidones. Application to the total synthesis and absolute configurational assignment of 3α,4α-epoxy-5β-pipermethystine†
Urbano
Osorio-Nieto‡
a,
Laura Y.
Vázquez-Amaya‡
a,
Herbert
Höpfl
 b,
Leticia
Quintero
a and
Fernando
Sartillo-Piscil
*a
b,
Leticia
Quintero
a and
Fernando
Sartillo-Piscil
*a
aCentro de Investigación de la Facultad de Ciencias Químicas, Benemérita Universidad Autónoma de Puebla (BUAP), 14 Sur Esq. San Claudio, Col. San Manuel, 72570, Puebla, México. E-mail: fernando.sartillo@correo.buap.mx; Fax: +52222 2454972; Tel: +52 222 2955500 ext. 7391
bCentro de Investigaciones Químicas, Instituto de Investigación en Ciencias Básicas y Aplicadas, Universidad Autónoma del Estado de Morelos, Avenida Universidad 1001, Col. Chamilpa, 62209, Cuernavaca, México
First published on 21st November 2017
Abstract
The substrate-controlled asymmetric total synthesis and absolute configurational assignment of biologically active 3α,4α-epoxy-5β-pipermethystine, a minor component in the aerial parts of kava, has been achieved by featuring, as a key step, the environmentally friendly and direct synthesis of 2,3-epoxyamides from allyl amines. By using the chiron approach, first a carbohydrate-derived dehydropiperidine was prepared and subjected to a stereoselective tandem C–H/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C oxidation reaction. In this attempt, the required α,α-trans-epoxy-2-piperidone skeleton of the kava metabolite precursor was not achieved, although the tandem oxidation was highly stereoselective. However, starting from non-carbohydrate 3-hydroxy-4,5-dehydropiperidine, and using the same tandem oxidation, the target intermediate was obtained in high yield and complete unprecedented anti-stereoselectivity. Since the proposed mechanistic course of this tandem oxidation implies the transient formation of an α,β-unsaturated amide followed by the subsequent epoxidation reaction, this second approach supports the previously established biotransformation proposal of (−)-pipermethystine to (−)-3α,4α-epoxy-5β-pipermethystine.
C oxidation reaction. In this attempt, the required α,α-trans-epoxy-2-piperidone skeleton of the kava metabolite precursor was not achieved, although the tandem oxidation was highly stereoselective. However, starting from non-carbohydrate 3-hydroxy-4,5-dehydropiperidine, and using the same tandem oxidation, the target intermediate was obtained in high yield and complete unprecedented anti-stereoselectivity. Since the proposed mechanistic course of this tandem oxidation implies the transient formation of an α,β-unsaturated amide followed by the subsequent epoxidation reaction, this second approach supports the previously established biotransformation proposal of (−)-pipermethystine to (−)-3α,4α-epoxy-5β-pipermethystine.
Introduction
With approximately 3000 species reported so far, the family of Piperaceae constitutes a major group of flowering plants.1 The Piperaceae family is divided into more than ten genera such as Arthanthe, Ottonia, Macropiper, Zippelia, Peperomia, Manekia, Piperanthera, Piper and others.2 The genus of Piper, and in particular, Piper methysticum, has been used for centuries in religious ceremonies and social drinking in the South Pacific and more recently in Europe and North America.3 It is also recognized as a widespread source of biologically active natural compounds such as lactones,4 terpenes,5 lignans,6 hydroquinones,7 steroids,8 (dihydro)chalcones,9 amides and alkaloids.10 While some of these compounds have antioxidant,7a,11 anticonceptive,12 anti-inflammatory,13 antimicrobial,14 antidiabetic,15 and other16 pharmacological benefits, others such as awaine (1),17 pipermethystine (2),18 and 3α,4α-epoxy-5β-pipermethystine (3),17 are associated with health problems, e.g. nausea and hepatotoxicity.3a The truth is that the pharmacological studies of these alkaloids 1–3 are unclear (Fig. 1).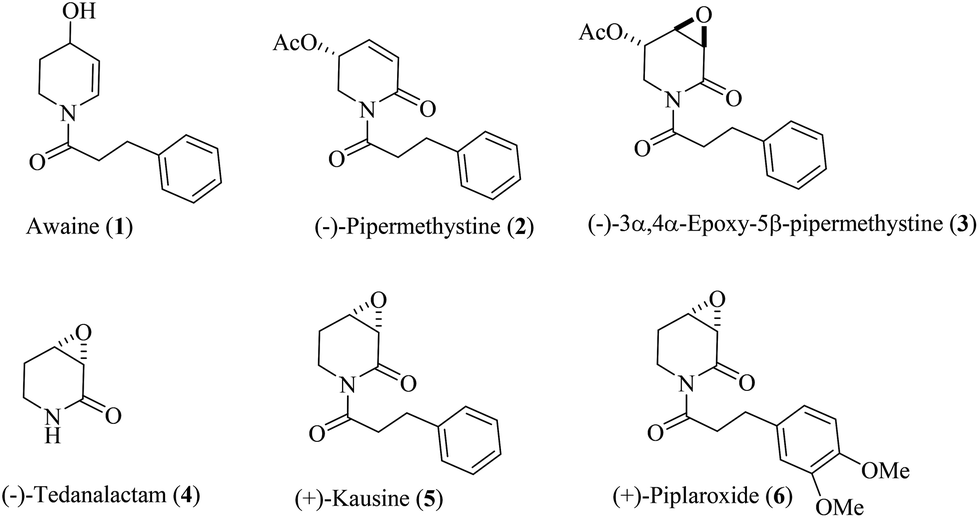 | ||
| Fig. 1 Piperidine alkaloids extracted from Piper: 1–3 from Piper methysticum, (−)-4 from Piper crassinervium, (+)-5 from Piper capense, and (+)-6 from Piper tuberculatum. | ||
Due to the low concentration of 3 in nature and the difficulty of plant collection in significant quantities, extensive biological tests have not been conducted so far. Unfortunately, addressing this problem by synthesis is not easy, especially the preparation in an enantiopure form. General methodologies for preparing 5-oxygenated-5,5-dihydro-2(1H)-pyridones19 or 5-oxygenated-3,4-epoxy-2-piperidones are not available. There is a single report from the group of Liebeskind20 revealing an enantiodivergent approach to 5-hydroxy-5,6-dihydro-2(1H)-pyridones, and the subsequent application to the total synthesis of the enantiomers of pipermethystine 2 in high optical purity and good overall yield (ee > 99%, 32%). However, subsequent efforts to transform pipermethystine (−)-2 into (−)-3α,4α-5β-epoxy-pipermethystine [(−)-3] via double-bond epoxidation have not been disclosed. This is probably due to the difficulty in achieving the epoxidation of α,β-unsaturated amides or lactams.21 A structurally less complex biologically active alkaloid such as (−)-tedanalactam (4)22 required synthesis routes of 7 or more than 10 steps.23 Others, such as (+)-kausine (5),24 which could be prepared by the acylation of tedanalactam, have not been synthesized in the laboratory, yet. Recently, by means of a direct chemical method to obtain 2,3-epoxyamides (glycidic amides) from allyl amines,25 the syntheses of (−)-4 and (+)-piplaroxide (6) have been achieved, not only in a few steps, but also by using an economical and eco-friendly approach.26 Although this methodology represents a convenient synthetic alternative for the epoxidation of unsaturated amides,27 the poor stereoselectivity associated with this protocol is an issue that needs to be addressed. To this end, the present work approaches this problem by replacing the chiral auxiliary strategy employed previously with a substrate-controlled strategy, in order to develop a protocol for the stereoselective synthesis of optically pure 5-oxygenated-3,4-epoxy-2-piperidones. In addition, this method was applied to achieve the first total synthesis and configurational assignment of (+)-3α,4α-epoxy-5β-pipermethystine [(−)-3].
Results and discussion
Previously, the chemical structure and relative configuration of (−)-3α,4α-epoxy-5β-pipermethystine [(−)-3] was established by Tang and co-workers based exclusively on NMR data analysis, revealing that the acetyloxy group at the C-5 position is anti-oriented to the epoxide ring.17 Accordingly, the carbohydrate-derived dehydropiperidine 7 might be a suitable precursor for the synthesis of either (−) or (+)-3. For this purpose, the chiron approach strategy was suggested because of two reasons. Firstly, the configurational integrity of the stereogenic center at C-4 can be preserved, which would provide certainty about the absolute configuration. Secondly, due to steric restrictions originating from the fused trans-dehydro-piperidine/furanose moiety and the substituent at the C-4 position, an anti-π-facial epoxidation reaction of 7 to 8 mediated by NaClO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 was anticipated. Finally, the synthesis of either (+) or (−)-3 would be accomplished by a sequential hydrolysis/oxidative C–C bond cleavage/decarboxylation protocol and acylation reactions (Scheme 1).
25 was anticipated. Finally, the synthesis of either (+) or (−)-3 would be accomplished by a sequential hydrolysis/oxidative C–C bond cleavage/decarboxylation protocol and acylation reactions (Scheme 1).
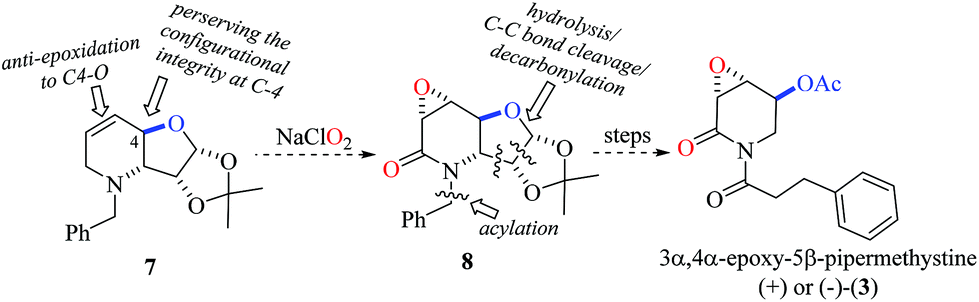 | ||
| Scheme 1 A chiron approach strategy to 3α,4α-epoxy-5β-pipermethystine [(+) or (−)-3]. | ||
To test the proposed synthetic strategy, starting from diacetone-D-glucose (DAG), chiron 7 was prepared in five-steps (Scheme 2). DAG was quantitatively oxidized to ketone 9 using aqueous NaOCl, TEMPO and Bu4NHSO4,28 and without further purification, ketone 9 was then subjected to reductive amination (benzylamine and TiCl4, followed by NaBH4) to give the secondary amine 10 in 76% overall yield. Applying the sequential SHOWO protocol29 (hydrolysis/oxidation/Wittig olefination), the respective allylamine 11 was obtained in moderate yield. Allylation of 11 with allyl bromide and Na2CO3 to yield diallyl amine 12, followed by the ring-closing metathesis reaction of 12·HCl with the Hoveyda–Grubbs 1st generation catalyst,30 afforded the desired carbohydrate-derived dehydropiperidine 7 (Scheme 2). Having prepared precursor 7, the direct tandem oxidation of the allyl amine function to the corresponding epoxyamide was explored.25
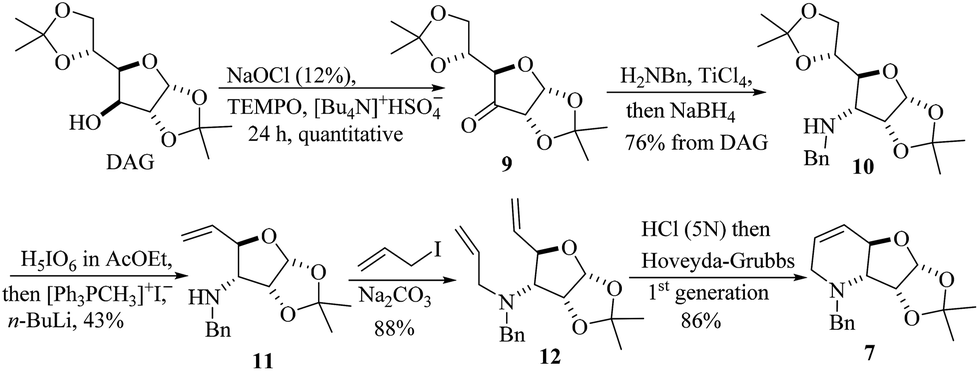 | ||
| Scheme 2 Synthesis of the carbohydrate-derived dehydropiperidine 7 starting from DAG. | ||
However, the stereoisomer obtained was not the expected product, but the reaction afforded the contrary stereoisomer in good chemical yield (75%) and absolute stereoselectivity. It seems, therefore, that the 1,2-O-isopropylidene moiety along with the axially oriented H-4, and not the C-4 substituent, is a determinant for the stereochemical outcome of the reaction. Thus, the intermediate A, which is presumably formed during the tandem reaction with NaClO2, undergoes preferential attack by ClO− on the exo-face for the epoxidation step to exclusively provide 13 (Scheme 3).
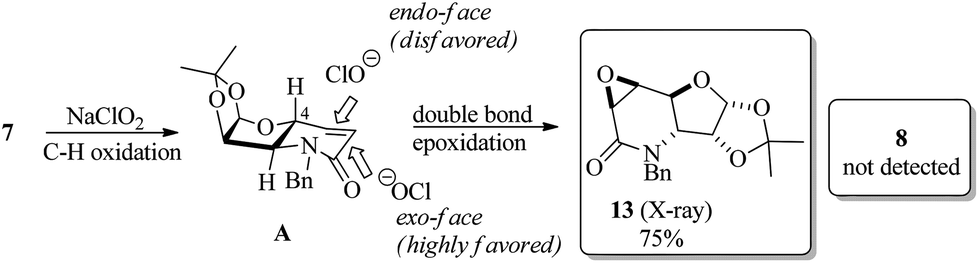 | ||
| Scheme 3 Substrate-controlled direct stereoselective synthesis of glycidic amide 13 from dehydropiperidine 7. | ||
Although the oxidation of 7 mediated by NaClO2 gave the undesired stereoisomer precursor for the total synthesis of 3α,4α-epoxy-5β-pipermethystine, the high stereoselectivity with which 13 was achieved indicates that the substrate-controlled strategy is effective for the stereoselective synthesis of the 3,4-epoxy-2-piperidones. Consequently, having in mind the total synthesis and absolute configuration determination of either (3R,4S,5S)-3 or (3S,4R,5R)-3, two further chiral dehydropiperidine precursors were prepared, i.e., (1′S,5R)-14 and (1′S,5S)-14. In this new approach, an anti-stereoselective epoxidation reaction (β-face) is expected due to the presence of the bulky tributyldimethylsilanoxy (OTBS) group in the allylic position. To enable the determination of the absolute configuration of the target natural product, (S)-4-methoxy-α-methylbenzylamine31 was employed as a chiral resolving agent (Scheme 4).
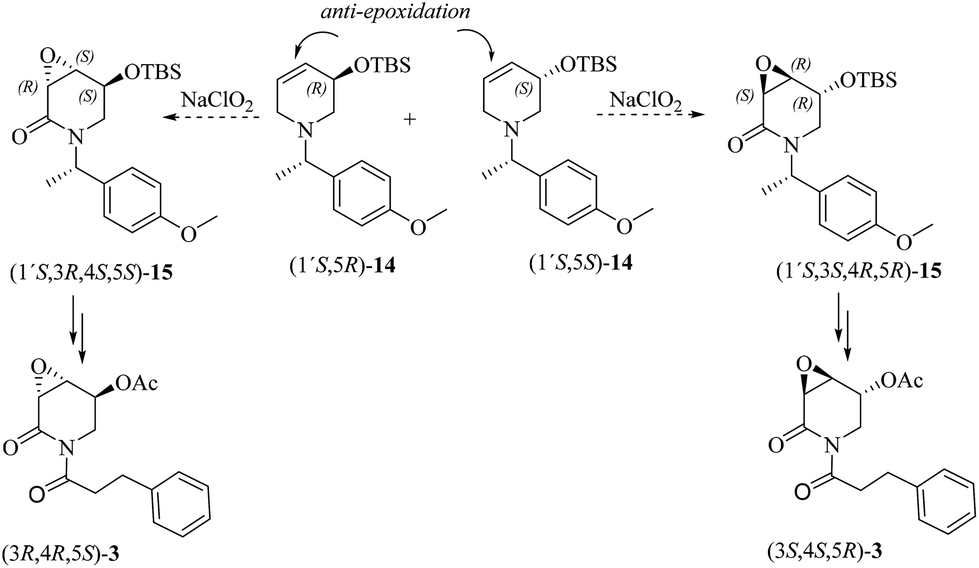 | ||
| Scheme 4 A second approach to the direct synthesis of 3α,4α-epoxy-5β-pipermethystines [(3R,4R,5S)-3 and (3S,4S,5R)-3] starting from 3-O-silyloxy-4,5-dehydropiperidines (1′S,5R)-14 and (1′S,5S)-14. | ||
The synthetic pathway to the 3-O-silyloxy-4,5-dehydropiperidines (1′S,5R)-14 and (1′S,5S)-14 is depicted in Scheme 5. trans-Cinnamaldehyde 16 was transformed into racemic α-silylnitrile (±)-17 upon treatment with TBSCl, KCN and ZnCl2.32 The conversion of (±)-17 to (1′S,5R)-14 and (1′S,5S)-14 was accomplished by following Brussee's synthetic route, which involves the transformation of unsaturated cyanohydrins into chiral unsaturated azaheterocycles via ring-closing metathesis.33 Accordingly, compound (±)-17 was first converted in good yield into the diastereomeric mixture of homoallyl amines (1′S)-18via a sequential protocol: (a) partial reduction of CN with DiBAL, (b) transimination with (S)-4-methoxy-α-methylbenzylamine [(S)-B] and NH4Br, and (c) imine reduction with NaBH4. Then, the diastereomeric mixture of homoallylic amines was subjected to a standard allylation reaction (allyl iodine, Na2CO3) to obtain the corresponding diastereomeric mixture of the tertiary unsaturated amines (1′S)-19, which were finally transformed into the desired 3-O-silyloxy-4,5-dehydropiperidines (1′S,5R)-14 and (1′S,5S)-14 by ring-closing metathesis using the Hoveyda–Grubbs 2nd generation catalyst (Scheme 5). At this stage, diastereoisomeric dehydropiperidines (1′S,5R)-14 and (1′S,5S)-14 were separated by chromatography.
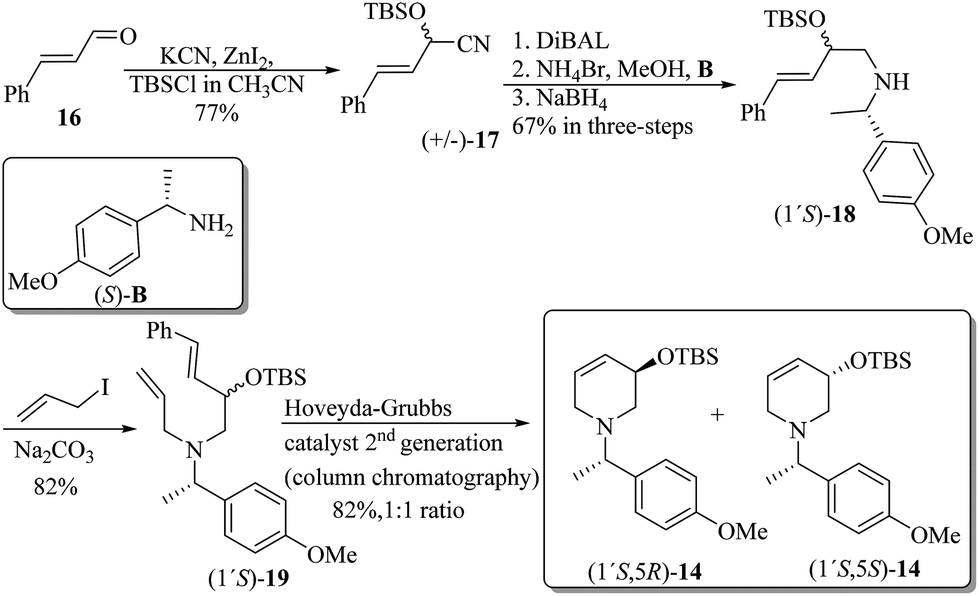 | ||
| Scheme 5 Synthesis of 3-O-silyloxy-4,5-dehydropiperidines (1′S,5R)-14 and (1′S,5S)-14 starting from trans-cinnamaldehyde 16. | ||
To further explore the oxidation reactions of the corresponding epoxyamides, allyl amines (1′S,5R)-14 and (1′S,5S)-14 were subjected to the above-described tandem oxidation reaction with NaClO2, to yield the respective 3α,4α-epoxy-5β-silyloxy-2-piperidones (1′S,3R,4S,5S)-15 and (1′S,3S,4R,5R)-15 in good yields and with the desired anti-stereoselectivity. Removal of the TBS group with tetrabutylammonium fluoride (TBAF) afforded the respective alcohols (1′S,3R,4R,5S)-20 and (1′S,3S,4S,5R)-20 as crystalline materials. Based on the chiral amine substituent of known configuration, single-crystal X-ray diffraction analysis enabled the assignment of the absolute configuration of the remaining stereogenic centers within the piperidine ring as depicted in Scheme 6. Interestingly, the 3,4-epoxy-5-hydroxypiperidin-2-ones (1′S,3R,4R,5S)-20 and (1′S,3S,4S,5R)-20 can also be obtained in similar yields by tandem oxidation with NaClO2 starting from (1′S,5R)-21 and (1′S,5S)-21, which are obtained by the removal of the TBS group from (1′S,5R)-14 and (1′S,5S)-14 (Scheme 6).
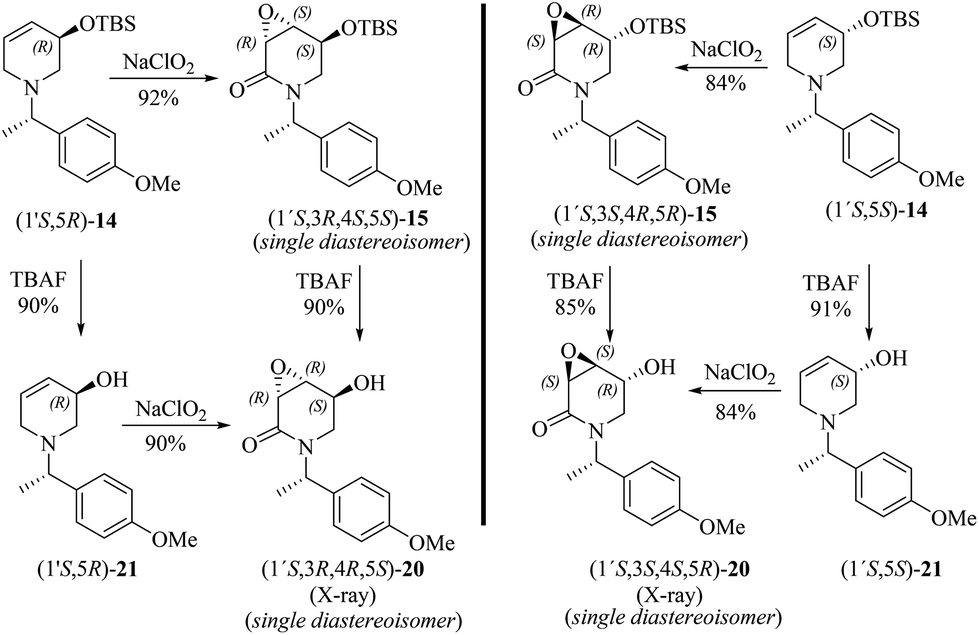 | ||
| Scheme 6 Highly diastereoselective tandem oxidation of both protected and unprotected 4,5-dehydropiperidines to the corresponding 3,4-epoxy-2-piperidones. | ||
Epoxidation reactions of allyl alcohols generally occur in a syn-stereoselective way with the hydroxyl group being responsible for the stereoinduction by means of hydrogen bonding with the oxidation reagent.34 As far as we know, the oxidation of (1′S,5R)-21 and (1′S,5S)-21 constitutes the first example of an epoxidation reaction of a six-membered cyclic allyl alcohol occurring in an anti-stereoselective manner.35 The lack of a hydrogen bonding interaction during the epoxidation step for (1′S,5R)-21 and (1′S,5S)-21 suggests that the half-chair conformers C and D, formed after the C–H oxidation step of 14 and 21, respectively, have the lowest energy when the OR substituent is oriented in a pseudo-equatorial position leaving thus the pseudo-axial position prone to be epoxidated. In this conformation, the presence of an intramolecular C–H⋯O hydrogen-bonding interaction between the C–H benzylic hydrogen atom and the oxygen atom from the dehydropiperidone carbonyl group might exert a further stabilization effect on the key intermediates C and D.36 This hypothesis is somewhat supported by the observation of short C–H⋯O intramolecular contacts in the molecular structures extracted from the SXRD analyses of (1′S,3R,4R,5S)-20 and (1′S,3S,4S,5R)-20. As also seen in Scheme 7, the corresponding O⋯H distances are 2.32 and 2.27 Å, being significantly shorter than the sum of the van der Waals radii of hydrogen and oxygen (2.72 Å).
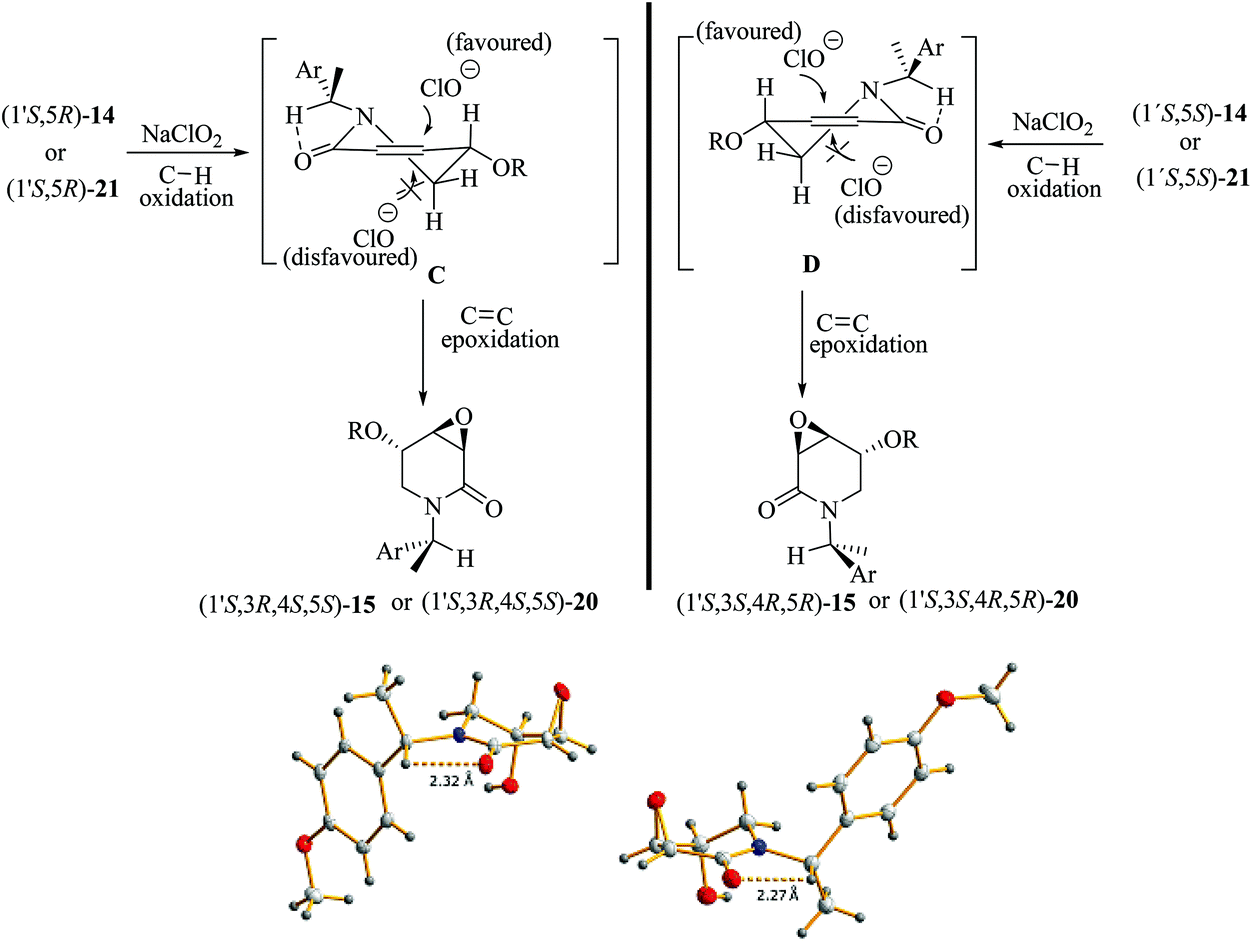 | ||
| Scheme 7 Diastereoselective epoxidation pathway of 14 and 21 to 15 and 20via the half-chair low energy conformers C and D. The molecular structures of (1′S,3R,4R,5S)-20 (left) and (1′S,3S,4S,5R)-20 (right) obtained by SXRD analysis exhibit short intramolecular C–H⋯O contacts. | ||
Starting from (1′S,3R,4R,5S)-20 and (1′S,3S,4S,5R)-20, the synthesis of 3α,4α-epoxy-5β-pipermethystine 3 was achieved in three steps. First, acetylation with acetic anhydride (AcO2) in basic media provided (1′S,3R,4R,5S)-22 and (1′S,3S,4S,5R)-22, which by oxidative debenzylation with ceric ammonium nitrate (CAN) gave (3R,4R,5S)-23 and (3S,4S,5R)-23, respectively. A second acylation with hydrocinnamoyl chloride (24) in the presence of n-BuLi yielded (−) and (+)-3α,4α-epoxy-5β-pipermethystine with ∼34% overall yield for this three-step sequence (Scheme 8). In addition, the stereoselective synthesis enabled establishing that the naturally occurring alkaloid (−)-3 possesses the absolute configuration 3R,4R,5S, by means of polarimetry. The NMR data of the enantiomers synthesized herein are in full agreement with the data and structure reported by the Tang group.17
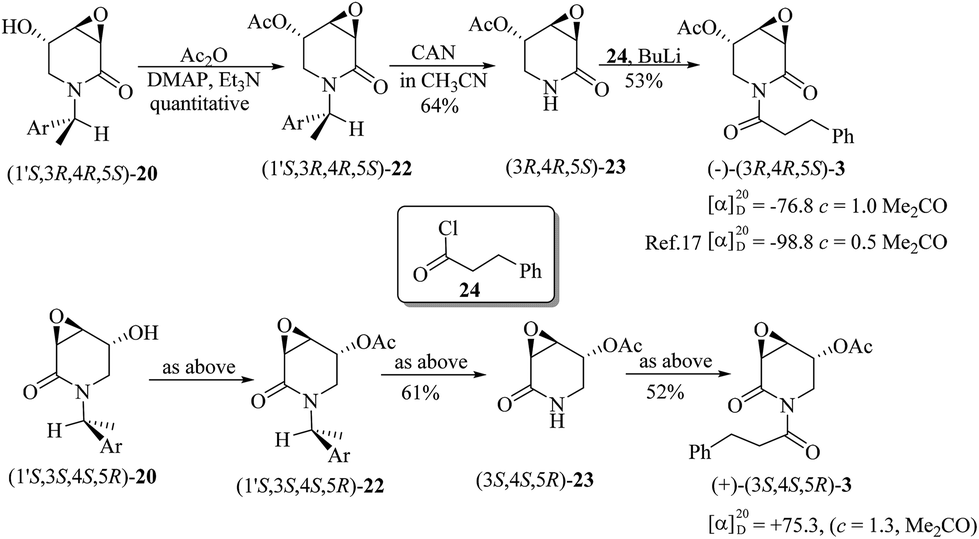 | ||
| Scheme 8 Completion of the total synthesis of (−) and (+)-3α,4α-epoxy-5β-pipermethystine, enabling the determination of the absolute configuration of (−)-3. | ||
Because of the close chemical structural relationship between (−)-pipermethystine 2 and (−)-3α,4α-epoxy-5β-pipermethystine 3, it has been suggested that the latter is biosynthesized from the former via enzymatic epoxidation.17 However, due to the intrinsic complications involved in achieving the epoxidation of α,β-unsaturated lactams by synthetic methods,21 this hypothesis has not been chemically proved yet. The finding that naturally occurring 3α,4α-epoxy-5β-pipermethystine 3 preserves the configurational integrity at C-5 of pipermethystine 2 provides the missing experimental proof for the hypothetical biosynthesis of (−)-3 from (−)-2 (Scheme 9).
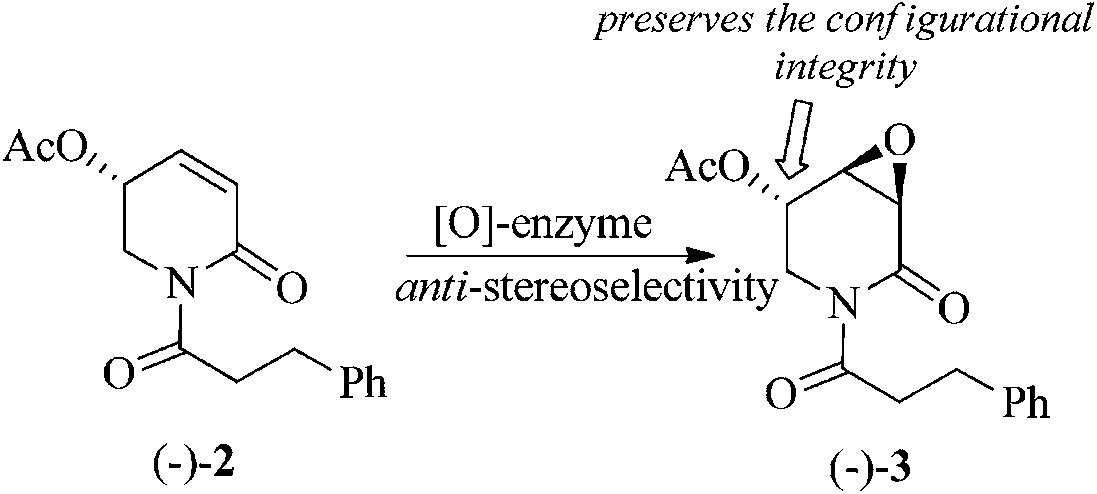 | ||
| Scheme 9 Proposal for the biosynthetic pathway to (−)-3 from the alkaloid (−)-2. | ||
Conclusions
A direct and stereoselective protocol for the synthesis of 5-oxygenated-3,4-epoxy-piperidin-2-ones from chiral dehydropiperidines was achieved. Another remarkable feature of this stereoselective reaction mediated by NaClO2 is that a π-facial anti-stereoselective epoxidation occurs, even though a free hydroxyl group is present at the allylic position. Based on the elementary synthetic tandem oxidation protocol established herein, the first total synthesis of (−)-3α,4α-epoxy-5β-pipermethystine was accomplished. In addition, the absolute configuration of this biologically active natural product was established, which allowed one to support the previously proposed biosynthetic enzyme-mediated oxidation of (−)-pipermethystine into (−)-3α,4α-epoxy-5β-pipermethystine.Experimental section
Commercially available reagents were used without further purification. Column chromatography (CC) was performed using silica gel (200–300 mesh) with solvents indicated in the text. The melting points were determined on an open capillary tube and were uncorrected. The optical rotations were measured with a digital polarimeter using the sodium D line (589 nm) and are reported as degrees at 20 °C. The concentrations are given as g per 100 mL. Unless otherwise stated, 1H NMR and 13C NMR spectra were obtained on 500 MHz equipment. All samples were analyzed in CDCl3 with TMS as the internal reference using a relative scale in parts per million (ppm) for the chemical shift (δ) and Hz for the coupling constant (J). The splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quartet; qu, quintet; m, multiplet; and br, broad. The high-resolution mass spectra (HRMS) were acquired in the electron-impact (EI) mode using a TOF mass analyzer or in fast-atom-bombardment (FAB) mode using a QMS mass analyzer.(3R)-N-Benzyl-(3-deoxy-1,2:5,6-di-O-isopropylidene-α-D-glucofuranose)-3-yl amine (10)
To a solution of DAG (5.00 g, 19.2 mmol), TEMPO (0.60 g, 3.84 mmol) and TBAHS (1.30 g, 3.84 mmol) in 50 mL of CH2Cl2 at 0 °C was added dropwise a solution of NaOCl (23.8 mL, 38.4 mmol) at 12% in water. The solution was stirred for 1 hour, whereafter four portions of a solution of NaOCl (11.9 mL, 19.2 mmol) were added within an interval of 2 h. The reaction was quenched after 24 hours by adding dropwise a solution of NaOH (5 mL, 1 N). After extraction with methylene chloride (4 × 10 mL), the combined organic phases were dried over Na2SO4 and concentrated at reduced pressure. The intermediate ketone 9 (6.00 g, 19.2 mmol) was obtained as a reddish crystalline material and subjected to the subsequent reaction step without further purification. To a balloon-bottom flask containing a solution of ketone 9 in dry THF (50 mL) at 0 °C TiCl4 (1.92 mL, 1.92 mmol, 1 M solution in toluene) was added slowly. Then, BnNH2 (2.51 mL, 23.04 mmol) was added dropwise and the mixture was allowed to stir at room temperature for 24 h. After evaporating the solvent under reduced pressure, the solid was washed with AcOEt until a white color was observed. The corresponding imine (7.2 g) was obtained as a reddish crystalline solid and was reduced without further purification. For the reduction, to a solution of the imine in MeOH (50 mL) at 0 °C was added a solution of NaBH4 (1.79 g, 48 mmol) in MeOH (10 mL) in three portions. After stirring at room temperature for 12 h, the reaction was quenched by adding a saturated aqueous solution of NH4Cl (5 mL) followed by AcOEt (30 mL) and water (5 mL). The solid was washed with AcOEt (4 × 10 mL), whereupon the combined organic portions were dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by column chromatography using a mixture of hexane :AcOEt (5:1, v/v). The secondary amine 10 was obtained as a colorless crystalline solid (5.09 g, 76%). Rf = 0.53 (2:1 hexane/AcOEt). Mp = 75–77 °C. [α]20D = +113.8 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38–7.31 (m, 5H), 5.78 (d, J = 4.0 Hz, 1H), 4.59 (t, J = 4.5 Hz, 1H), 4.36 (td, J = 7.0, 3.0 Hz, 1H), 4.19 (t, J = 8.0 Hz, 1H), 3.97 (d, J = 13.0 Hz, 1H), 3.93 (dd, J = 8.0, 6.5 Hz, 1H), 3.84 (dd, J = 9.5, 3.5 Hz, 1H), 3.76 (d, J = 13.0 Hz, 1H), 3.07 (dd, J = 9.5, 4.5 Hz, 1H), 1.56 (s, 1H), 1.53 (s, 3H), 1.40 (s, 3H), 1.38 (s, 3H), 1.35 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ (ppm): 140.0 (ipso), 128.4 (ortho), 128.1 (meta), 127.1 (para), 112.0 [(CH3)2
:AcOEt (5:1, v/v). The secondary amine 10 was obtained as a colorless crystalline solid (5.09 g, 76%). Rf = 0.53 (2:1 hexane/AcOEt). Mp = 75–77 °C. [α]20D = +113.8 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38–7.31 (m, 5H), 5.78 (d, J = 4.0 Hz, 1H), 4.59 (t, J = 4.5 Hz, 1H), 4.36 (td, J = 7.0, 3.0 Hz, 1H), 4.19 (t, J = 8.0 Hz, 1H), 3.97 (d, J = 13.0 Hz, 1H), 3.93 (dd, J = 8.0, 6.5 Hz, 1H), 3.84 (dd, J = 9.5, 3.5 Hz, 1H), 3.76 (d, J = 13.0 Hz, 1H), 3.07 (dd, J = 9.5, 4.5 Hz, 1H), 1.56 (s, 1H), 1.53 (s, 3H), 1.40 (s, 3H), 1.38 (s, 3H), 1.35 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ (ppm): 140.0 (ipso), 128.4 (ortho), 128.1 (meta), 127.1 (para), 112.0 [(CH3)2![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) –], 109.5 [(CH3)2–], 104.4 (C1), 78.9 (C4), 77.8 (C2), 75.6 (C5), 65.0 (C6), 61.0 (C3), 52.2 (PhCH2–), 26.7 (–CH3), 26.5 (–CH3), 26.2 (–CH3), 25.4 (–CH3). HRMS-EI m/z 349.1880 (calcd for C19H27NO5, 349.1889).
–], 109.5 [(CH3)2–], 104.4 (C1), 78.9 (C4), 77.8 (C2), 75.6 (C5), 65.0 (C6), 61.0 (C3), 52.2 (PhCH2–), 26.7 (–CH3), 26.5 (–CH3), 26.2 (–CH3), 25.4 (–CH3). HRMS-EI m/z 349.1880 (calcd for C19H27NO5, 349.1889).
(3aR,5R,6R,6aR)-N-Benzyl-2,2-dimethyl-5-vinyltetrahydrofuro[2,3-d][1,3]dioxol-6-amine (11)
To a solution of 10 (0.75 g, 2.14 mmol) in dry AcOEt (15 mL) at 0 °C was added H5IO6 (0.97 g, 4.28 mmol). The mixture was stirred at 0 °C for ten minutes and then for an additional 90 minutes at room temperature. The reaction was quenched by adding a saturated aqueous solution of NaHCO3 (1.5 mL), whereupon the organic phase was extracted with AcOEt (4 × 5 mL). After drying the combined organic phases over Na2SO4 and concentrating under reduced pressure, the residue was dissolved in dry THF and added dropwise to a solution of the corresponding Wittig reagent, which was prepared from methyltriphenylphosphonium iodide (1.32 g, 3.1 mmol) and n-BuLi (2.58 mL, 3.1 mmol, 1.2 M solution in hexane). The reaction mixture was allowed to react for 10 h before adding 3 mL of a saturated aqueous solution of NaHCO3. The organic phase was separated, and the aqueous phase was extracted with AcOEt (4 × 10 mL). The combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. Purification by column chromatography (SiO2, hexane/AcOEt, 10 :1, v/v) yielded 0.253 g (43%) of 11 as a viscous yellow oil. Rf = 0.47 (3:1 hexane/AcOEt). [α]20D = +76.51 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.37–7.30 (m, 4H), 7.27–7.23 (m, 1H), 5.86 (ddd, J = 17.0, 10.5, 6.5 Hz, 1H), 5.78 (d, J = 4.0 Hz, 1H), 5.45 (dt, J = 17.0, 1.5 Hz, 1H), 5.26 (ddd, J = 10.5, 1.5, 1.0 Hz, 1H), 4.56 (t, J = 4.0 Hz, 1H), 4.12 (dd, J = 9.5, 6.5 Hz, 1H), 3.95 (d, J = 13.0 Hz, 1H), 3.82 (d, J = 13.5 Hz, 1H), 2.80 (dd, J = 9.5, 4.5 Hz, 1H), 1.89 (br, 1H), 1.54 (s, 3H), 1.35 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 139.9 (ipso), 135.3 (C5), 128.3 (ortho), 128.0 (meta), 127.0 (para), 118.2 (C6), 111.8 [(CH3)2–], 104.2 (C1), 80.7 (C4), 77.7 (C2), 65.1 (C3), 51.9 (PhCH2–), 26.6 (–CH3), 26.4 (–CH3). HRMS-EI m/z 276.1596 (calcd for C16H22NO3, 276.1599).
:1, v/v) yielded 0.253 g (43%) of 11 as a viscous yellow oil. Rf = 0.47 (3:1 hexane/AcOEt). [α]20D = +76.51 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.37–7.30 (m, 4H), 7.27–7.23 (m, 1H), 5.86 (ddd, J = 17.0, 10.5, 6.5 Hz, 1H), 5.78 (d, J = 4.0 Hz, 1H), 5.45 (dt, J = 17.0, 1.5 Hz, 1H), 5.26 (ddd, J = 10.5, 1.5, 1.0 Hz, 1H), 4.56 (t, J = 4.0 Hz, 1H), 4.12 (dd, J = 9.5, 6.5 Hz, 1H), 3.95 (d, J = 13.0 Hz, 1H), 3.82 (d, J = 13.5 Hz, 1H), 2.80 (dd, J = 9.5, 4.5 Hz, 1H), 1.89 (br, 1H), 1.54 (s, 3H), 1.35 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 139.9 (ipso), 135.3 (C5), 128.3 (ortho), 128.0 (meta), 127.0 (para), 118.2 (C6), 111.8 [(CH3)2–], 104.2 (C1), 80.7 (C4), 77.7 (C2), 65.1 (C3), 51.9 (PhCH2–), 26.6 (–CH3), 26.4 (–CH3). HRMS-EI m/z 276.1596 (calcd for C16H22NO3, 276.1599).
(3aR,5R,6R,6aR)-N-Allyl-N-benzyl-2,2-dimethyl-5-vinyltetrahydrofuro-[2,3-d]-[1,3]dioxol-6-amine (12)
To a mixture of 11 (0.18 g, 0.66 mmol) and Na2CO3 (0.33 g, 1.98 mmol) in dry MeCN (10 mL) was added dropwise a solution of allyl iodide (0.121 mL, 1.98 mmol) in MeCN (1 mL). The reaction mixture was stirred overnight at room temperature followed by reflux for two additional hours. After the removal of the solvent by distillation under reduced pressure, the resulting solid was washed with AcOEt (5 × 5 mL). The combined organic phases were filtered through Na2SO4, concentrated and purified by column chromatography (SiO2, hexane/AcOEt, 17 :1, v/v) to obtain 0.183 g of 12 (88%) as a viscous yellow liquid. Rf = 0.55 (8:1 hexane/AcOEt). [α]20D = +66.4 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.35–7.20 (m, 5H), 5.87–5.78 (m, 2H), 5.69 (d, J = 4.0 Hz, 1H), 5.42 (dt, J = 17.0, 1.5 Hz, 1H), 5.30 (dt, J = 10.5, 1.0 Hz, 1H), 5.19 (ddd, J = 17.0, 3.5, 1.5 Hz, 1H), 5.10 (ddd, J = 10.5, 3.5, 2.0 Hz, 1H), 4.67 (t, J = 4.0 Hz 1H), 4.64 (dd, J = 10.0, 7.0 Hz, 1H), 4.04 (d, J = 14.5 Hz, 1H), 3.83 (d, J = 14.5 Hz, 1H), 3.42 (dd, J = 14.5, 5.5 Hz, 1H), 3.34 (dd, J = 14.5, 7.0 Hz, 1H), 2.93 (dd, J = 10.0, 4.0 Hz, 1H), 1.58 (s, 3H), 1.34 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 140.2 (ipso), 136.8 (C8), 135.9 (C5), 128.3 (ortho), 128.1 (meta), 126.7 (para), 118.7 (C6), 116.9 (C9), 112.1 [(CH3)2–], 103.8 (C1), 79.1 (C2), 76.5 (C4), 66.7 (C3), 55.2 (PhCH2–), 54.8 (C7), 26.6 (–CH3), 26.2 (–CH3). HRMS-EI m/z 315.1825 (calcd for C19H25NO3, 315.1834).
:1, v/v) to obtain 0.183 g of 12 (88%) as a viscous yellow liquid. Rf = 0.55 (8:1 hexane/AcOEt). [α]20D = +66.4 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.35–7.20 (m, 5H), 5.87–5.78 (m, 2H), 5.69 (d, J = 4.0 Hz, 1H), 5.42 (dt, J = 17.0, 1.5 Hz, 1H), 5.30 (dt, J = 10.5, 1.0 Hz, 1H), 5.19 (ddd, J = 17.0, 3.5, 1.5 Hz, 1H), 5.10 (ddd, J = 10.5, 3.5, 2.0 Hz, 1H), 4.67 (t, J = 4.0 Hz 1H), 4.64 (dd, J = 10.0, 7.0 Hz, 1H), 4.04 (d, J = 14.5 Hz, 1H), 3.83 (d, J = 14.5 Hz, 1H), 3.42 (dd, J = 14.5, 5.5 Hz, 1H), 3.34 (dd, J = 14.5, 7.0 Hz, 1H), 2.93 (dd, J = 10.0, 4.0 Hz, 1H), 1.58 (s, 3H), 1.34 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 140.2 (ipso), 136.8 (C8), 135.9 (C5), 128.3 (ortho), 128.1 (meta), 126.7 (para), 118.7 (C6), 116.9 (C9), 112.1 [(CH3)2–], 103.8 (C1), 79.1 (C2), 76.5 (C4), 66.7 (C3), 55.2 (PhCH2–), 54.8 (C7), 26.6 (–CH3), 26.2 (–CH3). HRMS-EI m/z 315.1825 (calcd for C19H25NO3, 315.1834).
(3aR,4aR,8aR,8bR)-8-Benzyl-2,2-dimethyl-3a,4a,7,8,8a,8b-hexahydro-[1,3]dioxolo[4′,5′:4,5]furo[3,2-b]pyridine (7)
A mixture of 12 (0.21 g, 0.68 mmol) and the Hoveyda–Grubbs 1st generation catalyst (0.017 mg, 0.027 mmol) in dry CH2Cl2 (10 mL) was refluxed for 2 h, whereupon the solvent was removed under reduced pressure. The residue was purified by column chromatography (SiO2, hexane/AcOEt, 15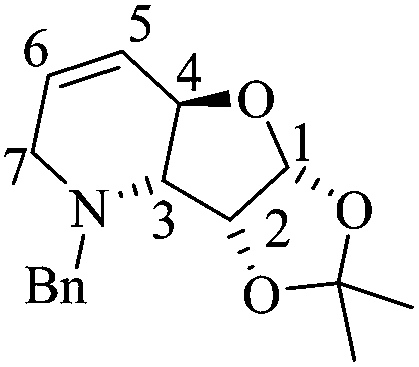 :1, v/v) to obtain 0.168 g of 7 (86%) as a colorless crystalline solid. Rf = 0.55 (4:1 hexane/AcOEt). Mp = 143–146 °C. [α]20D = +46.3 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38–7.25 (m, 5H), 6.02 (ddd, J = 10.5, 3.5, 1.5 Hz, 1H), 5.91 (d, J = 3.5 Hz, 1H), 5.59 (ddt, J = 10.0, 4.0, 2.5 Hz, 1H), 4.80 (td, J = 4.0, 1.0 Hz, 1H), 4.62–4.58 (m, 1H), 4.27 (d, J = 12.5 Hz, 1H), 3.36 (d, J = 12.5 Hz, 1H), 3.28 (ddt, J = 17.5, 3.5, 2.5 Hz, 1H), 2.79 (ddt, J = 17.5, 4.0, 2.5 Hz, 1H), 2.30 (dd, J = 9.0, 4.0 Hz, 1H), 1.60 (s, 3H), 1.37 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 137.2 (ipso), 129.4 (ortho), 128.2 (meta), 127.5 (C6), 127.3 (para), 125.4 (C5), 112.6 [(CH3)2–], 106.1 (C1), 77.2 (C2), 74.0 (C4), 68.6 (C3), 59.1 (PhCH2–), 54.1 (C7), 26.2 (–CH3), 26.0 (–CH3). HRMS-FAB m/z [M + H]+: 288.1608 (calcd for C17H22NO3, 288.1600).
:1, v/v) to obtain 0.168 g of 7 (86%) as a colorless crystalline solid. Rf = 0.55 (4:1 hexane/AcOEt). Mp = 143–146 °C. [α]20D = +46.3 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38–7.25 (m, 5H), 6.02 (ddd, J = 10.5, 3.5, 1.5 Hz, 1H), 5.91 (d, J = 3.5 Hz, 1H), 5.59 (ddt, J = 10.0, 4.0, 2.5 Hz, 1H), 4.80 (td, J = 4.0, 1.0 Hz, 1H), 4.62–4.58 (m, 1H), 4.27 (d, J = 12.5 Hz, 1H), 3.36 (d, J = 12.5 Hz, 1H), 3.28 (ddt, J = 17.5, 3.5, 2.5 Hz, 1H), 2.79 (ddt, J = 17.5, 4.0, 2.5 Hz, 1H), 2.30 (dd, J = 9.0, 4.0 Hz, 1H), 1.60 (s, 3H), 1.37 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 137.2 (ipso), 129.4 (ortho), 128.2 (meta), 127.5 (C6), 127.3 (para), 125.4 (C5), 112.6 [(CH3)2–], 106.1 (C1), 77.2 (C2), 74.0 (C4), 68.6 (C3), 59.1 (PhCH2–), 54.1 (C7), 26.2 (–CH3), 26.0 (–CH3). HRMS-FAB m/z [M + H]+: 288.1608 (calcd for C17H22NO3, 288.1600).
(1aS,3aR,3bR,6aR,7aS,7bS)-3-Benzyl-5,5-dimethylhexahydro-[1,3]dioxolo[4′,5′:4,5]furo[3,2-b]oxireno[2,3-d]pyridin-2(1aH)-one (13)
To a solution of 7 (0.05 g, 0.17 mmol) and NaH2PO4 (0.24 g, 1.29 mmol) at 0 °C in a mixture of THF/t-BuOH/H2O (3 mL/1.29 mL/1.29 mL) was added NaClO2 (0.12 g, 1.03 mmol) dissolved in H2O (1 mL). The mixture was stirred for 20 minutes before adding dropwise a saturated solution of NaOH (1 mL). After washing with brine (2 × 5 mL) and extracting the aqueous layer with AcOEt (4 × 10 mL), the combined organic phases were dried over Na2SO4 and concentrated under vacuum. The crude was purified by column chromatography (SiO2, hexane/AcOEt, 5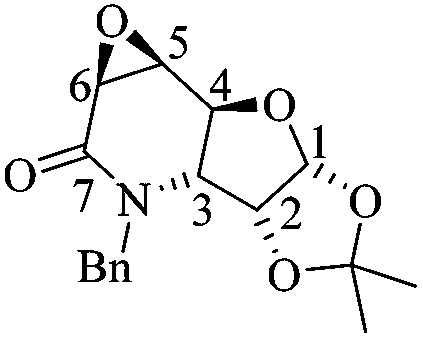 :1, v/v) to obtain 0.040 g of 13 (75%) as a white solid. Rf = 0.51 (1:1 hexane/AcOEt). Mp = 148–150 °C. [α]20D = +4.86 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38–7.23 (m, 5H), 5.77 (d, J = 3.5 Hz, 1H), 5.53 (dd, J = 15.5, 1.0 Hz, 1H), 4.44 (d, J = 9.5 Hz, 1H), 4.40 (td, J = 3.5, 1.0 Hz, 1H), 4.05 (d, J = 15.5 Hz, 1H), 3.86 (dt, J = 4.5, 0.5 Hz, 1H), 3.70 (d, J = 4.5 Hz, 1H), 3.40 (dd, J = 9.5, 3.5 Hz, 1H), 1.57 (s, 3H), 1.34 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 166.9 (C7), 136.2 (ipso), 128.9 (meta), 127.6 (para), 127.3 (ortho), 113.4 [(CH3)2–], 106.1 (C1), 76.4 (C4), 75.8 (C2), 55.4 (C3), 51.6 (C5), 51.0 (C6), 46.7 (PhCH2–), 26.1 (–CH3), 26.1 (–CH3). HRMS-FAB m/z [M + H]+: 318.1329 (calcd for C17H20NO5, 318.1341).
:1, v/v) to obtain 0.040 g of 13 (75%) as a white solid. Rf = 0.51 (1:1 hexane/AcOEt). Mp = 148–150 °C. [α]20D = +4.86 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38–7.23 (m, 5H), 5.77 (d, J = 3.5 Hz, 1H), 5.53 (dd, J = 15.5, 1.0 Hz, 1H), 4.44 (d, J = 9.5 Hz, 1H), 4.40 (td, J = 3.5, 1.0 Hz, 1H), 4.05 (d, J = 15.5 Hz, 1H), 3.86 (dt, J = 4.5, 0.5 Hz, 1H), 3.70 (d, J = 4.5 Hz, 1H), 3.40 (dd, J = 9.5, 3.5 Hz, 1H), 1.57 (s, 3H), 1.34 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 166.9 (C7), 136.2 (ipso), 128.9 (meta), 127.6 (para), 127.3 (ortho), 113.4 [(CH3)2–], 106.1 (C1), 76.4 (C4), 75.8 (C2), 55.4 (C3), 51.6 (C5), 51.0 (C6), 46.7 (PhCH2–), 26.1 (–CH3), 26.1 (–CH3). HRMS-FAB m/z [M + H]+: 318.1329 (calcd for C17H20NO5, 318.1341).
2-((tert-Butyldimethylsilyl)oxy)-4-phenylbut-3-en-nitrile [(±)-17]
To a mixture of KCN (1.95 g, 30 mmol), ZnI2 (0.03 g, 0.10 mmol) and TBSCl (2.26 g, 15 mmol) in dry MeCN (6 mL) at room temperature was added trans-cinnamaldehyde (1.05 mL, 10 mmol). The reaction mixture was refluxed for 10 h and then stirred for 38 hours at room temperature. After removing the solvent under reduced pressure, the residue was washed with AcOEt (3 × 15). The organic layer was concentrated under reduced pressure and purified by column chromatography (SiO2, hexane/AcOEt, 98 :2, v/v) to obtain 3.15 g of (±)-1737 (77%) as a yellow greenish oil. Rf = 0.60 (9:1 hexane/AcOEt). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.42–7.29 (m, 5H), 6.81 (dd, J = 15.5, 1.5 Hz, 1H), 6.18 (dd, J = 16.0, 6.0 Hz, 1H), 5.13 (dd, J = 6.0, 1.5 Hz, 1H), 0.94 (s, 9H), 0.22 (s, 3H), 0.19 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 135.0 (ipso), 133.6 (C3), 128.7 (Ph), 126.9 (Ph), 123.6 (C4), 118.4 (–CN), 62.6 (C2), 25.5 [(H3)3C–], 18.1 [(H3C)3–], −4.9 (–CH3), −5.0 (–CH3).
:2, v/v) to obtain 3.15 g of (±)-1737 (77%) as a yellow greenish oil. Rf = 0.60 (9:1 hexane/AcOEt). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.42–7.29 (m, 5H), 6.81 (dd, J = 15.5, 1.5 Hz, 1H), 6.18 (dd, J = 16.0, 6.0 Hz, 1H), 5.13 (dd, J = 6.0, 1.5 Hz, 1H), 0.94 (s, 9H), 0.22 (s, 3H), 0.19 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 135.0 (ipso), 133.6 (C3), 128.7 (Ph), 126.9 (Ph), 123.6 (C4), 118.4 (–CN), 62.6 (C2), 25.5 [(H3)3C–], 18.1 [(H3C)3–], −4.9 (–CH3), −5.0 (–CH3).
N-((S)-1-(4-Methoxyphenyl)ethyl)-4-phenyl-2-((tert-butyl dimethylsilyl)oxy)but-3-en-1-amines [(1′S)-18]
To a solution of 17 (0.10 g, 0.36 mmol) in dry THF at −70 °C was added dropwise diisobutylaluminum hydride (1.82 mL, 1.82 mmol, 1 M solution in hexane). The reaction mixture was stirred for 3 h before adding NH4Cl (0.05 g, 0.92 mmol) and dry MeOH (1.5 mL). Then the reaction mixture was stirred for 1 hour, whereafter (S)-(−)-4-methoxy-α-methylbenzylamine (0.14 g, 0.91 mmol) dissolved in dry MeOH (1 mL) was added. After stirring the mixture for 1 h at room temperature, NaBH4 (0.034 g, 0.915 mmol) was added at 0 °C. The reaction mixture was allowed to react overnight and quenched by the addition of a 1 :4 (v/v) solution of THF/HCl (1 N, 3 mL). The combined phases were separated and the aqueous phase was extracted with EtOAc (4 × 10 mL). A saturated solution of NaOH (3 mL) was added to the aqueous phase and extracted with AcOEt (4 × 10 mL). The combined organic portions were concentrated under reduced pressure and the product was purified by column chromatography (SiO2, hexane/AcOEt, as a gradient mixture from 97:3 to 90:10, v/v) to obtain 0.10 g of the diastereomeric mixture of (1′S)-18. Reported as a diastereomeric mixture: Rf = 0.60 (2:1 hexane/AcOEt). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.36–7.21 (m, 16H), 6.88–6.85 (m, 4H), 6.52 (d, J = 16.0 Hz, 1H), 6.49 (d, J = 16.0 Hz, 1H), 6.13 (dd, J = 16.0, 6.5 Hz, 1H), 6.13 (dd, J = 16.0, 6.5 Hz, 1H), 4.44–4.37 (m, 2H), 3.81 (s, 3H), 3.80 (s, 3H), 3.79–3.73 (m, 2H), 2.62–2.49 (m, 4H), 1.79 (br, 2H), 1.34 (d, J = 6.5 Hz, 3H), 1.33 (d, J = 6.5 Hz, 3H), 0.92 (s, 9H), 0.92 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H), 0.06 (s, 3H), 0.05 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.4 (para Ar), 137.6 (ipso Ph), 136.8 (ipso Ar), 131.4 (C3), 131.3 (C3), 130.2 (C4), 130.2 (C4), 128.5 (meta Ph), 127.6 (para Ph), 127.5 (ortho Ar), 127.4 (para Ph), 126.3 (ortho Ph), 113.7 (meta Ar), 113.6 (meta Ar), 73.3 (C2), 72.8 (C2), 57.5 (C1′), 57.0 (C1′), 56.2 (–OCH3), 54.2 (C1), 54.0 (C1), 25.9 [(H3)3C–], 25.8 [(H3)3C–], 24.5 (C2′), 18.2 [(H3C)3–], 18.2 [(H3C)3–], −4.0 (–CH3), −4.0 (–CH3), −4.7 (–CH3), −4.8 (–CH3). HRMS-FAB m/z [M + H]+: 412.2654 (calcd for C25H38NO2Si, 412.2672).
:4 (v/v) solution of THF/HCl (1 N, 3 mL). The combined phases were separated and the aqueous phase was extracted with EtOAc (4 × 10 mL). A saturated solution of NaOH (3 mL) was added to the aqueous phase and extracted with AcOEt (4 × 10 mL). The combined organic portions were concentrated under reduced pressure and the product was purified by column chromatography (SiO2, hexane/AcOEt, as a gradient mixture from 97:3 to 90:10, v/v) to obtain 0.10 g of the diastereomeric mixture of (1′S)-18. Reported as a diastereomeric mixture: Rf = 0.60 (2:1 hexane/AcOEt). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.36–7.21 (m, 16H), 6.88–6.85 (m, 4H), 6.52 (d, J = 16.0 Hz, 1H), 6.49 (d, J = 16.0 Hz, 1H), 6.13 (dd, J = 16.0, 6.5 Hz, 1H), 6.13 (dd, J = 16.0, 6.5 Hz, 1H), 4.44–4.37 (m, 2H), 3.81 (s, 3H), 3.80 (s, 3H), 3.79–3.73 (m, 2H), 2.62–2.49 (m, 4H), 1.79 (br, 2H), 1.34 (d, J = 6.5 Hz, 3H), 1.33 (d, J = 6.5 Hz, 3H), 0.92 (s, 9H), 0.92 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H), 0.06 (s, 3H), 0.05 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.4 (para Ar), 137.6 (ipso Ph), 136.8 (ipso Ar), 131.4 (C3), 131.3 (C3), 130.2 (C4), 130.2 (C4), 128.5 (meta Ph), 127.6 (para Ph), 127.5 (ortho Ar), 127.4 (para Ph), 126.3 (ortho Ph), 113.7 (meta Ar), 113.6 (meta Ar), 73.3 (C2), 72.8 (C2), 57.5 (C1′), 57.0 (C1′), 56.2 (–OCH3), 54.2 (C1), 54.0 (C1), 25.9 [(H3)3C–], 25.8 [(H3)3C–], 24.5 (C2′), 18.2 [(H3C)3–], 18.2 [(H3C)3–], −4.0 (–CH3), −4.0 (–CH3), −4.7 (–CH3), −4.8 (–CH3). HRMS-FAB m/z [M + H]+: 412.2654 (calcd for C25H38NO2Si, 412.2672).
N-Allyl-2-((tert-butyldimethylsilyl)oxy)-N-((S)-1-(4-methoxyphenyl)ethyl)-4-phenylbut-3-en-1-amines [(1′S)-19]
To a mixture of diastereomeric (1′S)-18 (0.49 g, 1.19 mmol) and Na2CO3 (0.15 g, 1.42 mmol) in dry MeCN (10 mL) at room temperature was added dropwise a solution of allyl iodide (0.13 mL, 1.42 mmol) in dry MeCN (1 mL). The reaction mixture was stirred overnight and then refluxed for 3 h. The solvent was removed under reduced pressure, whereupon the solid was washed with AcOEt (4 × 5 mL). After evaporation of the organic phase under reduced pressure, the residue was purified by column chromatography (SiO2, hexane/AcOEt, 98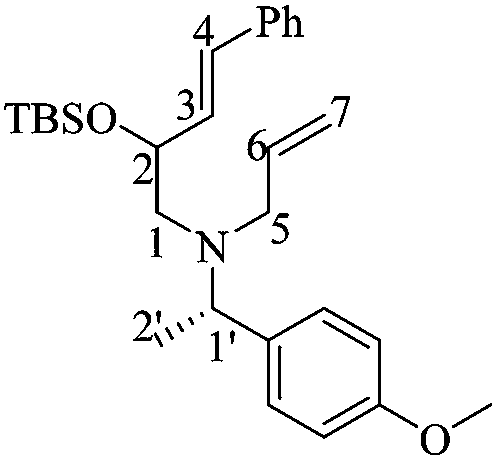 :2, v/v) to obtain 0.44 g of diastereomeric (1′S)-19 (82%) as a yellow oil. Reported as a diastereomeric mixture: Rf = 0.66 (10:1 hexane/AcOEt). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.37–7.21 (m, 16H), 6.82–6.76 (m, 4H), 6.49 (d, J = 15.5 Hz, 1H), 6.45 (d, J = 16.0 Hz, 1H), 6.27 (dd, J = 16.0, 6.5 Hz, 1H), 6.12 (dd, J = 16.0, 6.0 Hz, 1H), 5.89–5.82 (m, 2H), 5.18–5.06 (m, 4H), 4.24 (qu, J = 6.0 Hz, 2H), 3.91 (q, J = 6.5 Hz, 1H), 3.89 (q, J = 6.0 Hz, 1H), 3.77 (s, 3H), 3.75 (s, 3H), 3.21–3.09 (m, 4H), 2.63–2.54 (m, 3H), 2.47 (dd, J = 13.0, 7.5 Hz, 1H), 1.31 (d, J = 7.0 Hz, 3H), 1.30 (d, J = 7.0 Hz, 3H), 0.90 (s, 9H), 0.89 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H), 0.01 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.2 (para Ar), 158.1 (para Ar), 137.4 (ipso Ph), 137.3 (ipso Ph), 137.3 (C6), 137.3 (C6), 135.9 (ipso Ar), 135.8 (ipso Ar), 132.8 (C3), 132.7 (C3), 128.9 (C4), 128.9 (ortho Ar), 128.8 (C4), 128.8 (ortho Ar), 128.4 (meta Ph), 128.4 (meta Ph), 127.0 (para Ph), 126.3 (ortho Ph), 126.3 (ortho Ph), 116.4 (C7), 116.3 (C7), 113.2 (meta Ar), 113.2 (meta Ar), 73.0 (C2), 72.6 (C2), 58.7 (C1′), 57.9 (C1′), 57.2 (C1), 56.7 (C1), 55.1 (–OCH3), 55.1 (–OCH3), 54.8 (C5), 54.7 (C5), 25.9 [(H3)3C–], 18.3 [(H3C)3–], 18.2 [(H3C)3–], 16.3 (C2′), 15.0 (C2′), −4.4 (–CH3), −4.4 (–CH3), −4.6 (–CH3), −4.6 (–CH3). HRMS-FAB m/z [M + H]+: 450.2816 (calcd for C28H40NO2Si, 450.2828).
:2, v/v) to obtain 0.44 g of diastereomeric (1′S)-19 (82%) as a yellow oil. Reported as a diastereomeric mixture: Rf = 0.66 (10:1 hexane/AcOEt). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.37–7.21 (m, 16H), 6.82–6.76 (m, 4H), 6.49 (d, J = 15.5 Hz, 1H), 6.45 (d, J = 16.0 Hz, 1H), 6.27 (dd, J = 16.0, 6.5 Hz, 1H), 6.12 (dd, J = 16.0, 6.0 Hz, 1H), 5.89–5.82 (m, 2H), 5.18–5.06 (m, 4H), 4.24 (qu, J = 6.0 Hz, 2H), 3.91 (q, J = 6.5 Hz, 1H), 3.89 (q, J = 6.0 Hz, 1H), 3.77 (s, 3H), 3.75 (s, 3H), 3.21–3.09 (m, 4H), 2.63–2.54 (m, 3H), 2.47 (dd, J = 13.0, 7.5 Hz, 1H), 1.31 (d, J = 7.0 Hz, 3H), 1.30 (d, J = 7.0 Hz, 3H), 0.90 (s, 9H), 0.89 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H), 0.01 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.2 (para Ar), 158.1 (para Ar), 137.4 (ipso Ph), 137.3 (ipso Ph), 137.3 (C6), 137.3 (C6), 135.9 (ipso Ar), 135.8 (ipso Ar), 132.8 (C3), 132.7 (C3), 128.9 (C4), 128.9 (ortho Ar), 128.8 (C4), 128.8 (ortho Ar), 128.4 (meta Ph), 128.4 (meta Ph), 127.0 (para Ph), 126.3 (ortho Ph), 126.3 (ortho Ph), 116.4 (C7), 116.3 (C7), 113.2 (meta Ar), 113.2 (meta Ar), 73.0 (C2), 72.6 (C2), 58.7 (C1′), 57.9 (C1′), 57.2 (C1), 56.7 (C1), 55.1 (–OCH3), 55.1 (–OCH3), 54.8 (C5), 54.7 (C5), 25.9 [(H3)3C–], 18.3 [(H3C)3–], 18.2 [(H3C)3–], 16.3 (C2′), 15.0 (C2′), −4.4 (–CH3), −4.4 (–CH3), −4.6 (–CH3), −4.6 (–CH3). HRMS-FAB m/z [M + H]+: 450.2816 (calcd for C28H40NO2Si, 450.2828).
(S)-3-((tert-Butyldimethylsilyl)oxy)-1-((S)-1-(4-methoxyphenyl)ethyl)-3,4-dehydropiperidine [(1′S,5R)-14] and (R)-3-((tert-butyldimethylsilyl)oxy)-1-((S)-1-(4-methoxyphenyl)ethyl)-3,4-dehydropiperidine [(1′S,5S)-14]
A mixture of (1′S)-19 (0.4 g, 0.90 mmol) and the Hoveyda–Grubbs 2nd generation catalyst (0.028 g, 0.04 mmol) in dry CH2Cl2 (20 mL) was refluxed for 7 h. After the removal of the solvent under reduced pressure, a black viscous liquid was obtained, which was purified by column chromatography (SiO2, hexane/AcOEt, 98:2, v/v) to obtain 0.26 g of a mixture of (1′S,5R)-14 and (1′S,5S)-14 as a yellow brownish oil (82% combined yield).
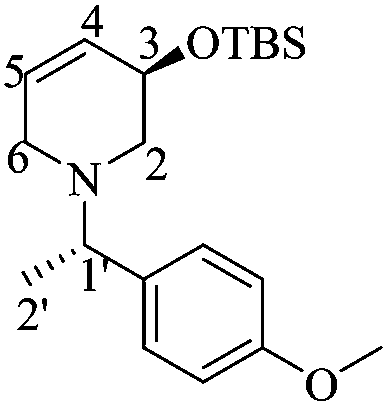 :1 hexane/AcOEt). [α]20D = −38.9 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.25–7.22 (m, 2H), 6.87–6.84 (m, 2H), 5.77–5.73 (m, 1H), 5.68–5.65 (m, 1H), 4.33–4.30 (m, 1H), 3.80 (s, 3H), 3.48 (q, J = 6.5 Hz, 1H), 3.21–3.17 (m, 1H), 2.88 (dd, J = 11.0, 5.5 Hz, 1H), 2.81 (ddd, J = 16.5, 5.5, 2.5 Hz, 1H), 2.01 (dd, J = 10.5, 8.5 Hz), 1.36 (d, J = 6.5 Hz, 3H), 0.84 (s, 9H), 0.006 (s, 3H), −0.01 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.4 (para), 135.7 (ipso), 130.1 (C4), 128.4 (ortho), 126.9 (C5), 113.5 (meta), 66.8 (C3), 63.2 (C1′), 55.2 (–OCH3), 54.7 (C2), 49.8 (C6), 25.8 [(H3)3C–], 19.4 (C2′), 18.2 [(CH3)3–], −4.6 (–CH3), −4.7 (–CH3). HRMS-EI m/z 347.2299 (calcd for C20H33NO2Si, 347.2281).
:1 hexane/AcOEt). [α]20D = −38.9 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.25–7.22 (m, 2H), 6.87–6.84 (m, 2H), 5.77–5.73 (m, 1H), 5.68–5.65 (m, 1H), 4.33–4.30 (m, 1H), 3.80 (s, 3H), 3.48 (q, J = 6.5 Hz, 1H), 3.21–3.17 (m, 1H), 2.88 (dd, J = 11.0, 5.5 Hz, 1H), 2.81 (ddd, J = 16.5, 5.5, 2.5 Hz, 1H), 2.01 (dd, J = 10.5, 8.5 Hz), 1.36 (d, J = 6.5 Hz, 3H), 0.84 (s, 9H), 0.006 (s, 3H), −0.01 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.4 (para), 135.7 (ipso), 130.1 (C4), 128.4 (ortho), 126.9 (C5), 113.5 (meta), 66.8 (C3), 63.2 (C1′), 55.2 (–OCH3), 54.7 (C2), 49.8 (C6), 25.8 [(H3)3C–], 19.4 (C2′), 18.2 [(CH3)3–], −4.6 (–CH3), −4.7 (–CH3). HRMS-EI m/z 347.2299 (calcd for C20H33NO2Si, 347.2281).
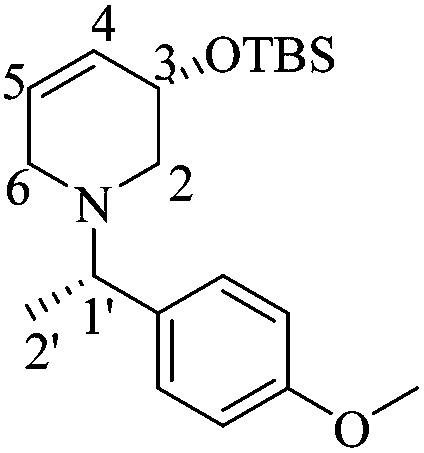 :1 hexane/AcOEt). [α]20D = +28.4 (c = 0.83, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.22–7.19 (m, 2H), 6.81–6.79 (m, 2H), 5.70 (dtd, J = 10.5, 3.0, 1.5 Hz, 1H), 5.63 (ddd, J = 10.0, 5.0, 1.5 Hz, 1H), 4.26–4.23 (m, 1H), 3.75 (s, 3H), 3.41 (q, J = 7.0 Hz, 1H), 2.95 (ddd, J = 16.5, 5.5, 2.5 Hz, 1H), 2.81–2.77 (m, 1H), 2.69 (dd, J = 11.0, 5.0 Hz, 1H), 2.78 (dd, J = 11.5, 6.5 Hz, 1H), 1.34 (d, J = 6.5 Hz, 3H), 0.83 (s, 9H), 0.00 (s, 3H), −0.001 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.4 (para), 135.4 (ipso), 128.9 (C4), 128.6 (ortho), 127.5 (C5), 113.5 (meta), 66.5 (C3), 63.2 (C1′), 55.2 (–OCH3), 54.9 (C2), 49.8 (C6), 25.9 [(H3)3C–], 19.5 (C2′), 18.3 [(CH3)3–], −4.6 (–CH3), −4.7 (–CH3). HRMS-EI m/z 347.2274 (calcd for C20H33NO2Si, 347.2281).
:1 hexane/AcOEt). [α]20D = +28.4 (c = 0.83, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.22–7.19 (m, 2H), 6.81–6.79 (m, 2H), 5.70 (dtd, J = 10.5, 3.0, 1.5 Hz, 1H), 5.63 (ddd, J = 10.0, 5.0, 1.5 Hz, 1H), 4.26–4.23 (m, 1H), 3.75 (s, 3H), 3.41 (q, J = 7.0 Hz, 1H), 2.95 (ddd, J = 16.5, 5.5, 2.5 Hz, 1H), 2.81–2.77 (m, 1H), 2.69 (dd, J = 11.0, 5.0 Hz, 1H), 2.78 (dd, J = 11.5, 6.5 Hz, 1H), 1.34 (d, J = 6.5 Hz, 3H), 0.83 (s, 9H), 0.00 (s, 3H), −0.001 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.4 (para), 135.4 (ipso), 128.9 (C4), 128.6 (ortho), 127.5 (C5), 113.5 (meta), 66.5 (C3), 63.2 (C1′), 55.2 (–OCH3), 54.9 (C2), 49.8 (C6), 25.9 [(H3)3C–], 19.5 (C2′), 18.3 [(CH3)3–], −4.6 (–CH3), −4.7 (–CH3). HRMS-EI m/z 347.2274 (calcd for C20H33NO2Si, 347.2281).
(R)-3-Hydroxy-1-((S)-1-(4-methoxyphenyl)ethyl)-3,4-dehydropiperidine [(1′S,5R)-21]
To a solution of (1′S,5R)-14 (0.23 g, 0.66 mmol) in dry THF (7 mL) at 0 °C was added dropwise TBAF (0.66 mL, 0.66 mmol, 1 M solution in THF). After 8 h, the reaction mixture was quenched by adding a saturated aqueous solution of NaHCO3 (1.0 mL). The phases were separated and the aqueous phase was extracted with AcOEt (3 × 10 mL) and CH2Cl2 (3 × 10 mL). After drying the organic phases over Na2SO4 and concentrating under reduced pressure, the residue was purified by column chromatography (SiO2, hexane/AcOEt, 1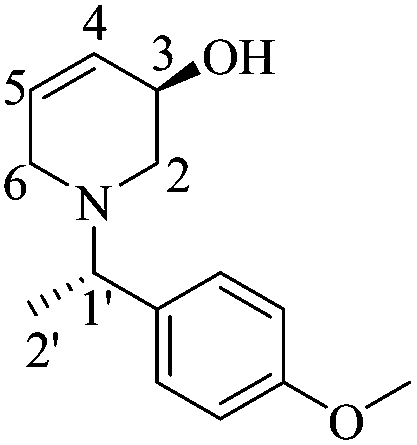 :5, v/v) to give 0.14 g (90%) of (1′S,5R)-21 as a yellow oil. Rf = 0.30 (AcOEt). [α]20D = −85.18 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.26–7.20 (m, 2H), 6.87–6.84 (m, 2H), 5.87–5.83 (m, 1H), 5.79–5.75 (m, 1H), 4.06–4.04 (m, 1H), 3.79 (s, 3H), 3.51 (q, J = 6.5 Hz, 1H), 2.99 (d, J = 17.0 Hz, 1H), 2.83–2.76 (m, 2H), 2.49 (dd, J = 11.0, 3.0 Hz, 1H), 1.38 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.6 (para), 135.3 (ipso), 129.1 (C5), 128.5 (ortho), 127.8 (C4), 113.5 (meta), 64.7 (C3), 63.0 (C1′), 55.2 (–OCH3), 54.4 (C2), 50.4 (C6), 18.9 (C2′). HRMS-EI m/z 233.1421 (calcd for C14H19NO2, 233.1416).
:5, v/v) to give 0.14 g (90%) of (1′S,5R)-21 as a yellow oil. Rf = 0.30 (AcOEt). [α]20D = −85.18 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.26–7.20 (m, 2H), 6.87–6.84 (m, 2H), 5.87–5.83 (m, 1H), 5.79–5.75 (m, 1H), 4.06–4.04 (m, 1H), 3.79 (s, 3H), 3.51 (q, J = 6.5 Hz, 1H), 2.99 (d, J = 17.0 Hz, 1H), 2.83–2.76 (m, 2H), 2.49 (dd, J = 11.0, 3.0 Hz, 1H), 1.38 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 158.6 (para), 135.3 (ipso), 129.1 (C5), 128.5 (ortho), 127.8 (C4), 113.5 (meta), 64.7 (C3), 63.0 (C1′), 55.2 (–OCH3), 54.4 (C2), 50.4 (C6), 18.9 (C2′). HRMS-EI m/z 233.1421 (calcd for C14H19NO2, 233.1416).
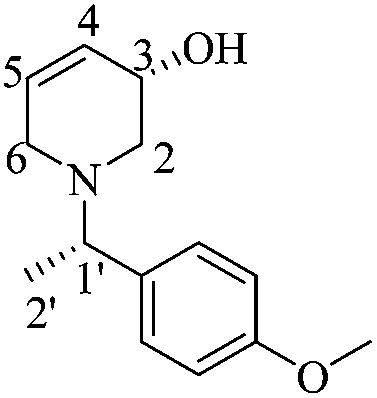
(S)-3-Hydroxy-1-((S)-1-(4-methoxyphenyl)ethyl)-3,4-dehydropiperidine [(1′S,5S)-21]
Following the same deprotection procedure as for (1′S,5R)-21, 0.06 g (91%) of (1′S,5S)-21 were obtained as a white crystalline solid. Rf = 0.34 (AcOEt). Mp = 84–86 °C [α]20D = +50.13 (c = 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.21 (dt, J = 10.0, 3.5 Hz, 2H), 6.86 (dt, J = 9.0, 3.0 Hz, 2H), 5.90–5.86 (m, 1H), 5.85–5.82 (m, 1H), 3.97–3.94 (m, 1H), 3.80 (s, 3H), 3.50 (q, J = 7.0 Hz, 1H), 3.27 (dd, J = 18.5, 4.0 Hz, 1H), 2.80–2.74 (m, 2H), 2.27 (dd, J = 11.5, 2.5 Hz, 1H), 1.39 (d, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): 158.6 (para), 135.2 (ipso), 129.3 (C5), 128.6 (ortho), 127.6 (C4), 113.6 (meta), 64.5 (C3), 63.2 (C1′), 55.2 (–OCH3), 54.4 (C2), 50.1 (C6), 19.3 (C2′). HRMS-EI m/z 233.1419 (calcd for C14H19NO2, 233.1416).
(3R,4S,5S)-5-((tert-Butyldimethylsilyl)oxy)-3,4-epoxy-1-((S)-1-(4-methoxyphenyl)ethyl)piperidin-2-one [(1′S,3R,4S,5S)-15]
To a solution of (1′S,5R)-14 (0.17 g, 0.49 mmol), 2-methyl-2-butene (0.52 mL, 4.9 mmol) and NaH2PO4 (0.68 g, 5.0 mmol) in THF/t-BuOH/H2O (11.5 mL/5 mL/5 mL) at 0 °C was added NaClO2 (0.36 g, 3.9 mmol) dissolved in H2O (1 mL). The solution was stirred for 20 minutes at 0 °C, whereupon a saturated solution of NaOH (1.5 mL) was added and the organic layer was extracted with AcOEt (4 × 10 mL). After drying the combined organic phases over Na2SO4 and concentrating under reduced pressure, the residue was purified by column chromatography (SiO2, hexane/AcOEt, 12 :1, v/v) to obtain 0.17 g of (1S′,3R,4S,5S)-15 (92%) as a white crystalline material. Rf = 0.28 (6:1 hexane/AcOEt). Mp = 91–93 °C. [α]20D = −77.7 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.24–7.21 (m, 2H), 6.84–6.81 (m, 2H), 5.94 (q, J = 7.0 Hz, 1H), 4.31 (dt, J = 2.5, 2.0 Hz, 1H), 3.78 (s, 3H), 3.59 (dd, J = 4.0, 0.5 Hz, 1H), 3.38 (ddd, J = 4.0, 2.5, 1.0 Hz, 1H), 3.22 (dd, J = 13.0, 2.5 Hz, 1H), 2.80 (dt, J = 13.0, 1.5 Hz, 1H), 1.43 (d, J = 7.0 Hz, 3H), 0.75 (s, 9H), −0.05 (s, 3H), −0.19 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 165.4 (C2), 158.8 (para), 131.3 (ipso), 128.9 (ortho), 113.7 (meta), 64.6 (C5), 55.2 (–OCH3), 54.3 (C4), 51.0 (C3), 49.7 (C1′), 42.6 (C6), 25.7 [(H3)3C–], 18.0 [(CH3)3–], 15.9 (C2′), −4.9 (–CH3), −5.1 (–CH3). HRMS-EI m/z 377.2029 (calcd for C20H31NO4Si, 377.2022).
:1, v/v) to obtain 0.17 g of (1S′,3R,4S,5S)-15 (92%) as a white crystalline material. Rf = 0.28 (6:1 hexane/AcOEt). Mp = 91–93 °C. [α]20D = −77.7 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.24–7.21 (m, 2H), 6.84–6.81 (m, 2H), 5.94 (q, J = 7.0 Hz, 1H), 4.31 (dt, J = 2.5, 2.0 Hz, 1H), 3.78 (s, 3H), 3.59 (dd, J = 4.0, 0.5 Hz, 1H), 3.38 (ddd, J = 4.0, 2.5, 1.0 Hz, 1H), 3.22 (dd, J = 13.0, 2.5 Hz, 1H), 2.80 (dt, J = 13.0, 1.5 Hz, 1H), 1.43 (d, J = 7.0 Hz, 3H), 0.75 (s, 9H), −0.05 (s, 3H), −0.19 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 165.4 (C2), 158.8 (para), 131.3 (ipso), 128.9 (ortho), 113.7 (meta), 64.6 (C5), 55.2 (–OCH3), 54.3 (C4), 51.0 (C3), 49.7 (C1′), 42.6 (C6), 25.7 [(H3)3C–], 18.0 [(CH3)3–], 15.9 (C2′), −4.9 (–CH3), −5.1 (–CH3). HRMS-EI m/z 377.2029 (calcd for C20H31NO4Si, 377.2022).
(3S,4R,5R)-5-((tert-Butyldimethylsilyl)oxy)-3,4-epoxy-1-((S)-1-(4-methoxyphenyl)ethyl)piperidin-2-one [(1′S,3S,4R,5R)-15]
Following the same procedure as for (1S′,3R,4S,5S)-15, 0.17 g of (1′S,3S,4R,5R)-15 (84%) were obtained as a yellow oil. Rf = 0.28 (6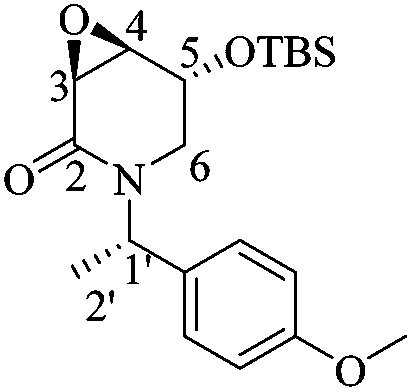 :1 hexane/AcOEt). [α]20D = −28.0 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.18–7.15 (m, 2H), 6.87–6.84 (m, 2H), 5.87 (q, J = 7.0 Hz, 1H), 4.30 (q, J = 1.0 Hz, 1H), 3.79 (s, 3H), 3.59 (dd, J = 4.0, 1.0 Hz, 1H), 3.41–3.39 (m, 1H), 2.86 (dd, J = 13.5, 2.5 Hz, 1H), 2.70 (dt, J = 14.0, 1.5 Hz, 1H), 1.42 (d, J = 6.5 Hz, 3H), 0.89 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 165.3 (C2), 158.8 (para), 131.4 (ipso), 128.5 (ortho), 113.8 (meta), 64.8 (C5), 55.2 (–OCH3), 54.5 (C4), 51.3 (C3), 49.9 (C1′), 44.3 (C6), 25.6 [(H3)3C–], 18.0 [(CH3)3–], 15.2 (C2′), −4.6 (CH3), −4.7 (CH3). HRMS-EI m/z 377.2035 (calcd for C20H31NO4Si, 377.2022).
:1 hexane/AcOEt). [α]20D = −28.0 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.18–7.15 (m, 2H), 6.87–6.84 (m, 2H), 5.87 (q, J = 7.0 Hz, 1H), 4.30 (q, J = 1.0 Hz, 1H), 3.79 (s, 3H), 3.59 (dd, J = 4.0, 1.0 Hz, 1H), 3.41–3.39 (m, 1H), 2.86 (dd, J = 13.5, 2.5 Hz, 1H), 2.70 (dt, J = 14.0, 1.5 Hz, 1H), 1.42 (d, J = 6.5 Hz, 3H), 0.89 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 165.3 (C2), 158.8 (para), 131.4 (ipso), 128.5 (ortho), 113.8 (meta), 64.8 (C5), 55.2 (–OCH3), 54.5 (C4), 51.3 (C3), 49.9 (C1′), 44.3 (C6), 25.6 [(H3)3C–], 18.0 [(CH3)3–], 15.2 (C2′), −4.6 (CH3), −4.7 (CH3). HRMS-EI m/z 377.2035 (calcd for C20H31NO4Si, 377.2022).
(3R,4R,5S)-5-Hydroxy-3,4-epoxy-1-((S)-1-(4-methoxyphenyl)ethyl)piperidin-2-one [(1′S,3R,4R,5S)-20]
To a solution of (1′S,3R,4S,5S)-15 (0.16 g, 0.46 mmol) in dry THF (15 mL) at 0 °C was added dropwise TBAF (0.5 mL, 0.4 mmol, 1 M solution in THF). After 2 h, the reaction mixture was quenched by adding a saturated aqueous solution of NaHCO3 (0.5 mL). The phases were separated and the aqueous phase was extracted with AcOEt (3 × 10 mL) and CH2Cl2 (3 × 10 mL). After drying the combined organic phases over Na2SO4 and concentrating under reduced pressure, the crude was purified by column chromatography (SiO2, hexane/AcOEt, 1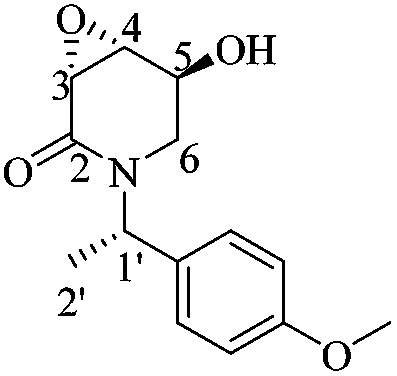 :1, v/v) to yield (1′S,3R,4R,5S)-20 (0.11 g, 90%) as a white crystalline solid. Rf = 0.57 (AcOEt). Mp = 181–183 °C. [α]20D = −86.3 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.25–7.22 (m, 2H), 6.87 (dt, J = 9.0, 3.0 Hz, 2H), 5.97 (q, J = 7.0 Hz, 1H), 4.29 (ddd, J = 7.5, 5.5, 2.5 Hz, 1H), 3.79 (s, 3H), 3.61 (dd, J = 4.0, 0.5 Hz, 1H), 3.52 (ddd, J = 4.5, 3.0, 1.5 Hz, 1H), 3.30 (dd, J = 13.5, 3.0 Hz, 1H), 2.84 (ddd, J = 13.5, 2.0, 1.5 Hz, 1H), 1.45 (d, J = 7.0 Hz, 3H), 1.12 (d, J = 7.5 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ (ppm): 165.2 (C2), 159.1 (para), 131.4 (ipso), 128.5 (ortho), 114.0 (meta), 64.1 (C5), 55.2 (–OCH3), 53.6 (C4), 51.1 (C3), 49.6 (C1′), 42.0 (C6), 15.6 (C2′).
:1, v/v) to yield (1′S,3R,4R,5S)-20 (0.11 g, 90%) as a white crystalline solid. Rf = 0.57 (AcOEt). Mp = 181–183 °C. [α]20D = −86.3 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.25–7.22 (m, 2H), 6.87 (dt, J = 9.0, 3.0 Hz, 2H), 5.97 (q, J = 7.0 Hz, 1H), 4.29 (ddd, J = 7.5, 5.5, 2.5 Hz, 1H), 3.79 (s, 3H), 3.61 (dd, J = 4.0, 0.5 Hz, 1H), 3.52 (ddd, J = 4.5, 3.0, 1.5 Hz, 1H), 3.30 (dd, J = 13.5, 3.0 Hz, 1H), 2.84 (ddd, J = 13.5, 2.0, 1.5 Hz, 1H), 1.45 (d, J = 7.0 Hz, 3H), 1.12 (d, J = 7.5 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ (ppm): 165.2 (C2), 159.1 (para), 131.4 (ipso), 128.5 (ortho), 114.0 (meta), 64.1 (C5), 55.2 (–OCH3), 53.6 (C4), 51.1 (C3), 49.6 (C1′), 42.0 (C6), 15.6 (C2′).
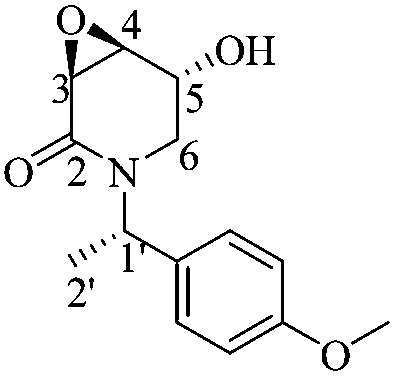
(3S,4S,5R)-5-Hydroxy-3,4-epoxy-1-((S)-1-(4-methoxyphenyl)ethyl)piperidin-2-one [(1′S,3S,4S,5R)-20]
Following the same procedure as for (1′S,3R,4R,5S)-20, 0.102 g of (1′S,3S,4S,5R)-20 (85%) were obtained as a white crystalline solid. Rf = 0.57 (AcOEt). Mp = 130–133 °C. [α]20D = −106.8 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.19–7.16 (m, 2H), 6.86 (dt, J = 8.5, 3.5 Hz, 2H), 5.89 (q, J = 7.0 Hz, 1H), 4.36 (ddd, J = 8.0, 5.0, 2.5 Hz, 1H), 3.79 (s, 3H), 3.61 (dd, J = 4.0, 1.0 Hz, 1H), 3.57 (dd, J = 4.5, 3.5 Hz, 1H), 2.90 (d, J = 2.5 Hz, 2H), 1.98 (d, J = 8.5 Hz, 1H), 1.46 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 165.2 (C2), 158.9 (para), 131.0 (ipso), 128.5 (ortho), 113.9 (meta), 64.5 (C5), 55.2 (–OCH3), 54.1 (C4), 51.1 (C3), 49.9 (C2′), 43.6 (C6), 15.3 (C1′).
(3R,4R,5S)-5-Acetyloxy-3,4-epoxy-1-((S)-1-(4-methoxyphenyl)ethyl)piperidin-2-one [(1′S,3R,4R,5S)-22]
To a solution of (1′S,3R,4R,5S)-20 (0.06 g, 0.23 mmol) and DMAP (0.008 g, 0.07 mmol) in dry MeCN (10 mL) was added dropwise a solution of triethylamine (0.05 mL, 0.35 mmol) in dry MeCN (1 mL). After the addition of a solution of acetic anhydride (0.033 mL, 0.35 mmol) in dry MeCN (1 mL), the solution was stirred for 1.5 hours at room temperature, whereafter NaHCO3 was added until a neutral pH was reached. The liquid phase was filtered, dried over Na2SO4, concentrated under reduced pressure and purified by column chromatography (SiO2, hexane/AcOEt, 5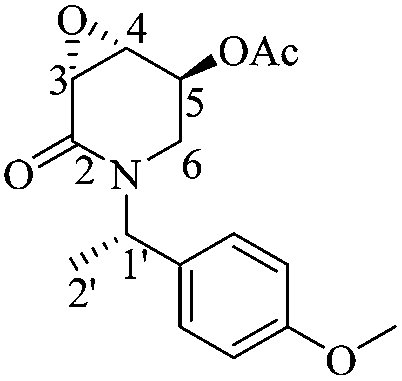 :1, v/v) to give 0.07 g of (1′S,3R,4R,5S)-22 (95%) as a white crystalline solid. Rf = 0.40 (1:1 hexane/AcOEt). Mp = 116–118 °C. [α]20D = −61.38 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.23–7.20 (m, 2H), 6.84 (dt, J = 9.5, 3.0 Hz, 2H), 5.96 (q, J = 7.0 Hz, 1H), 5.29 (dd, J = 5.5, 3.0 Hz, 1H), 3.78 (s, 3H), 3.64 (dd, J = 4.0, 0.5 Hz, 1H), 3.54 (ddd, J = 4.5, 3.0, 1.5 Hz, 1H), 3.30 (dd, J = 14.0, 2.5 Hz, 1H), 2.99 (ddd, J = 14.0, 2.0, 1.5 Hz, 1H), 1.74 (s, 3H), 1.43 (d, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 169.6 (MeO2), 164.8 (C2), 158.9 (para), 131.1 (ipso), 128.8 (ortho), 113.5 (meta), 65.8 (C5), 55.2 (–OCH3), 51.5 (C4), 51.0 (C3), 49.6 (C1′), 39.1 (C6), 20.5 (H3CO2–), 15.5 (C2′). HRMS-EI m/z 305.1266 (calcd for C16H19NO5, 305.1263).
:1, v/v) to give 0.07 g of (1′S,3R,4R,5S)-22 (95%) as a white crystalline solid. Rf = 0.40 (1:1 hexane/AcOEt). Mp = 116–118 °C. [α]20D = −61.38 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.23–7.20 (m, 2H), 6.84 (dt, J = 9.5, 3.0 Hz, 2H), 5.96 (q, J = 7.0 Hz, 1H), 5.29 (dd, J = 5.5, 3.0 Hz, 1H), 3.78 (s, 3H), 3.64 (dd, J = 4.0, 0.5 Hz, 1H), 3.54 (ddd, J = 4.5, 3.0, 1.5 Hz, 1H), 3.30 (dd, J = 14.0, 2.5 Hz, 1H), 2.99 (ddd, J = 14.0, 2.0, 1.5 Hz, 1H), 1.74 (s, 3H), 1.43 (d, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 169.6 (MeO2), 164.8 (C2), 158.9 (para), 131.1 (ipso), 128.8 (ortho), 113.5 (meta), 65.8 (C5), 55.2 (–OCH3), 51.5 (C4), 51.0 (C3), 49.6 (C1′), 39.1 (C6), 20.5 (H3CO2–), 15.5 (C2′). HRMS-EI m/z 305.1266 (calcd for C16H19NO5, 305.1263).
(3S,4S,5R)-5-Acetyloxy-3,4-epoxy-1-((S)-1-(4-methoxyphenyl)ethyl)piperidin-2-one [(1′S,3S,4S,5R)-22]
Following the same procedure as for (1′S,3R,4R,5S)-22, 0.06 g of (1′S,3S,4S,5R)-22 were obtained (98%) as a greenish viscous liquid. Rf = 0.40 (1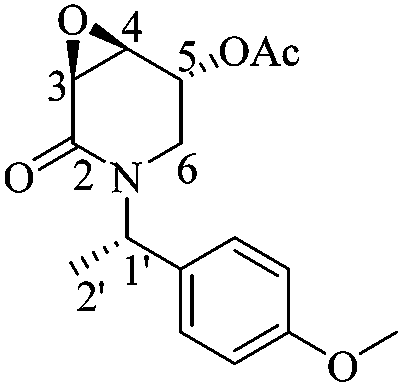 :1 hexane/AcOEt). [α]20D = −20.91 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.17–7.13 (m, 2H), 6.85 (dt, J = 9.5, 3.0 Hz, 2H), 5.87 (q, J = 7.0 Hz, 1H), 5.34 (dd, J = 4.5, 2.0 Hz, 1H), 3.79 (s, 3H), 3.64 (dd, J = 3.5, 1.0 Hz, 1H), 3.62 (ddd, J = 4.0, 3.0, 1.0 Hz, 1H), 2.99–2.92 (m, 2H), 2.10 (s, 3H), 1.38 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 170.0 (MeO2), 164.9 (C2), 158.9 (para), 130.7 (ipso), 128.5 (ortho), 113.9 (meta), 66.5 (C5), 55.2 (–OCH3), 51.9 (C4), 51.2 (C3), 50.0 (C1′), 40.76 (C6), 20.8 (H3CO2–), 14.9 (C2′). HRMS-EI m/z 305.1257 (calcd for C16H19NO5, 305.1263).
:1 hexane/AcOEt). [α]20D = −20.91 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.17–7.13 (m, 2H), 6.85 (dt, J = 9.5, 3.0 Hz, 2H), 5.87 (q, J = 7.0 Hz, 1H), 5.34 (dd, J = 4.5, 2.0 Hz, 1H), 3.79 (s, 3H), 3.64 (dd, J = 3.5, 1.0 Hz, 1H), 3.62 (ddd, J = 4.0, 3.0, 1.0 Hz, 1H), 2.99–2.92 (m, 2H), 2.10 (s, 3H), 1.38 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 170.0 (MeO2), 164.9 (C2), 158.9 (para), 130.7 (ipso), 128.5 (ortho), 113.9 (meta), 66.5 (C5), 55.2 (–OCH3), 51.9 (C4), 51.2 (C3), 50.0 (C1′), 40.76 (C6), 20.8 (H3CO2–), 14.9 (C2′). HRMS-EI m/z 305.1257 (calcd for C16H19NO5, 305.1263).
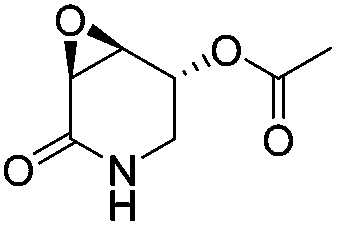
(3R,4R,5S)-5-Acetyloxy-3,4-epoxypiperidin-2-one [(3R,4R,5S)-23]
To a solution of (1′S,3R,4R,5S)-22 (0.06 g, 0.186 mmol) in MeCN (7 mL) at 0 °C was added a solution of ceric ammonium nitrate (0.31 g, 0.56 mmol) in water (1.5 mL). The mixture was stirred for 6 h before adding 1 mL of H2O. After extracting the product with AcOEt (4 × 5 mL), the combined organic phases were dried over Na2SO4 and the solvent was removed under reduced pressure. The crude was purified by column chromatography (SiO2, hexane/AcOEt, 2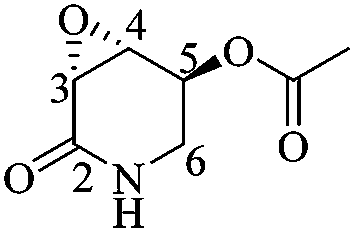 :1, v/v) to obtain 0.02 g (64%) of (3R,4R,5S)-23 as a white crystalline solid. Rf = 0.51 (AcOEt). Mp = 101–104 °C. [α]20D = +46.3 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 5.72 (br, 1H), 5.45–5.44 (m, 1H), 3.67 (ddd, J = 4.0, 3.0, 1.5 Hz, 1H), 3.56 (dd, J = 14.0, 2.5 Hz, 1H), 3.51 (dd, J = 3.0, 2.5 Hz, 1H), 3.26 (ddt, J = 14.0, 6.0, 1.5 Hz, 1H), 2.12 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 170.2 (MeO2), 167.4 (C2), 65.8 (C5), 52.2 (C4), 50.5 (C3), 41.0 (C6), 20.8 (H3CO2–). HRMS-FAB m/z [M + H]+: 172.0617 (calcd for C7H10NO4, 170.0610).
:1, v/v) to obtain 0.02 g (64%) of (3R,4R,5S)-23 as a white crystalline solid. Rf = 0.51 (AcOEt). Mp = 101–104 °C. [α]20D = +46.3 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 5.72 (br, 1H), 5.45–5.44 (m, 1H), 3.67 (ddd, J = 4.0, 3.0, 1.5 Hz, 1H), 3.56 (dd, J = 14.0, 2.5 Hz, 1H), 3.51 (dd, J = 3.0, 2.5 Hz, 1H), 3.26 (ddt, J = 14.0, 6.0, 1.5 Hz, 1H), 2.12 (s, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 170.2 (MeO2), 167.4 (C2), 65.8 (C5), 52.2 (C4), 50.5 (C3), 41.0 (C6), 20.8 (H3CO2–). HRMS-FAB m/z [M + H]+: 172.0617 (calcd for C7H10NO4, 170.0610).
(3S,4S,5R)-5-Acetyloxy-3,4-epoxypiperidin-2-one [(3S,4S,5R)-23]
Following the same procedure as for (3R,4R,5S)-23, 0.02 g of (3S,4S,5R)-23 were obtained (61%) as a white crystalline solid. Rf = 0.51 (AcOEt), Mp = 101–104 °C. [α]20D = −46.36 (c = 1.0, CHCl3). HRMS-FAB m/z [M + H]+: 172.0621 (calcd for C7H10NO4, 172.0610).
(3R,4R,5S)-5-Acetyloxy-3,4-epoxy-(1-hydrocinnamoyl)piperidin-2-one [(−)-(3R,4R,5S)-3]
A mixture of cinnamic acid (0.74 g, 5.00 mmol) and Pd(OH)2/C 5 wt% (0.034 mg) in AcOEt under H2 (100 psi) was stirred for 22 h at room temperature. Then the solution was passed through a pad of silica gel using AcOEt as the mobile phase. The resulting hydrocinnamic acid was isolated as a colorless crystalline solid in quantitative yield. To a solution of the hydrocinnamic acid (0.12 g, 0.79 mmol) in dry CH2Cl2 at 0 °C was added a solution of DMF (0.06 mL, 0.79 mmol) in dry CH2Cl2 (0.3 mL) followed by small portions of oxalyl chloride (0.07 mL, 0.79 mmol). After stirring for 1 hour at room temperature, the solvent was removed to yield the corresponding acyl halide. For the final step, to a solution of (3R,4R,5S)-23 (0.045 g, 0.26 mmol) in dry THF (8.0 mL) at −78 °C was added dropwise n-BuLi (0.21 mL, 0.34 mmol, 1 M solution in hexane). After stirring for 20 minutes, the previously freshly prepared hydrocinnamic acid chloride was dissolved in dry THF (2.0 mL) and added through a cannula. The reaction mixture was allowed to react for 2 h before adding H2O (1.0 mL). The aqueous phase was extracted with AcOEt (4 × 10 mL). After drying the combined organic phases over Na2SO4 and concentrating under reduced pressure, the product was purified by column chromatography (SiO2, hexane/AcOEt, 9 :1, v/v) to obtain 0.42 g (53%) of the naturally occurring alkaloid (−)-(3R,4R,5S)-3 as a yellow oil. Rf = 0.34 (2:1 hexane/AcOEt). [α]20D = −76.8 (c = 1.0, Me2CO), lit.17 [α]20D = −98.8 (c = 0.5, Me2CO). 1H NMR [500 MHz, (CD3)2CO] δ (ppm): 7.29–7.24 (m, 4H), 7.19–7.16 (m, 1H), 5.64 (ddd, J = 3.0, 2.5, 0.5 Hz, 1H), 4.46 (ddd, J = 15.0, 2.5, 1.5 Hz, 1H), 3.89 (ddd, J = 4.0, 3.0, 1.5 Hz, 1H), 3.67 (dd, J = 4.0, 1.0 Hz, 1H), 3.34 (dd, J = 14.5, 2.5 Hz, 1H), 3.25 (ddd, J = 17.5, 9.0, 6.5 Hz, 1H), 3.18 (ddd, J = 17.5, 8.5, 6.5 Hz, 1H), 2.96–2.86 (m, 2H), 2.02 (s, 3H). 13C-NMR [125 MHz, (CD3)2CO] δ (ppm): 175.0 (C7), 170.3 (MeO2), 169.2 (C2), 142.1 (ipso), 129.2 (Ph), 129.1 (Ph), 126.8 (Ph), 66.8 (C5), 53.4 (C4), 52.5 (C3), 41.8 (C6), 41.6 (C8), 31.4 (C9), 20.6 (H3CO2–).
:1, v/v) to obtain 0.42 g (53%) of the naturally occurring alkaloid (−)-(3R,4R,5S)-3 as a yellow oil. Rf = 0.34 (2:1 hexane/AcOEt). [α]20D = −76.8 (c = 1.0, Me2CO), lit.17 [α]20D = −98.8 (c = 0.5, Me2CO). 1H NMR [500 MHz, (CD3)2CO] δ (ppm): 7.29–7.24 (m, 4H), 7.19–7.16 (m, 1H), 5.64 (ddd, J = 3.0, 2.5, 0.5 Hz, 1H), 4.46 (ddd, J = 15.0, 2.5, 1.5 Hz, 1H), 3.89 (ddd, J = 4.0, 3.0, 1.5 Hz, 1H), 3.67 (dd, J = 4.0, 1.0 Hz, 1H), 3.34 (dd, J = 14.5, 2.5 Hz, 1H), 3.25 (ddd, J = 17.5, 9.0, 6.5 Hz, 1H), 3.18 (ddd, J = 17.5, 8.5, 6.5 Hz, 1H), 2.96–2.86 (m, 2H), 2.02 (s, 3H). 13C-NMR [125 MHz, (CD3)2CO] δ (ppm): 175.0 (C7), 170.3 (MeO2), 169.2 (C2), 142.1 (ipso), 129.2 (Ph), 129.1 (Ph), 126.8 (Ph), 66.8 (C5), 53.4 (C4), 52.5 (C3), 41.8 (C6), 41.6 (C8), 31.4 (C9), 20.6 (H3CO2–).
(3S,4S,5R)-5-Acetyloxy-3,4-epoxy-(1-hydrocinnamoyl)piperidin-2-one [(+)-(3S,4S,5R)-3]
Following the same procedure as for (−)-(3R,4R,5S)-3, 0.04 g of (+)-(3S,4S,5R)-3 (52%) were obtained as a yellow oil. Rf = 0.34 (2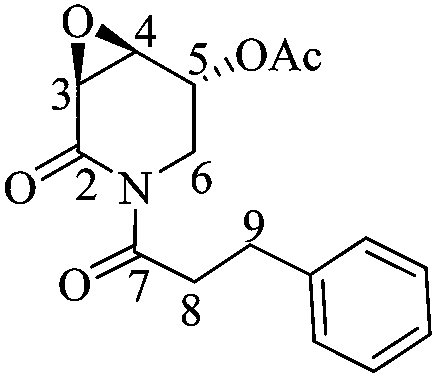 :1 hexane/AcOEt). [α]20D = +75.3 (c = 1.3, Me2CO).
:1 hexane/AcOEt). [α]20D = +75.3 (c = 1.3, Me2CO).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by CONACyT (project number: 255891) and the Marcos Moshinsky Foundation. U. O. N. acknowledges CONACyT for a graduate scholarship (grant number: 383339) and LYVA BUAP-VIEP for an undergraduate scholarship. Access to the X-ray facilities of the Chemical Research Center at Morelos State University (LANEM) is gratefully acknowledged.Notes and references
- (a) J. C. do Nascimento, V. F. de Paula, J. M. David and J. P. David, Quim. Nova, 2012, 35, 2288–2311 CrossRef CAS; (b) P. S. Soltis, D. E. Soltis and M. W. Chase, Nature, 1999, 402, 402–404 CrossRef CAS PubMed; (c) A. J. Bornstein, J. Arnold Arbor., Harv. Univ., 1989, 70, 1–55 CrossRef.
- http://www.theplantlist.org/browse/A/ .
- (a) L. R. Olsen, M. P. Grillo and C. Skonberg, Chem. Res. Toxicol., 2011, 24, 992–1002 CrossRef CAS PubMed; (b) Y. N. Singh and N. N. Singh, CNS Drugs, 2002, 16, 731–743 CrossRef PubMed; (c) S. A. Norton and P. Ruze, J. Am. Acad. Dermatol., 1994, 31, 89–97 CrossRef CAS PubMed.
- (a) T. D. Xuan, A. A. Elzaawely, M. Fukuta and S. Tawata, J. Agric. Food Chem., 2006, 54, 720–725 CrossRef CAS PubMed; (b) A. R. Bilia, L. Scalise, M. C. Bergonzi and F. F. Vincieri, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 812, 203–214 CrossRef CAS; (c) A. Denham, M. McIntyre and J. Whitehouse, J. Altern. Complement. Med., 2002, 8, 237–263 CrossRef PubMed.
- (a) I. M. Scott, H. R. Jensen, B. J. R. Philogene and J. T. Arnason, Phytochem. Rev., 2008, 7, 65–75 CrossRef CAS; (b) D. L. Moreira, E. F. Guimaraes and M. A. C. Kaplan, Phytochemistry, 1998, 48, 1075–1077 CrossRef CAS.
- (a) C. S. Ramos, S. A. Vanin and M. J. Kato, Phytochemistry, 2008, 69, 2157–2161 CrossRef CAS PubMed; (b) G. Zhao, Y. Hui, J. K. Pupprecht, J. L. McLaughlin and K. V. Wood, J. Nat. Prod., 1992, 55, 347–356 CrossRef CAS PubMed.
- (a) L. F. Yamaguchi, J. H. G. Lago, T. M. Tanizaki, P. D. Mascio and M. J. Kato, Phytochemistry, 2006, 67, 1838–1843 CrossRef CAS PubMed; (b) A. P. Danelutte, J. H. G. Lago, M. C. M. Young and M. J. Kato, Phytochemistry, 2003, 64, 555–559 CrossRef CAS PubMed.
- (a) V. A. Facundo and S. M. Morais, Biochem. Syst. Ecol., 2003, 31, 111–113 CrossRef CAS; (b) V. S. Parmar, S. C. Jain, S. Gupta, S. Talwar, V. K. Rajwanshi, R. Kumar, A. Azim, S. Malhotra, N. Kumar, R. Jain, N. K. Sharma, O. D. Tyagi, S. J. Lawrie, W. Errington, O. W. Howarth, C. E. Olsen, S. K. Singh and J. Wengel, Phytochemistry, 1998, 49, 1069–1078 CrossRef CAS.
- (a) B. Portet, N. Fabre, R. Rozenberg, J. L. H. Jiwan, C. Moulis and J. Q. Leclercq, J. Chromatogr., A, 2008, 1210, 45–54 CrossRef CAS PubMed; (b) B. Portet, N. Fabre, V. Roumy, H. Gornitzka, G. Bourdy, S. Chevalley, M. Sauvain, A. Valentin and C. Moulis, Phytochemistry, 2007, 68, 1312–1320 CrossRef CAS PubMed; (c) J. Orjala, A. D. Wright, C. A. J. Erdelmeier, O. Sticher and T. Rali, Helv. Chim. Acta, 1993, 76, 1481–1488 CrossRef CAS.
- (a) V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CAS; (b) V. S. Parmar, S. C. Jain, K. S. Bisht, R. Jain, P. Taneja, A. Jha, O. D. Tyagi, A. K. Prasad, J. Wengel, C. E. Olsen and P. M. Boll, Phytochemistry, 1997, 46, 597–673 CrossRef CAS.
- T. K. Tabopda, J. Ngoupayo, J. Liu, A. C. Mitaine-Offer, S. A. K. Tanoli, S. N. Klan, M. S. Ali, B. T. Ngadjui, E. Tsamo, M. A. Lacaille-Dubois and B. Luu, Phytochemistry, 2008, 69, 1726–1731 CrossRef CAS PubMed.
- Z. A. Zakaria, H. Patahuddin, A. S. Mohamad, D. A. Israf and M. R. Sulaiman, J. Ethnopharmacol., 2010, 128, 42–48 CrossRef CAS PubMed.
- (a) D. R. Silva, S. Barone, A. E. Svidzinski, C. A. Bersani-Amado and D. A. G. Cortz, J. Ethnopharmacol., 2008, 116, 569–573 CrossRef PubMed; (b) L. C. Lin, C. C. Shen and T. H. Tsai, J. Nat. Prod., 2006, 69, 842–844 CrossRef CAS PubMed.
- (a) A. Guerrini, G. Sacchetti, D. Rossi, G. Paganetto, M. Muzzoli, E. Andreotti, M. Tognolini, M. E. Maldonado and R. Bruni, Environ. Toxicol. Pharmacol., 2009, 27, 39–48 CrossRef CAS PubMed.
- L. S. R. Arambewela, L. D. A. M. Arawwawala and W. D. Ratnasooriya, J. Ethnopharmacol., 2005, 102, 239–245 CrossRef CAS PubMed.
- (a) H. Zhang, H. Matsuda, S. Nakamura and M. Yoshikawa, Bioorg. Med. Chem. Lett., 2008, 18, 3272–3277 CrossRef CAS PubMed; (b) P. H. Evans, W. S. Bowers and E. J. Funk, J. Agric. Food Chem., 1984, 32, 1254–1256 CrossRef CAS; (c) H. Huang, C. M. Morgan, R. N. Asolkar, M. E. Koivunen and P. G. Marrone, J. Agric. Food Chem., 2010, 58, 9994–10000 CrossRef CAS PubMed; (d) H. Matsuda, K. Ninomiya, T. Morikawa, D. Yasuda, I. Yamaguchi and M. Yoshikawa, Bioorg. Med. Chem., 2009, 17, 7313–7323 CrossRef CAS PubMed; (e) A. Quílez, B. Berenguer, G. Gilardoni, C. Souccar, S. de Mendonça, L. F. S. Oliveira, M. J. Martín-Calero and G. Vidari, J. Ethnopharmacol., 2010, 128, 583–589 CrossRef PubMed.
- K. Dragull, W. Y. Yoshida and C.-S. Tang, Phytochemistry, 2003, 63, 193–198 CrossRef CAS PubMed.
- R. M. Smith, Tetrahedron, 1979, 35, 437–439 CrossRef CAS.
- (a) H. K. Lee, J. S. Chun and C. S. Pak, Tetrahedron Lett., 2001, 42, 3483–3486 CrossRef CAS; (b) H.-J. Altenbach and K. Himmeldirk, Tetrahedron: Asymmetry, 1995, 6, 1077–1080 CrossRef CAS.
- R. G. Arrayás, A. Alcudia and L. S. Liebeskind, Org. Lett., 2001, 3, 3381–3383 CrossRef.
- Because of the lower reactivity, epoxidation of α,β-unsaturated amides has been achieved so far with the aid of transition metals. See (a) Y. Chu, X. Liu, W. Li, X. Hu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 1996–2000 RSC; (b) T. Nemoto, H. Kakei, V. Gnanadesikan, S.-Ya. Tosaki, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 14544–14545 CrossRef CAS PubMed however; Li and Smith reported that only the five-membered epoxy lactams can be prepared by epoxidation: (c) B. Li and M. B. Smith, Synth. Commun., 1995, 25, 1265–1275 CrossRef CAS.
- (a) J. H. G. Lago and M. J. Kato, Nat. Prod. Res., 2007, 21, 910–914 CrossRef CAS PubMed; (b) J. M. L. Cronan and J. H. Cardellina II, Nat. Prod. Lett., 1994, 5, 85–88 CrossRef CAS.
- (a) S. Konda, B. Kurva, L. Nagarapu and A. M. Dattatray, Tetrahedron Lett., 2015, 56, 834–836 CrossRef CAS; (b) M. S. Majik, P. S. Parameswaran and S. G. Tilve, J. Org. Chem., 2009, 74, 6378–6381 CrossRef CAS PubMed.
- A. K. Mohamed, M. L. Valerie, C. Cecile, D. S. H. Laurent and A. E. O. Nadine, Fitoterapia, 2010, 81, 632–635 CrossRef PubMed.
- L. Fuentes, U. Osorio, L. Quintero, H. Höpfl, N. Vázquez-Cabrera and F. Sartillo-Piscil, J. Org. Chem., 2012, 77, 5515–5524 CrossRef CAS PubMed.
- J. Romero-Ibañez, L. Xochicale-Santana, L. Quintero, L. Fuentes and F. Sartillo-Piscil, J. Nat. Prod., 2016, 79, 1174–1178 CrossRef PubMed.
- While attempts to epoxidize an α,β-unsaturated amide failed (see M. B. Johansen, A. B. Leduc and M. A. Kerr, Synlett, 2007, 2593–2595 CAS ), the direct synthesis of epoxyamides from allyl amines was successful for the preparation of epoxyamide precursors of the enantiomers of norbalasubramide; see L. Fuentes, M. Hernandez-Juárez, J. L. Terán, L. Quintero and F. Sartillo-Piscil, Synlett, 2013, 878–882 Search PubMed.
- T. Okada, T. Asawa, Y. Sugiyama, M. Kirihara, T. Iwai and Y. Kimura, Synlett, 2014, 596–598 CAS.
- F. Sartillo-Piscil, M. Vargas, C. Anaya de Parrodi and L. Quintero, Tetrahedron Lett., 2003, 44, 3919–3921 CrossRef CAS.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS.
- The (S)-4-methoxy-α-methylbenzylamine was selected instead of (S)-α-methylbenzylamine because the former can be removed under oxidative conditions, which are compatible with the epoxide ring functionality. Removal of the chiral substituent by reductive conditions was not effective.
- J. K. Rasmussen and S. M. Heilmann, Org. Synth., 1990, 7, 521 Search PubMed.
- (a) J. Brussee and A. van der Gen, Recl. Trav. Chim. Pays-Bas, 1991, 110, 25–26 CrossRef CAS; (b) A. M. C. H. van den Nieuwendijk, A. B. T. Ghisaidoobe, H. S. Overkleeft, J. Brussee and A. van der Gen, Tetrahedron, 2004, 60, 10385–10396 CrossRef CAS.
- Representative examples: (a) K. Kamata, T. Hirano, S. Kusuya and N. Mizuno, J. Am. Chem. Soc., 2009, 131, 6997–7004 CrossRef CAS PubMed; (b) W. Adam, P. L. Alsters, R. Neumann, C. R. Saha-Möller, D. Sloboda-Rozner and R. Zhang, J. Org. Chem., 2003, 68, 1721–1728 CrossRef CAS PubMed; (c) W. Adam, C. M. Mitchell and C. R. Saha-Möller, Eur. J. Org. Chem., 1999, 785–790 CrossRef CAS; (d) D. Prat, B. Delpech and R. Lett, Tetrahedron Lett., 1986, 27, 711–714 CrossRef CAS.
- Due to steric interactions in the conformer with the minimal transannular strain, the epoxidation of cyclooctenol affords a trans stereoselective product. See ref. 34b and c.
- (a) J. Sandoval-Lira, L. Fuentes, L. Quintero, H. Höpfl, J. M. Hernández-Pérez, J. L. Terán and F. Sartillo-Piscil, J. Org. Chem., 2015, 80, 4481–4490 CrossRef CAS PubMed; (b) S. Mejía, J. Hernández-Pérez, J. Sandoval-Lira and F. Sartillo-Piscil, Molecules, 2017, 22, 361 CrossRef PubMed.
- B. W. H. Turnbull, S. Oliver and P. E. Evans, J. Am. Chem. Soc., 2015, 137, 15374–15377 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The 1H and 13C NMR spectra of new and relevant products. The X-ray crystallographic data for 13, (1′S,3R,4R,5S)-20 and (1′S,3S,4S,5R)-20. CCDC 1576887–1576889. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob02700a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |