
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced structural diversity in terpenoid biosynthesis: enzymes, substrates and cofactors
Abith
Vattekkatte
 a,
Stefan
Garms
a,
Wolfgang
Brandt
b and
Wilhelm
Boland
*a
a,
Stefan
Garms
a,
Wolfgang
Brandt
b and
Wilhelm
Boland
*a
aDepartment of Bioorganic Chemistry, Max Planck Institute for Chemical Ecology, Beutenberg Campus, Hans-Knöll-Strasse 8, D-07745 Jena, Germany. E-mail: boland@ice.mpg.de; Fax: +49(0)3641 571 202
bDepartment of Bioorganic Chemistry, Leibniz Institute of Plant Biochemistry, Weinberg 3, 06120 Halle (Saale), Germany
First published on 18th December 2017
Abstract
The enormous diversity of terpenes found in nature is generated by enzymes known as terpene synthases, or cyclases. Some are also known for their ability to convert a single substrate into multiple products. This review comprises monoterpene and sesquiterpene synthases that are multiproduct in nature along with the regulation factors that can alter the product specificity of multiproduct terpene synthases without genetic mutations. Variations in specific assay conditions with focus on shifts in product specificity based on change in metal cofactors, assay pH and substrate geometry are described. Alterations in these simple cellular conditions provide the organism with enhanced chemodiversity without investing into new enzymatic architecture. This versatility to modulate product diversity grants organisms, especially immobile ones like plants with access to an enhanced defensive repertoire by simply altering cofactors, pH level and substrate geometry.
 Abith Vattekkatte | Abith Vattekkatte received his initial graduate education in chemistry from the University of Mumbai, India. He then moved to SUNY Albany, USA to finish his MS in organic chemistry in 2010, his thesis under Prof. Eric Block focused on chemistry of Allium (garlic and onion) species. He then joined the group of Prof. Wilhelm Boland at the Max Planck Institute for Chemical Ecology as a doctoral student with dissertation focused on multiproduct terpene biosynthesis. Abith defended his PhD thesis from Friedrich Schiller Universität, Germany in November 2015 with summa cum laude honors and as a postdoctoral fellow is involved in further developing effective enzymatic routes towards novel molecules. |
 Stefan Garms | Stefan Garms studied Chemistry at the Friedrich Schiller University in Jena with focus on organic chemistry. In 2004 he joins the Max-Planck institute of chemical Ecology in Jena for the preparation of his diploma thesis. Stefan Garms continued his work at the Max-Planck institute focusing on elucidation of enzymatic reaction cascades catalyzed by plant derived terpene synthases. After receiving his Ph.D from the University of Jena in 2011 he moved to the chemical industry, where he works as an analytical scientist in Research & Technology department at the Lonza AG in Visp, Switzerland. |
 Wolfgang Brandt | Wolfgang Brandt studied chemistry at the Martin-Luther-University Halle-Wittenberg in Halle (Germany) from 1974 till 1979. He received his Ph.D. in 1982 from the University of Halle for work on structure activity relationships of auxins under the supervision of Professor Alfred Barth. From 1985 till 1986 he was scientific co-worker at the Institute of Neurobiology and Brain Research Academy of Science of GDR in Magdeburg. In 1987 he became head of the research group “Molecular Modelling – Drug Design” at the Department of Biochemistry/Biotechnology of the University of Halle. In 1992 he was half a year in sabbatical leave at the Clinical Research Institute of Montreal, Canada (Prof. Dr P. W. Schiller). He received his “Habilitation” in 1997. In August 2001 he moved to the Leibniz Institute of Plant Biochemistry in Halle and became a group leader for computational chemistry. His research interests cover all fields of computational chemistry and molecular modelling. His work is published in over 170 scientific papers, books and some licenced patents. |
 Wilhelm Boland | Wilhelm Boland studied Chemistry and received his Ph.D. from the University of Cologne in 1978. He became Research Associate at the Institute of Biochemistry (University of Cologne) until 1987 and moved then to the University of Karlsruhe as a Professor for Organic Chemistry. In 1994 he became Full Professor for Bioorganic Chemistry at the University of Bonn. In 1997 he joined the Max Planck Society and became Director of the Department of Bioorganic Chemistry in the Max Planck Institute for Chemical Ecology in Jena. Professor Boland was the 1st Simeone-Silverstein lecturer (1995) of the International Society of Chemical Ecology and president of the society from 2008 to 2009. |
Introduction
Enzymes are potent catalysts that can enhance the reaction rates up to 20 orders of magnitude higher than uncatalyzed reactions.1–3 Most enzymes are extremely precise in nature with exceptional control over regio- and stereoselectivity for product generation.4 Evolution of enzymes over time has led to biosynthetic timelines that are in sync with cellular processes. However, reaction control and specificity take priority over rate enhancement in case of complex molecules.4,5 Moreover, the remarkable chemical diversity in nature can be attributed to the combinatorial biosynthetic chemistry that starts from a small number of simple precursors (Fig. 1).6 This chemodiversity can easily be appreciated by following synthetic challenges faced during terpene biosynthesis catalyzed by enzymes known as terpene synthases and cyclases. These biocatalysts transform simple acyclic substrates into the enormous variety of cyclic as well as acyclic compounds (Fig. 1).7–9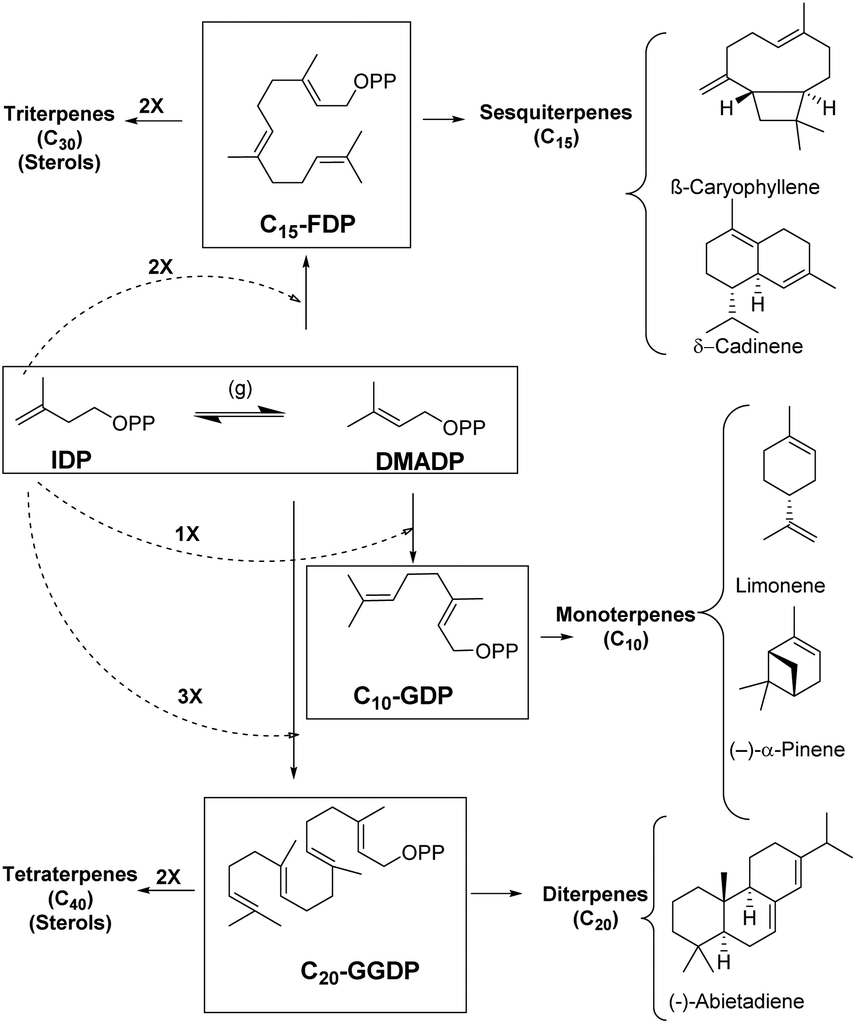 | ||
| Fig. 1 Biosynthetic pathways behind terpenoid diversity. | ||
Isopentenyl diphosphate (IDP) and dimethylallyl diphosphate (DMADP) are the principal substrates underlying the entire terpenoid diversity. Further combinatorial chain elongation generates geranyl diphosphate (GDP, C10), leading to monoterpenes (C10). Reaction with another IDP leads to chain elongation providing farnesyl diphosphate (FDP, C15), the precursor of the sesquiterpenes (C15), and, further elongation by addition of a C5 moiety leads to geranylgeranyl diphosphate (GGDP, C20), the substrate for the diterpene family (C20). GDP, FDP and GGDP perform the role of substrates for reactions in metal-mediated cleavage of pyrophosphate moiety and further intramolecular cyclization; these reactions generate diverse compounds with complex ring systems (Fig. 1).
Terpenes hold the distinction for the largest diversity among natural products, about 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds have been identified so far, among these monoterpenes, sesquiterpenes, and diterpenes represent nearly 400 distinct structural families.6,10–14 These ubiquitous natural products have been identified from insects, microorganisms and plants of both terrestrial and marine origins.15–19 Most terpenoids originate from plants and are known as precursors of many essential compounds like vitamins, hormones and medicines, apart from providing plants with their distinct fragrance and flavor properties.20 Essentially all terpenoids are derived by the condensation of multiple units of IDP and its isomer DMADP (Fig. 1) by prenyl converting enzymes, further conjugation or cyclization steps significantly enhance the chemodiversity in nature.21
000 compounds have been identified so far, among these monoterpenes, sesquiterpenes, and diterpenes represent nearly 400 distinct structural families.6,10–14 These ubiquitous natural products have been identified from insects, microorganisms and plants of both terrestrial and marine origins.15–19 Most terpenoids originate from plants and are known as precursors of many essential compounds like vitamins, hormones and medicines, apart from providing plants with their distinct fragrance and flavor properties.20 Essentially all terpenoids are derived by the condensation of multiple units of IDP and its isomer DMADP (Fig. 1) by prenyl converting enzymes, further conjugation or cyclization steps significantly enhance the chemodiversity in nature.21
A major factor behind terpenoid diversity is the ability of these highly substrate promiscous enzymes to synthesize multiple products from a single substrate through a complex reaction cascade (Fig. 2). Besides the main product, almost half of identified mono- and sesquiterpene synthases generate substantial amounts of different products with potential to be classified as multiproduct synthases.20,22 It is believed that the ability to form a large number of products from single substrate is mainly due to the electrophilic reaction cascades controlled by these enzymes.20 At the same time, this productivity reflects the tendency of nature to form mechanisms that maximize the number of products formed using the least number of biosynthetic steps.11,23,24 γ-Humulene synthase with 52 products and δ-selinene synthase with 34 products from Abies grandis are the current record holders for producing different sesquiterpenes from acyclic FDP.25–28 Rapid improvements in detection techniques have proven that these enzymes are even more promiscuous than previously thought.29,30
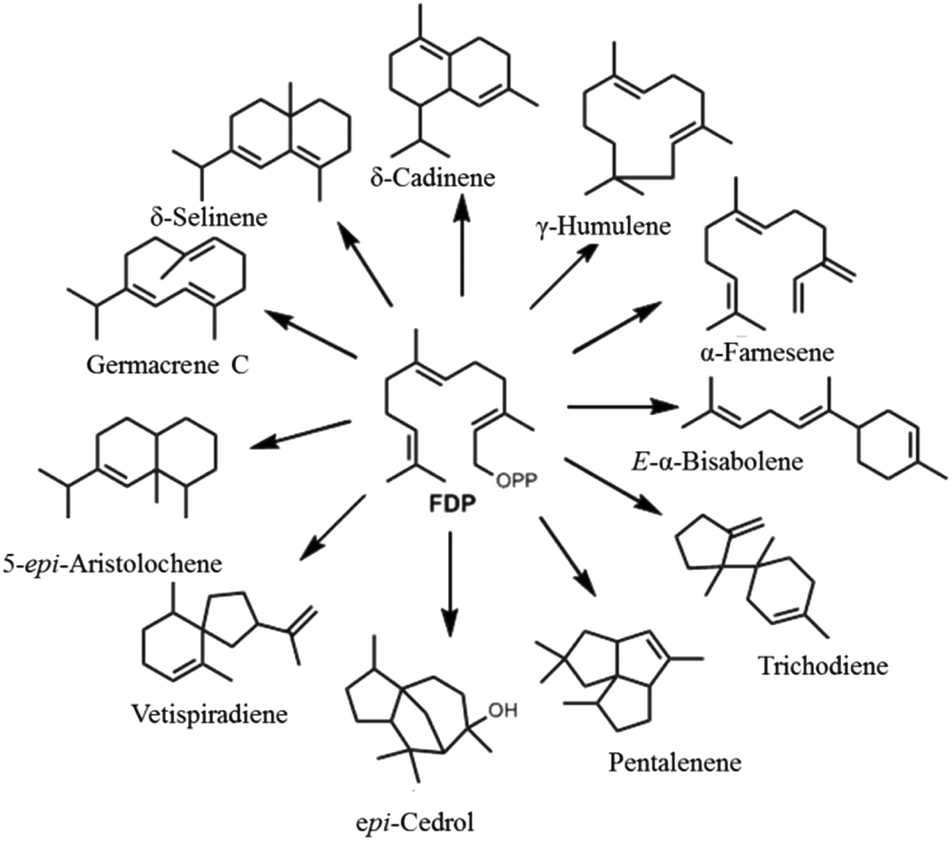 | ||
| Fig. 2 Representative figure of multiproduct terpene synthases converting a single substrate (FDP) into a bouquet of cyclic and acyclic products. | ||
This mini-review focuses on the role of reaction conditions in terpene synthase activity, such as metal cofactors, stereochemistry of the substrates and pH level. We list mono-, sesqui- and triterpene synthases that have been reported to generate multiple products. We also briefly summarize the current knowledge about the reaction mechanisms of the multiproduct terpene synthases.
Terpene synthase structure and multiple products
Many of the monoterpene and sesquiterpene synthases possess the ability to produce multiple products. This multiproduct nature may be due to their ability to allow various conformations leading to stabilization of the reactive carbocationic intermediates. In contrast, single product enzymes focus their entire catalytic activity onto a single product and, hence, give clear indication that multiple product formation is not accidentally but completely under catalytic control of the enzyme.20 Specific terpene synthases have distinctive ways of generating multiple products. Hence, the active site architecture plays a critical role in the tight control of reaction cascade. For e.g., the γ-humulene synthase of A. grandis needs two DDxxD motifs across each other at the entrance of active site to produce 52 distinct products from FDP. In the absence of one DDxxD motif, mutagenesis studies have shown fewer products indicating two distinct conformations of binding substrates in the motifs generate the different products. The presence of NSE/DTE motif at identical location as the second DDxxD motif in some terpene synthases leads to augmentation of multiple product formation.20 Whereas, terpene synthase TPS4 of Zea mays, which generates 7-epi-sesquithujene and β-bisabolene as major products, as well as 12 other terpenes in minor amounts is known to possess only a single DDxxD motif for multiproduct activity.TPS4 was used to study the correlation between active site architecture and the multiple product formation.31 Modelling of the active site cavity and docking simulations with substrate revealed the presence of two different pockets in the active site controlling the conformational change in the carbocation intermediate.32 The flexibility of conformations in the active site could also be a major factor in multiple product formation due to the possibility of numerous intermediates leading to more products. However, the active site has to ensure the correct substrate conformation so as to prevent the highly reactive carbocation from interacting with other electron-rich areas and premature quenching by water molecules. Such effects are termed as “negative catalysis” wherein the catalytic selectivity is the result of blocking side reactions over promoting desired product catalysis.33 The active site mechanism thus has to ensure the precise folding to allow conformational access to the right geometry and exclude any nucleophilic access to the carbocation. Hence, even though the multitude of products indicate the possibility of an unrestricted reaction cascade, in reality, the high stereospecificity, especially in the case of MtTPS5 from Medicago truncatula, shows strict folding and conformationally controlled access to the carbocation; all of the different products are highly enantiomerically pure.34 The major conformational variations in the intermediates are not feasible in short duration involving cationic cyclizations.35,36 Thermodynamically preferred conformations explain this phenomenon as opposed to be part of the natural reaction mechanism course, based on the co-crystallization and structural elucidation of trichodiene synthase with an intermediate analog.13,37 This leads to conclusion that the reaction cascade in terpene synthases are most likely controlled by kinetic factors instead of thermodynamic factors.35
Detailed investigation of terpene synthase structure–function relationship is absolutely essential to correlate the enzymatic features that catalyze the multiple product biosynthesis. The lack of multiproduct terpene synthase crystal structures and definitive structural assignments still impedes much headway for this domain. It would be critical to identify the amino acid residues that stabilize the concluding steps of the reaction cascade by structural elucidation via co-crystallization candidates.38–40 Single product generating 5-epi-aristolochene synthase (TEAS) cocrystallized with a substrate analog farnesyl hydroxylphosphonate shows that a catalytic triad with a tyrosine (Tyr520) and two aspartates (Asp444 and Asp525) is responsible for a critical reprotonation step.41 In the case of MtTPS5 enzyme which follows a similar pathway to multiple products, a single amino acid substitution leads to a significant shift in the product profile; the exchange of tyrosine (Y526) with phenylalanine prevents the formation of the key intermediate germacrene D (cf.Fig. 6). Many such mutagenesis studies have shown alterations in product profiles for other enzymes.34 Although, alterations by single amino acid mutations present an interesting prospect, dramatic shifts in both qualitative and quantitative terms can also be observed with simple changes in substrates, metal cofactors and assay pH.
We present below a list of terpene synthases with known ability to generate multiple products from a single substrate. Monoterpene synthases (Table 1) and sesquiterpene synthases (Table 2) are known to be multiproduct in nature, but recently also few examples of diterpene synthases (Table 3) have been reported which can channel the longer chain substrate in its active site to multiple products.
| Gene bank accession no. | Main product(s)a (%) | Designationb | Species | Ref. |
|---|---|---|---|---|
| a The amount of major products is approximately based on the composition of total terpene compounds in enzymatic assay. b The designation has been retained as in the original publication. | ||||
| AAB70707 | (−)-Camphene (54) | Ag6 | Abies grandis | 42 |
| AAF61453 | (−)-(4S)-β-Phellandrene (52) | Ag8 | Abies grandis | 42 |
| AAF61454 | Terpinolene (42) | Ag9 | Abies grandis | 42 |
| AAB70907 | (−)-(4S)-Limonene (∼70) | Ag10 | Abies grandis | 26 |
| AAF61455 | (−)-(4S)-Limonene (35) | Ag11 | Abies grandis | 42 |
| AGU92266 | Limonene | Abies grandis | 25 | |
| AGU92267 | Limonene | Abies grandis | 25 | |
| KC145534 | Myrcene | AmGAS | Achillea millefolium | 43 |
| EF433761 | Linalool | AmNES/LIS1 | Antirrhinum majus | 44 |
| EF433762 | Linalool | AmNES/LIS2 | Antirrhinum majus | 44 |
| AAG09310 | Myrcene (56) | AtTPS10 | Arabidopsis thaliana | 45 |
| AAU01970 | 1,8-Cineole (52) | AtTPS-Cin | Arabidopsis thaliana | 46 |
| BT053763 | α-Pinene | At3g25810 | Arabidopsis thaliana | 46 |
| KF987083 | Camphene (52) | AaTPS5 | Artemisia annua | 47 |
| KF987084 | 1,8-Cineole (59) | AaTPS6 | Artemisia annua | 47 |
| AY640154 | Limonene | Cucumissativus | 48 | |
| AY787633 | Linalool | Malus domestica | 49 | |
| ABB73044 | (+)-(4R)-Limonene (∼39) | LaLIMS | Lavandula angustivolia | 50 |
| DQ263742 | α-Pinene | LaBERS | Lavandula angustifolia | 50 |
| AAX69064 | β-Myrcene (∼50) | LeMTS2 | Lycopersicon esculentum | 51 |
| AY763425 | Limonene | MtTPS1 | Medicago truncatula | 52 |
| DQ188184 | Limonene | MtTPS5 | Medicago truncatula | 52 |
| AY787633 | Linalool | Malus × domestica | 49 | |
| ABP88782 | 1,8-Cineole (∼50) | CIN | Nicotiana suaveolens | 53 |
| AAV63792 | Terpinolene (∼50) | TES | Ocimum basilicum | 54 |
| AAV63790 | Fenchol (∼50) | FES | Ocimum basilicum | 54 |
| AY693646 | α-Thujene (79) | ZIS | Ocimum basilicum | 54 |
| EU596452 | β-Myrcene | Os08g07100 | Oryzasativa cv. Nipponbare | 55 |
| AAF76186 | Myrcene (∼54) | PTS-5526 | Perilla frutescens | 56 |
| AAS47692 | (−)-β-Pinene (57) | PaTPS-Pin | Picea abies | 57 |
| AAP72020 | (−)-α-Pinene (62.5) | PsTPS2 | Picea sitchensis | 58 |
| AAO61225 | (−)-α-Pinene (79) | Pt1 | Pinus taeda | 59 |
| AAO61227 | α-Terpineol (57.3) | Pt10 | Pinus taeda | 59 |
| AAX07267 | (−)-α-Pinene (∼40) | PmeTPS1 | Pseudotsuga menziesii | 60 |
| AAX07264 | Terpinolene (∼40) | PmeTPS2 | Pseudotsuga menziesii | 60 |
| ABH07677 | 1,8-Cineole (72) | Sf-CinS1 | Salvia fruticosa | 61 |
| AAC26018 | (+)-Sabinene (63) | SSS | Salvia officinalis | 62 |
| AAC26017 | (+)-Bornyl diphosphate (75) | SBS | Salvia officinalis | 62 |
| AAM89254 | (+)-3-Carene (73) | — | Salvia stenophylla | 63 |
| ACF24767 | α-Terpineol (∼44) | SamonoTPS1 | Santalum album | 64 |
| HQ343276 | Linalool | SaSSy | Santalum album | 65 |
| HQ343278 | Linalool | SspiSSy | Santalum spicatum | 65 |
| HQ343277 | Linalool | SauSSy | Santalum austrocaledonicum | 65 |
| AAG01140 | (+)-(4R)-Limonene (∼75) | dLMS | Schizonepeta tenuifolia | 66 |
| AAS79351 | (−)-α-Terpineol (50.1) | VvTPS1891 | Vitis vinifera | 67 |
| AAS79352 | (−)-α-Terpineol (50.1) | VvTPS4568 | Vitis vinifera | 67 |
| AAL59230 | (−)-α-Terpineol (∼40) | STC1-B73 | Zea mays | 68 |
| AY518310 | (S)-Limonene | TPS4 | Zea mays | 31 |
| AY518313 | (S)-Limonene | TPS5 | Zea mays | 31 |
| ABR09292 | (−)-α-Terpineol (∼60) | TPS26-B73 | Zea mays | 68 |
| Gene bank accession no. | Main product(s)a (%) | Designationb | Species | Ref. |
|---|---|---|---|---|
| a The amount of major products is approximately based on the composition of total terpene compounds in enzymatic assay. b The designation has been retained as in the original publication. | ||||
| AAC05727 | δ-Selinene (∼25)° | Ag4 | Abies grandis | 25 |
| AAC05728 | γ-Humulene (∼29) | Ag5 | Abies grandis | 25 |
| KC145534 | Germacrene A | AmGAS | Achillea millefolium | 43 |
| AAX59990 | (+)-α-Barbatene (∼27) | At5g44630 | Arabidopsis thaliana | 69 |
| EF433761 | Nerolidol | AmNES/LIS1 | Antirrhinum majus | 44 |
| EF433762 | Nerolidol | AmNES/LIS2 | Antirrhinum majus | 44 |
| ABX83200 | δ-Cadinene (∼70) | CmTPSNY | Cucumis melo | 70 |
| EAU85540 | δ-Cadinene | Cop4 | Coprinus cinereus | 71 |
| ABB73046 | (E)-α-Bergamotene (∼74) | LaBERS | Lavandula angustivolia | 50 |
| AAG41889 | δ-Elemene (∼50) | SSTLE1 | Lycopersicon esculentum | 72 |
| AAG41890 | δ-Elemene (∼75) | SSTLE2 | Lycopersicon esculentum | 72 |
| AAC39432 | Germacrene C (64) | — | Lycopersicon esculentum | 73 |
| AAG41892 | Germacrene D (∼70) | SSTLH2 | Lycopersicon hirsutum | 72 |
| ACC66281 | β-Cubebene (∼24) | Mg25 | Magnolia grandiflora | 74 |
| ABB01625 | (−)-Cubebol (30) | MtTPS5 | Medicago truncatula | 52 |
| CAH10288 | (Z)-Muurola-3,5-diene (45) | MxpSS1 | Mentha × piperita | 75 |
| AAV63787 | γ-Cadinene (∼30) | CDS | Ocimum basilicum | 76 |
| AAV63785 | β-Selinene (∼30) | SES | Ocimum basilicum | 76 |
| AAV63788 | α-Zingiberene (∼40) | ZIS | Ocimum basilicum | 76 |
| EU596452 | Zingiberene (∼25) | Os08g07100 | Oryza sativa | 55 |
| EU596454 | (E)-β-Caryophyllene (∼47) | Os08g04500 | Oryza sativa | 55 |
| ABJ16553 | (E)-β-Caryophyllene (∼46) | OsTPS3 | Oryza sativa | 77 |
| AAS47695 | Longifolene (61) | PaTPS-Lon | Picea abies | 57 |
| AAS86322 | (−)-Germacrene D (∼60) | PatTpsB15 | Pogostemon cablin | 78 |
| AAS86323 | (−)-Patchoulol (∼50) | PatTPS177 | Pogostemon cablin | 78 |
| ACF24768 | Germacrene D-4-ol (∼39) | SasesquiTPS1 | Santalum album | 64 |
| HQ343276 | α-Santalene | SaSSy | Santalum album | 65 |
| HQ343278 | α-Santalene | SspiSSy | Santalum spicatum | 65 |
| HQ343277 | Linalool | SauSSy | Santalum austrocaledonicum | 65 |
| AAO18435 | (E,E)-Farnesol (∼45) | TPS1-B73 | Zea mays | 79 |
| AAS88571 | (S)-β-Bisabolene (∼29) | TPS4-B73 | Zea mays | 31 |
| AAS88574 | Sesquithujene (∼28) | TPS5-Del1 | Zea mays | 31 |
| AAX99146 | (E)-β-Farnesene (∼50) | TPS10-B73 | Zea mays | 80 |
| AAX40665 | (+)-Germacrene D (∼50) | — | Zingiber officinale | 81 |
| BAG12021 | β-Eudesmol (63) | ZSS2 | Zingiber zerumbet | 82 |
| Gene bank accession no. | Main product(s)a (%) | Designationb | Species | Ref. |
|---|---|---|---|---|
| a The amount of major products is approximately based on the composition of total terpene compounds in enzymatic assay. b The designation has been retained as in the original publication. | ||||
| AT4G15370 | Baruol (∼90)° | BARS1 | Arabidopsis thaliana | 30 |
| AK070534 | Achilleol B (∼90%) | — | Oryza sativa | 83 |
Catalytic mechanism
The catalytic reactions involve complex carbocationic cascades, which are initiated in two ways and based on the initial steps terpene synthases are classified as class I or II based on the initial steps. Class I terpene synthases initiate the catalysis by the heterolytic cleavage of the diphosphate ester bond, while class II terpene synthases rely on a protonation as the first step. Most of the multiproduct terpene synthases mentioned in this review belong to the class I family. The metal-mediated cleavage of the diphosphate anion in the substrate initiates the reaction cascade of a terpene synthase. The metal cations also neutralize the negative charge on the diphosphate over the course of cascade to prevent the quenching of the cation.84–89 The biosynthetic cascade is further divided into partial reactions which logically separate catalytic events. The cleavage of the diphosphate anion generates a highly reactive transoid farnesyl carbocation, which undergoes various cyclizations and rearrangements, such as methyl- or hydride shifts. Due to the constrained geometry of the transoid farnesyl cation, it can only react with the distant C10–C11 double bond, leading to the formation of the (E,E)-germacrene-11-yl cation or the (E,E)-humul-10-yl cation (Fig. 3). Some terpene synthases overcome this geometrical constraint by rotation to the cisoid farnesyl cation and subdue the energy barrier of the isomerization around C2–C3 double bond by the release and migration of diphosphate anion.90–92 The accepted mechanism proceeds by the recapture at the C3 position of diphosphate anion producing the nerolidyl diphosphate intermediate. A C2–C3 sigma bond isomerization and the subsequent dephosphorylation generates the desired (Z,E)-farnesyl cation (Fig. 3).93 In the case of sesquiterpenes, the bisabolyl cation, cycloheptenyl cation, (2Z,6E)-germacrene-11yl cation and (2Z,6E)-10-humulyl cation lead to the corresponding bisabolane, daucane, cadalane and himachalane products. The end products are released after proton elimination or by quenching with water from the hydrophobic active site.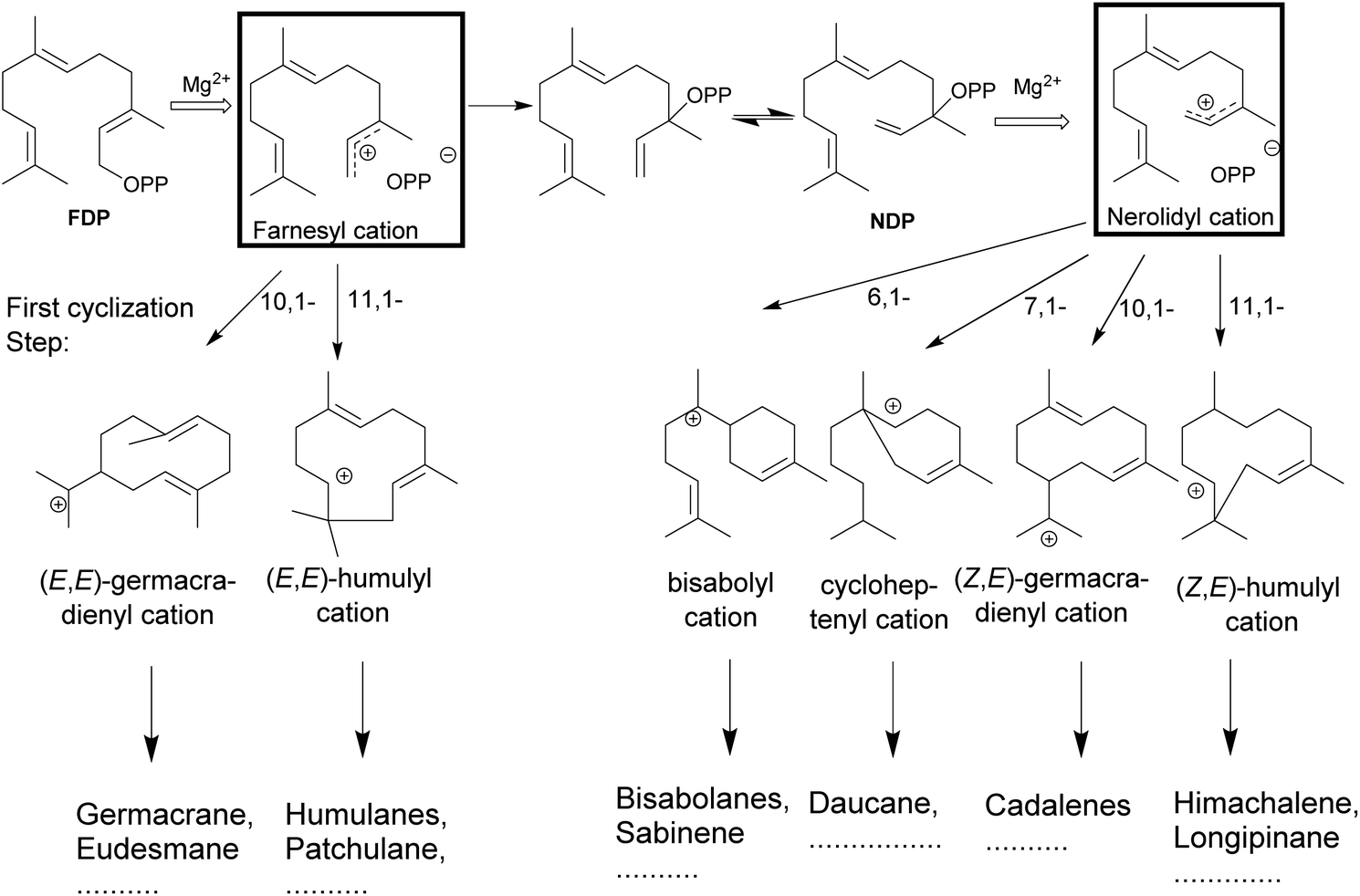 | ||
| Fig. 3 Possible cyclizations of (2E,6E)- or (2Z,6E)-farnesyl cations and some resulting carbon skeletons of sesquiterpenes. The loss of the diphosphate anion initiates the formation of a highly reactive transoid farnesyl cation, which after isomerization undergoes various cyclizations and rearrangements, methyl- or hydride shifts. Due to its constrained geometry, the electrophilic transoid farnesyl cation can attack only the distant C10–C11 double bond. However, the C2–C3 double bond isomerization to (2Z,6E)-farnesyl cations, the geometrically suitable conformation leading to other cyclic products. | ||
The available structural information suggests that the multiproduct formation depends on variations in conformation of substrate controlling the first bond formation followed by movement of the reactive intermediate allowing additional bond formation, hydrogen- or methyl group shifts, eliminations or reactions with nucleophiles to generate a neutral product. Thus enzymatic response to various mimics of reaction intermediates such as C2–C3 substrate isomers on incubation with multiproduct terpene synthases was of natural curiosity.
Substrate isomers
The universality of (E) isomers as substrates for terpene synthases was challenged by the discovery of a tomato monoterpene synthase that prefers neryl diphosphate, the (Z)-isomer of GDP as a substrate.94 In addition, a sesquiterpene synthase from wild tomato Solanum habrochaites was described, that accepts (Z,Z)-FDP as a substrate over the usual (E,E)-FDP to start sesquiterpene biosynthesis of multiple terpenoids.95 The isolation of a (Z,E)-FDP synthase from Mycobacterium tuberculosis96,97 and, more recently, a novel terpene synthase from trans-isoprenyl diphosphate synthases in the striped flea beetle98 suggest that the concept of isomeric prenyl diphosphates as natural substrates may be more prevalent than previously anticipated.Isomeric prenyl diphosphates like the cis–trans isomer of FDP (Z,E)-FDP have been used as a mimic for the cisoid neryl intermediate in various studies. This analog has been useful in the study of steps following the isomerization of C2–C3 double bond in the cyclization cascade (Fig. 3) in various terpene synthases.31 In 2010, Cop4 and Cop6 from Coprinus cinereus were investigated with both geometric isomers. The conversion of (E,E)-FDP in the case of Cop6 continues through a (6R)-β-bisabolyl carbocation and in the case of Cop4 through an (E,E)-germacradienyl carbocation. However, in the case of (Z,E)-FDP as substrate, both Cop4 and Cop6, the cascade continues via the (6S)-β-bisabolene cation.99
Interestingly, the same substrate showed dramatic shifts in both, quantitative and qualitative terms with other multiproduct terpene synthases from Zea mays and Medicago truncatula as discussed below.
Maize multiproduct terpene synthase enzymes TPS4-B73 and TPS5-Delprim, were incubated with various isotopomers of natural substrates to study the effects of change in the double bond geometry and final deprotonation reaction (Fig. 4A). Hence (Z)-[2-2H]- and [2,4,4,9,9,9-2H6]-geranyl diphosphates (GDP) and (Z,E)-[2-2H] and [2,4,4,13,13,13-2H6]-farnesyl diphosphates (FDP) were synthesized as substrate analogs with similarity to presumptive reaction intermediates. Remarkably, strong decrease in acyclic products resulting from the deuterated substrates was revealed on comparison with those from unlabeled (E)-GDP and (E,E)-FDP (Fig. 4C). Additionally, TPS4 and TPS5 exhibit higher turnover when incubated with (2Z)-substrates, due to the lack of kinetic isotope effects of primary deuterium atoms for terminating deprotonations and secondary deuterium atoms resulting from the hyperconjugation stabilization of the reaction intermediates (Fig. 4B). This observation of isotopically sensitive branching of product formation in maize supports the enzymatic biosynthesis of mono- and sesquiterpene volatiles from a shared carbocationic precursor for branched cascade in multiproduct synthases.100,101
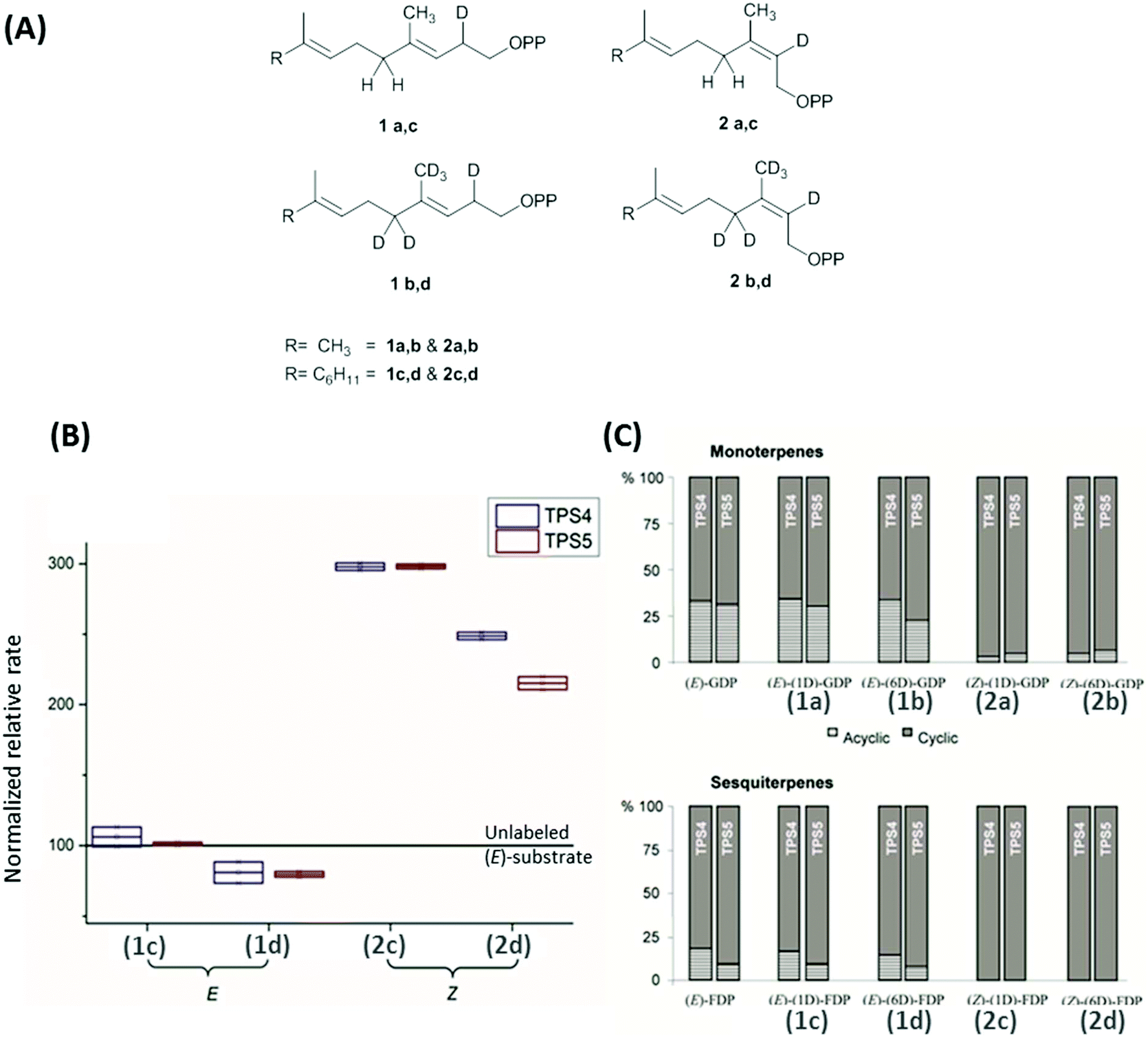 | ||
| Fig. 4 (A) Monodeuterated and hexadeuterated (2E)-GDP, (2E)-FDP (1a–d) and (2Z)-GDP, (2Z)-FDP (2a–d) used for isotope sensitive branching studies. (B) Comparison of total rates of sesquiterpene formation and (C) ratio acyclic/cyclic volatile formation with the incubation of deuterated (E)/(Z)-GDP and (E)/(Z)-FDP with TPS4 and TPS5. (B) Comparison of relative rates of sesquiterpene volatile formation from monodeuterated substrates (1c, 2c) and hexadeuterated substrates (1d, 2d). Volatile production improved by ∼200% and ∼150% with the mono and hexadeuterated substrate on comparison with unlabelled substrate (baseline at 100). (C) There was a distinct absence of acyclic products in the substrate with (2Z)-substrates seen in both monoterpenes (2a, 2b) and sesquiterpenes (2c, 2d).100 | ||
TPS4 and TPS5 from Zea mays showed major turnover enhancements with isomers as substrates although with the similar product profile as compared with natural substrates.100 However, the examination of MtTPS5 enzyme from Medicago truncatula for substrate promiscuity with (Z,E)-FDP showed massive variations in product profile. Surprisingly, completely new products with no similarity to previous products was observed with the cis-substrate analog.102 The substrate isomer initiated a novel cyclization pathway of sesquiterpenes resulting in humulane, amorphene and himachalane based products (Fig. 5). The absolute configuration of each product was determined, which helped in reconstruction of the exact stereochemical path followed by the reaction cascade. Interestingly, only one enantiomer of each product was observed indicating that a high stereoselective control of the enzyme over the reaction cascade is retained. Substrate promiscuity observed in terpene synthases provides living organisms with access to optically pure chemical blends without any optimization or mutations of available enzymatic architecture. The possible occurrence of this alternate cascade in nature was shown by the presence of these new products in natural volatiles of Medicago truncatula.102
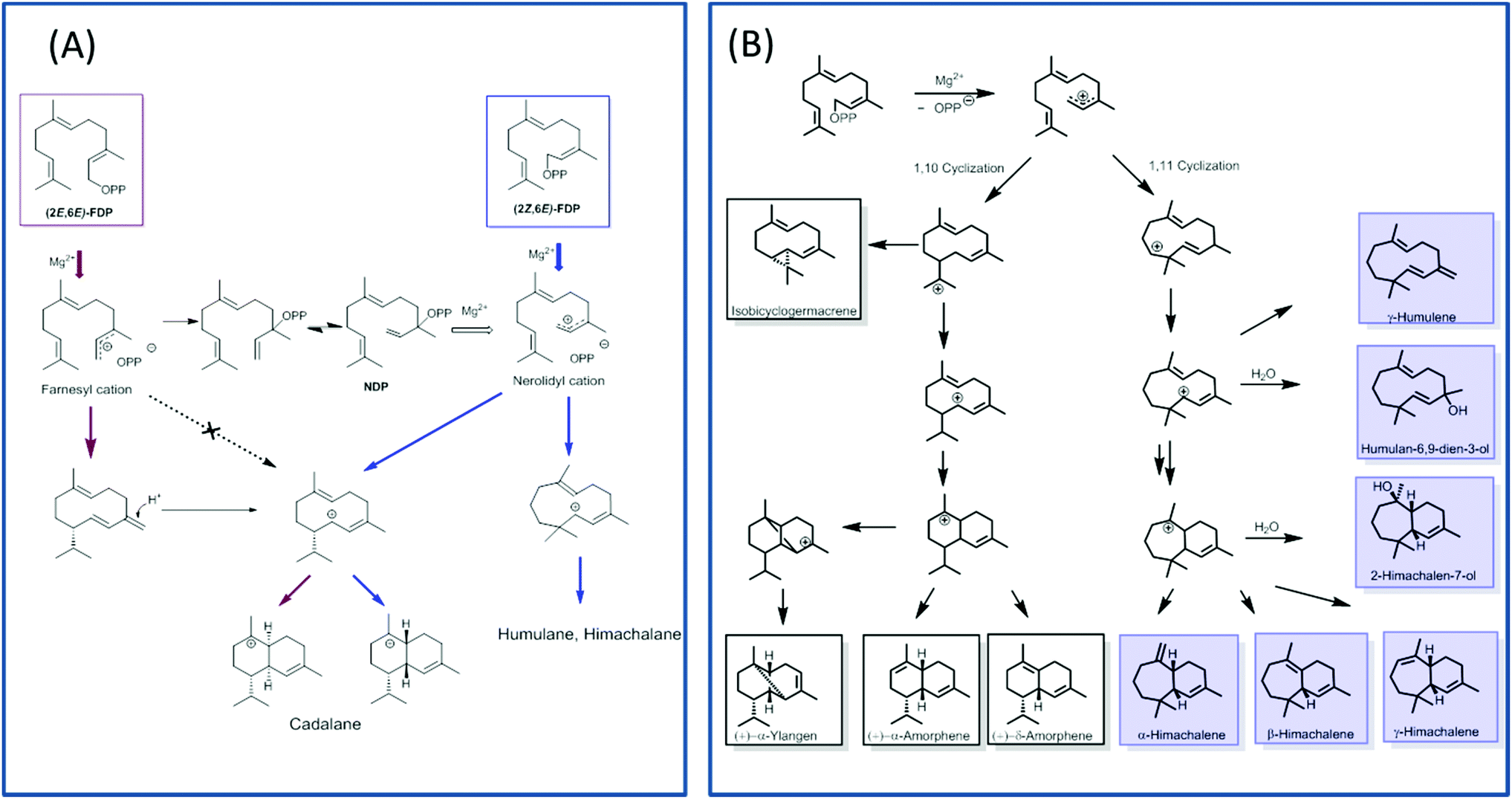 | ||
| Fig. 5 Representative figure depicting alternate cyclization pathways of MtTPS5 with isomeric farnesyldiphosphates. (A) Cyclization initiated by (2E,6E)-FDP (red arrows) or (2Z,6E)-FDP (blue arrows). (B) The cyclization pathway catalysed by MtTPS5 on incubation with (2Z,6E)-FDP prefers the (1,11) cyclization leading to humulene and himachalane products (blue) over the (1,10) cyclization producing cadalanes. | ||
In order to understand the major shift in product profile in the MtTPS5 enzyme, docking studies were performed with both substrates ((E,E)-FDP (Fig. 6A) and (Z,E)-FDP (Fig. 6B)). Interestingly, in (E,E)-FDP (Fig. 6A), cyclic conformations showed the methyl group at C3 in the direction of the postulated catalytic triad (D450, Y526, N530), which is responsible for the proton transfer in the later course of the reaction that forms germacrene D. The proximity of the potential proton source and the acceptor underpin the important part played by the hydroxyl group of tyrosine (Y526) in the transfer of proton, a role that in our previous work was demonstrated by mutagenesis and labeling studies.34 However, the tyrosine exchange by phenylalanine group does not completely deactivate the reprotonation step, indicating the presence of a water molecule as a Brønsted acid.28,34 The Y526F mutant generates almost exclusively germacranes (80%) leading to dominant cadalanes being reduced to 15% of product profile. The critical part played by the hydroxyl group of tyrosine (Y526) leading to intermediate germacrene D is supported by increase in the precursor related products.
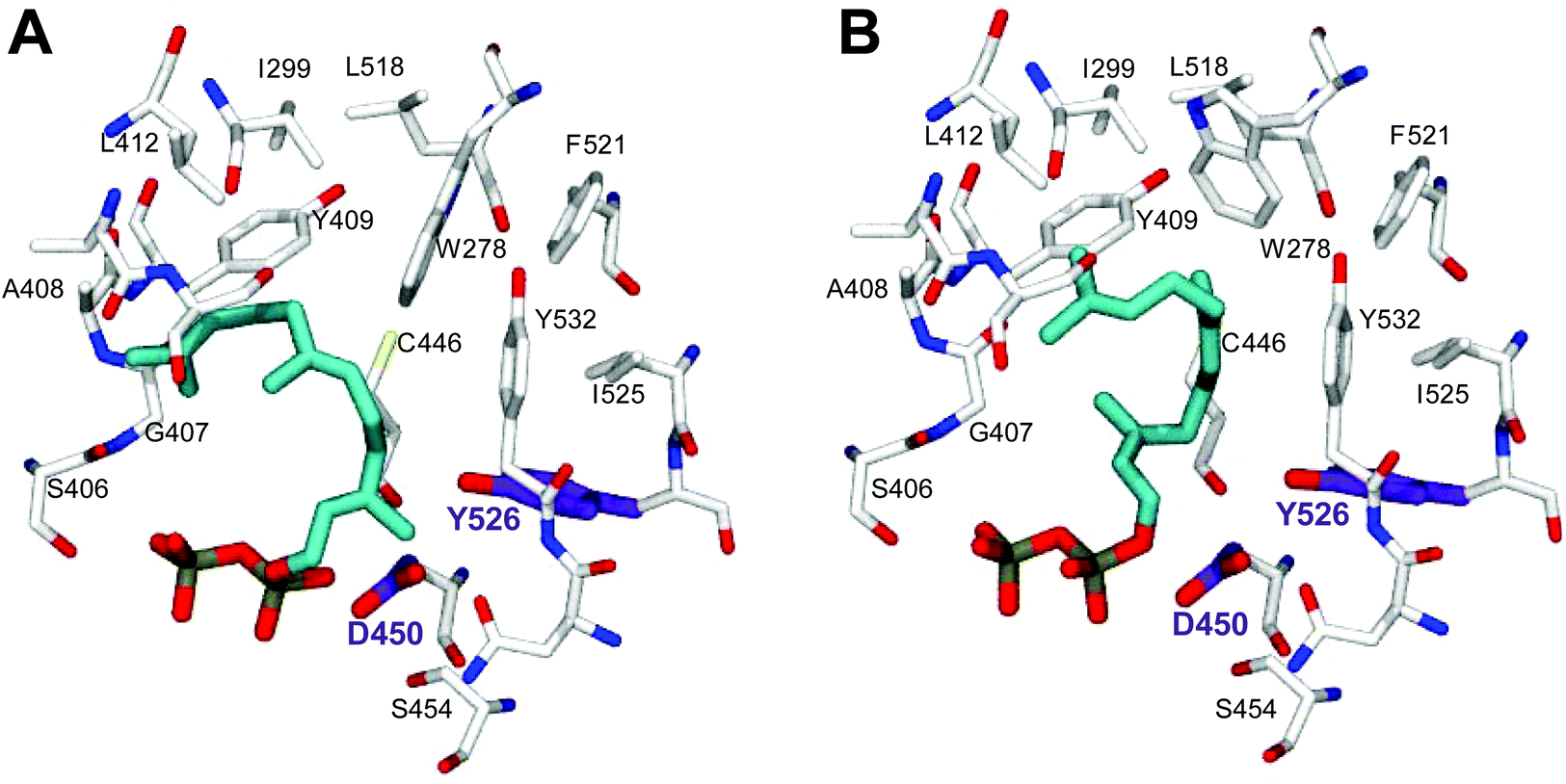 | ||
| Fig. 6 Docking studies of the MtTPS5 active site with (2E,6E)-FDP (A) and (2Z,6E)-FDP (B). The conformations were selected on the basis of the products absolute configurations. The amino acid involved in proton transfer (tyrosine, Y526) is highlighted in violet, the substrate in turquoise. | ||
In contrast, no conformations in the in silico analysis of (Z,E)-FDP, could justify the cyclized products in which the methyl group at C3 was localized in the vicinity of Y526. This finding is consistent with D2O-labeling experiments in which no labeled products were found. A total of 7 conformations were identified that allowed cyclization to a 10- or 11-membered ring (C1–C10 and C1–C11). Only one of these conformations led to the observed stereochemistry of the bridgehead hydrogen atoms of himachalane and amorphane (Fig. 6B). Hence, on the basis of these models it can be safely inferred that different starting conformations lead to differences in interactions corresponding to active site amino acid residues. In multiproduct terpene synthases with substrate isomers, these differences initiate an alternate cyclization cascade.102
Thus, geometrical isomers of substrates provide terpene synthases with the advantage to utilize the existing active site architecture for turnover advantage but also to initiate the biosynthesis of novel enzymatic chemistry. These observations are interesting, because with the upstream regulation of terpene biosynthesis the substrate geometry can be easily altered, giving rise to a possible new range of substrate isomers. This promiscuity with substrate geometry offers organism access to products which are more cost-effective than evolutionarily expensive mutations. However, the product specificity of terpene synthases may also depend on local factors such as the concentrations of metal cofactors and the pH level of the assays.
Metal cofactors as modulators of terpene biosynthesis
Multiple factors including some unknown factors govern the induction and regulation of terpenoid pathways.103 Metal cofactors play a critical role in initiation of the catalytic cascade with cleavage of the diphosphate moiety. Sesquiterpene synthases have been reported to prefer Mg2+ as a cofactor in vitro, but they also accept Mn2+ at low concentrations.104–107 In general, monoterpene synthases seem to be less selective in their divalent cation requirements.91 Insights into the mechanistic roles for the metal ions have been obtained from the crystallographic studies of 5-epi-aristolochene synthase (TEAS),41 pentalenene synthase,23 bornyl diphosphate synthase108 trichodiene synthase109 and more recently, sesquiterpene cyclase epi-isozizaene synthase (EIZS) from Streptomyces coelicolor.110 Interestingly, examination of the metal requirement of an avian prenyltransferase,111 supported the presence of two divalent metal ions in binding site for the highly charged diphosphate group, leading to the formation of a substrate complex with the enzyme. At least a single DDxxD motif appears in nearly all type I terpene synthases and these aspartates residues direct the substrate binding via the coordination of magnesium ions by forming salt bridges with the diphosphate group. This binding mechanism has been confirmed by X-ray crystallography studies of trichodiene synthase,109 bornyl diphosphate synthase108 and farnesyl diphosphate synthase.112A recent research work by Frick et al.113 involving a prenyl diphosphate synthase from the leaf beetle Phaedon cochleariae demonstrates that regulation of the local concentration of particular metal ions can induce a major switch in product specificity (Fig. 7). The P. cochleariae isoprenyl diphosphate synthase 1 (PcIDS1) on incubation with metal cofactors Co2+ or Mn2+, produces 96% GDP and merely 4% FDP, whereas in the presence of Mg2+, PcIDS1 yields 18% GDP and 82% FDP.
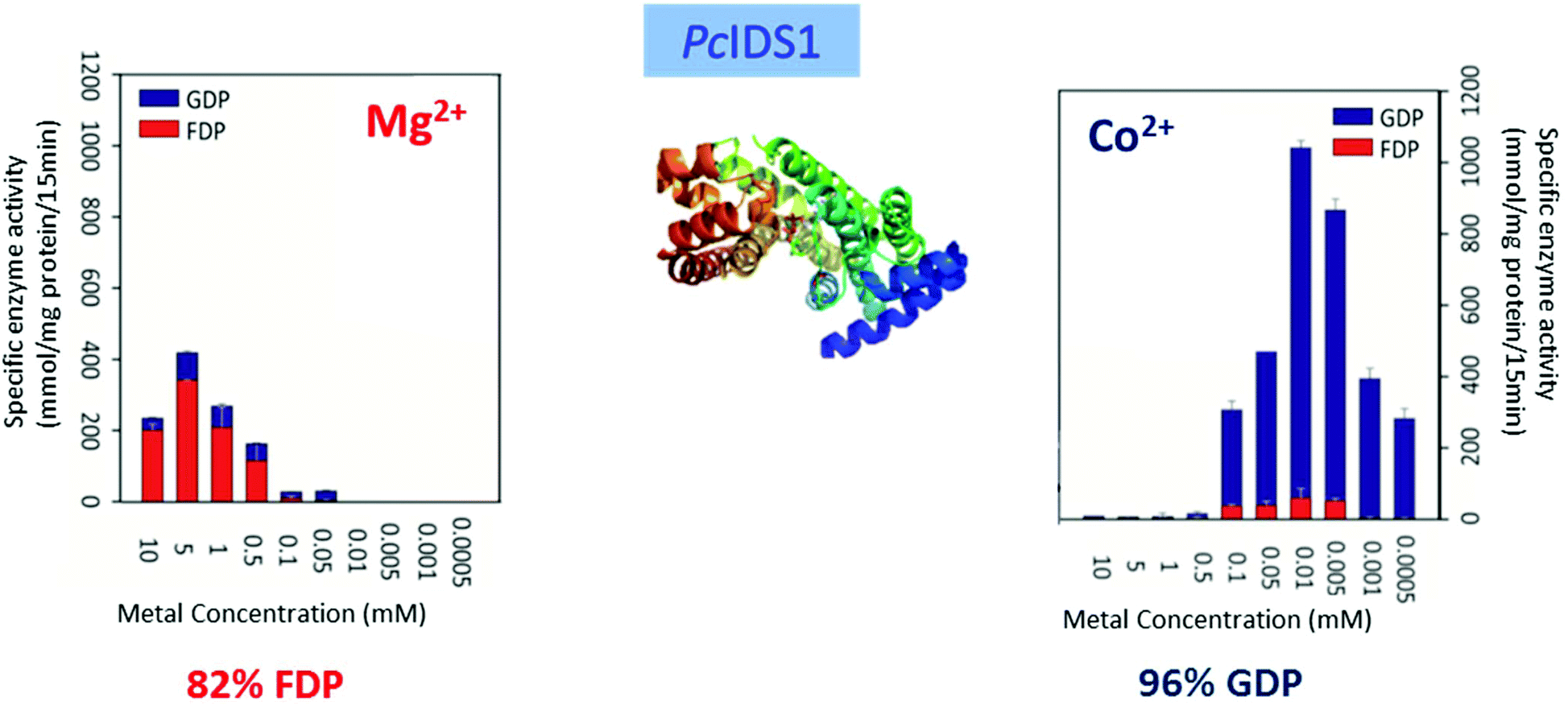 | ||
| Fig. 7 Comparison of P. cochleariae isoprenyl diphosphate synthase 1 (PcIDS1) with Co2+ or Mg2+ cations as metal cofactors. | ||
A detailed study involving in vitro PcIDS1 assays tested the activity of allylic substrates (IDP, DMADP and GDP) with many divalent cations (Co2+, Mg2+, Mn2+, Ni2+ and Zn2+). The combined IDP and DMADP based assays showed maximum PcIDS1 activity with Co2+ as the metal cofactor. The importance of these results lies in the observation that with Co2+, PcIDS1 favorably condenses IDP and DMADP to generate GDP which is critical for insect defense. Whereas, PcIDS1 with Mg2+ as a cofactor, prefers combination of IDP and GDP resulting in FDP, which in turn produces sesquiterpenes important for the insect's development.113 Hence a switch in the metal cofactor could be used by same enzyme for different products. Farnesyl diphosphate synthase (FPPS) in Aedes aegypti (AaFPPS) expressed in the corpora allata acts in diverse ways in the company of different metal ions: Mg2+ ions prefers the production of FDP, whereas Co2+ ions generates mostly GDP.114 The metal cations Mg2+ and Mn2+ are the most common initiators of the ionization of FDP in sesquiterpene synthases by binding with the diphosphate moiety of the substrate.115–117 However, there are examples of some plant sesquiterpene synthases which show catalytic activity in the presence of other metal ions.117,118 Apart from cleavage of diphosphate moiety, metal ions are involved in other roles such as the initiation of terpene synthases; like in case of apple α-farnesene synthase in which the catalytic activity is triggered by a monovalent potassium cation within the binding site.119 Δ6-Protoilludene synthases from a fungus usually proceeds through specific 1,11 cyclization pathway to produce the trans-humulyl cation derived Δ6-protoilludene. However, heterologous expression of the Δ6-protoilludene synthases show that different divalent ions, in particular Ca2+, can alter cyclization specificity by a switch between dual 1,10 and 1,11 cyclization pathways.120 Ca2+ plays a significant role in fungal physiology and the significant switch shown by divalent metal ion indicates a probable role for the metal ion in signaling and defense mechanisms in fungi through terpenes. The product specificity was also observed in case of AaTPS2 and AaTPS5, in presence of Mn2+ the enzymes showed the production of linalool which was absent in Mg2+ based assays.47
In terms of multiproduct synthases, a range of metal ions was tested for activity with TPS4 and TPS5 from maize and MtTPS5 from Medicago truncatula. Because more than one product is generated, variations in product concentration can be observed with each divalent metal ion.
Even though the change is not as dramatic as in insect terpene synthases, there are distinct differences in total product concentrations as well as individual product concentrations in multiproduct terpene synthases. In TPS4 and TPS5, the maximum activity is observed with Mg2+ cations. Detailed examination of individual product concentration revealed that almost all the products showed variation in product concentrations.31 In the case of MtTPS5 from Medicago truncatula, both Mg2+ cations and Mn2+ cations showed comparable maximum activity but at different concentrations (Fig. 8). However, cubebol loses its status as the major product to germacrene D with the switch from Mg2+ cations to Mn2+ ions.34 Hence, these examples point to an interesting case of local metal ion concentrations dependent mechanism which regulates the metabolic pathways into which single enzymes can allocate its resources to their defense or growth. Metal cations play a critical role in the terpene biosynthesis of both single product and multiproduct terpene synthases.
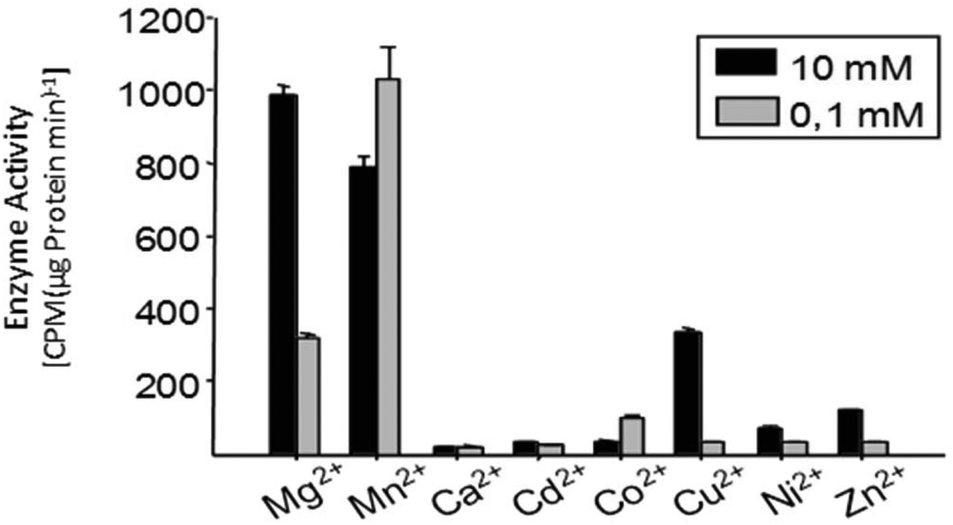 | ||
| Fig. 8 Comparison of enzyme activity of MtTPS5 from Medicago truncatula with different divalent metal ions. The dependence of the enzyme activity on various divalent metal ions was tested after incubation of the high-purity protein MtTPS5 with [1-H3]-FDP; the reaction products were extracted with pentane and quantified with a scintillation counter. | ||
Effect of pH on product specificity
Multiproduct terpene synthases are proteins associated with membranes, and the huge diversity of their products allows us to study the effect of pH level on product concentrations. An interesting shift was observed in MtTPS5 from Medicago truncatula: when the pH level increased, there was a distinct preference for products based on cadalane over germacerene based products. At higher pH values, there was complete domination of germacrene based products (Fig. 9).34 Whether this pH-dependent shift of product composition has a biological significance remains to be established.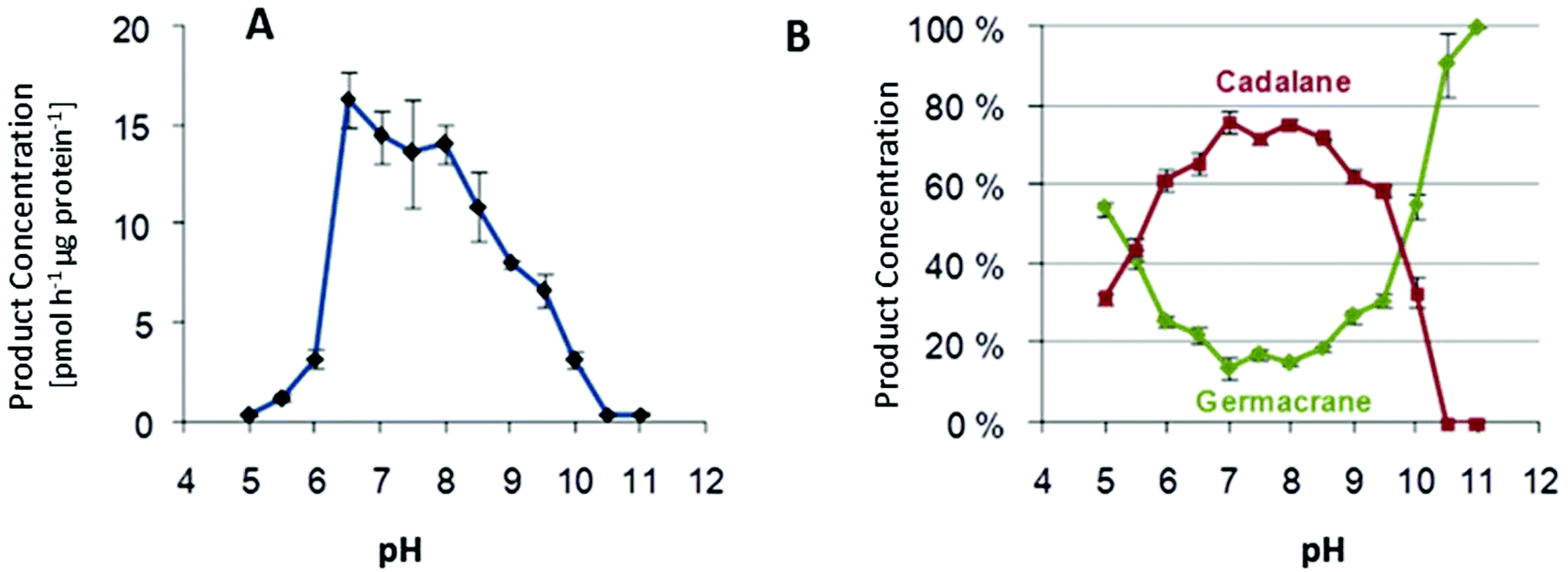 | ||
| Fig. 9 (A) Variation of total enzymatic activity of the MtTPS5 over a wide pH range on incubation with (2E,6E)-FDP. (B) Variation in cyclization preference over a range of pH. The highly purified protein by gel filtration was incubated at different pHs with (A): [1-3H]-(2E,6E)-FDP and the products extracted with pentane were quantified by a scintillation counter or (B): with unlabelled FDP and the products analyzed by GC-MS. Shown are the sums of the percentages of germacrane (green) and cadalane (red) on the total product amount. | ||
An interesting trend continues with multiproduct Cop4 and Cop6 from Coprinus cinereus, but they both show completely opposite behavior on pH variation. Cop6 demonstrates identical product profile under all conditions and 98% of α-cuprenene is generated from FDP. In case of Cop4, in both alkaline and acidic conditions it shows a preference for single major product down from three major compounds generated under neutral assay conditions. The cyclization cascade ends at pH 10 to produce (−)-germacrene D (91%) with the hydride shift and deprotonation of the germacradienyl cation, whereas at pH 5.0 cyclization continues besides (−)-germacrene D through a 1,6-ring closure to produce also a δ-cadinene (12%).99
In ecological terms, multiproduct terpene synthases provide organisms with a huge ecological advantage. The biosynthesis of pheromones requires a highly specific enzyme for a specific catalytic product along with a corresponding specific receptor in another organism. However, during the biosynthesis of plant defensive compounds, a plant can utilize this as an adaptive mechanism to protect itself from damage by a larger variety of herbivores. An ideal example of multiple terpene volatiles acting together as indirect defense is observed in maize as a response to S. littoralis feeding, as these volatiles attract the parasitic Cotesia marginiventris wasps which oviposits eggs into the S. littoralis leading to their eventual death (Fig. 10). Multiproduct TPS10 enzyme from maize on incubation with farnesyl diphosphate, produces (E)-β-farnesene, (E)-α-bergamotene, and additional S. littoralis-triggered sesquiterpenes.121 Olfactometric experiments with transgenic Arabidopsis plants overexpressing TPS10 enzymes showed that female parasitoid C. marginiventris used the TPS10 sesquiterpenes to trace their hosts with associative learning. The overexpression of the herbivore-induced (E)-β-caryophyllene synthase TPS23 in Arabidopsis showed that the volatile (E)-β-caryophyllene is associated with host presence by C. marginiventris (Fig. 10).122 Whereas in opposite scenario, under the soil TPS23 from maize produces a single compound (E)-β-caryophyllene, which is involved in indirect defense by attracting entomopathogenic nematodes to maize roots by Diabrotica. v. virgifera (Fig. 10). The changes in individual components in a blend of sesquiterpenes were too broad to identify different genotypes of maize. Parasitic wasps like C. marginiventris, can associate an effective oviposition of eggs with the terpenoid blends they come across in maize. During the course of field experiments, wasps show recognition ability to differentiate between diverse grass species. As the parasitic wasps use long term volatile-based instinctive reactions and associative learning, they can use this strategy toward the discerning the more beneficial plants.123,124 Interplanting maize with Melinis minutiflora, considerably diminished intensity of stem borers infestation by enhanced the larval parasitism by Cotesia sesamiae.125 This strategy can prove to be effective pest management tool by manipulating the terpene volatile blends in crop plants by attracting enemies of herbivores. With better understanding and successful elucidation of terpene biosynthetic pathways crop plants can be engineered to emit strong volatiles and their regulation with multiple factors.
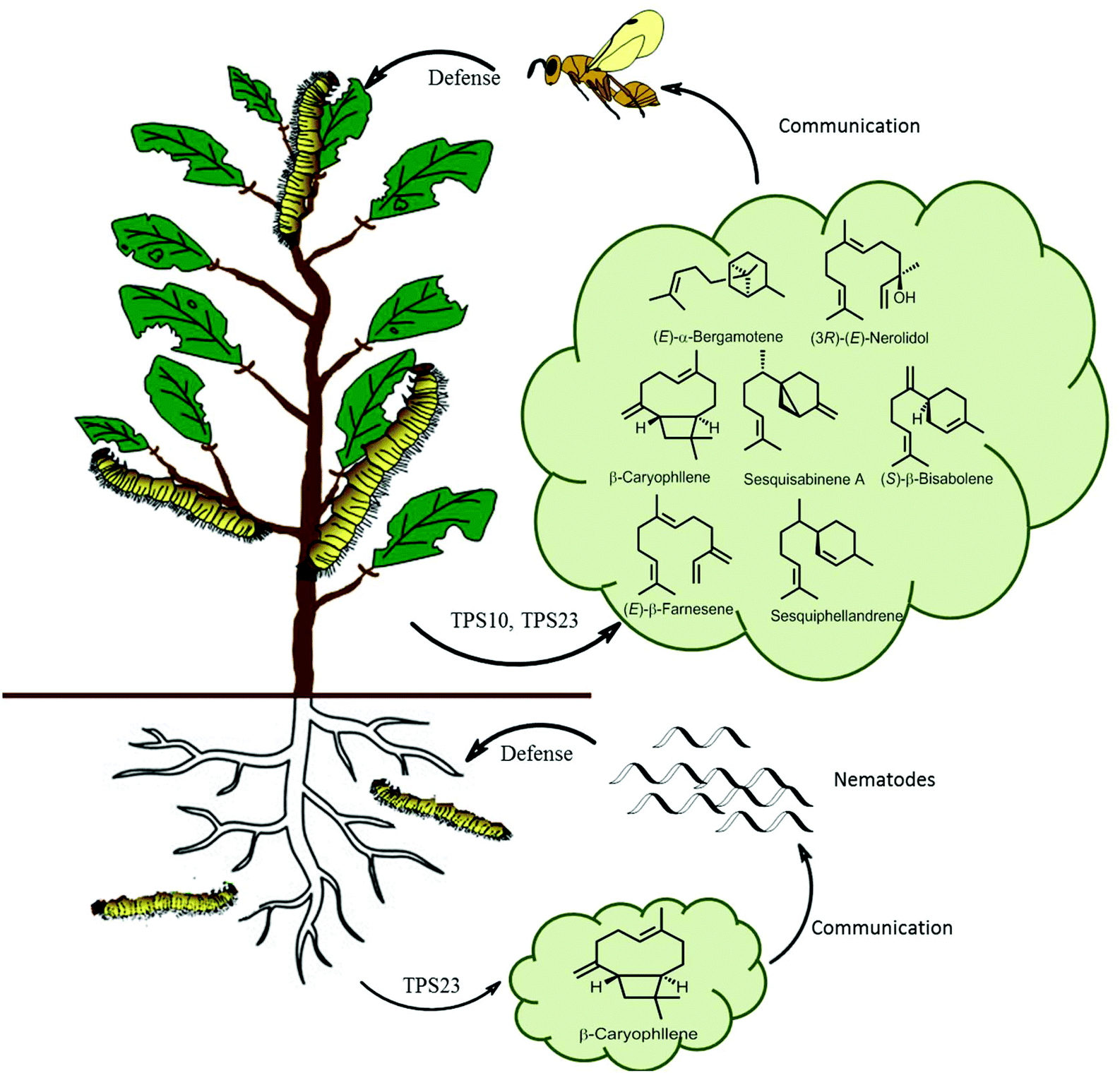 | ||
| Fig. 10 Scheme of sesquiterpene-based communication of a maize plant both above and below ground. Lepidopteran herbivores feeding of maize leaves activates TPS10 and TPS23, producing a volatile terpene blend which attracts parasitic wasps. Root damage by D. v. virgifera activates the terpene synthase TPS23 which attracts pathogenic nematodes.126 | ||
In this review, we have shown examples in which multiple products generated by multiproduct terpene synthases can be modulated by assay conditions. This modulation involves a dramatic shift in product specificity by pH level and metal cofactors and, more interestingly, the biosynthesis of novel products by substrate isomers using the alternative cyclizations cascade. Our hypothesis suggests that simple changes in assay conditions may lead an enzyme to shift its native preferences, allowing the organism to altering product preferences without associated costly changes to enzymatic structure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the Max Planck Society for support. We are also thankful to Dr Maritta Kunert and Kerstin Ploss for help with proofreading of manuscript. Open Access funding provided by the Max Planck Society.References
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed
.
- A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 5250–5254 CrossRef CAS
- J. Gao, S. Ma, D. T. Major, K. Nam, J. Pu and D. G. Truhlar, Chem. Rev., 2006, 106, 3188–3209 CrossRef CAS PubMed
- J. Clardy and C. Walsh, Nature, 2004, 432, 829–837 CrossRef CAS PubMed
- D. T. Major, Y. Freud and M. Weitman, Curr. Opin. Chem. Biol., 2014, 21, 25–33 CrossRef CAS PubMed
- J. Gershenzon and N. Dudareva, Nat. Chem. Biol., 2007, 3, 408–414 CrossRef CAS PubMed
- D. Tholl, Curr. Opin. Plant Biol., 2006, 9, 297–304 CrossRef CAS PubMed
- E. Oldfield and F. Y. Lin, Angew. Chem., Int. Ed., 2012, 51, 1124–1137 CrossRef CAS PubMed
- J. Bohlmann, G. Meyer-Gauen and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4126–4133 CrossRef CAS
-
J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman & Hall, New York, 1992 Search PubMed
- J. C. Sacchettini and C. D. Poulter, Science, 1997, 277, 1788–1789 CrossRef CAS PubMed
- Y. Yamada, T. Kuzuyama, M. Komatsu, K. Shin-ya, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 857–862 CrossRef CAS PubMed
- D. W. Christianson, Science, 2007, 316, 60–61 CrossRef PubMed
- H. V. Thulasiram, H. K. Erickson and C. D. Poulter, Science, 2007, 316, 73–76 CrossRef CAS PubMed
- S. C. Roberts, Nat. Chem. Biol., 2007, 3, 387–395 CrossRef CAS PubMed
- B. M. Fraga, Nat. Prod. Rep., 2011, 28, 1580–1610 RSC
- B. M. Fraga, Nat. Prod. Rep., 2009, 26, 1125–1155 RSC
- B. M. Fraga, Nat. Prod. Rep., 2000, 17, 483–504 RSC
- T. Sato, H. Yamaga, S. Kashima, Y. Murata, T. Shinada, C. Nakano and T. Hoshino, ChemBioChem, 2013, 14, 822–825 CrossRef CAS PubMed
- J. Degenhardt, T. G. Köllner and J. Gershenzon, Phytochemistry, 2009, 70, 1621–1637 CrossRef CAS PubMed
- W. Brandt, L. Bräuer, N. Günnewich, J. Kufka, F. Rausch, D. Schulze, E. Schulze, R. Weber, S. Zakharova and L. Wessjohann, Phytochemistry, 2009, 70, 1758–1775 CrossRef CAS PubMed
- H. Gambliel and R. Croteau, J. Biol. Chem., 1984, 259, 740–748 CAS
- C. A. Lesburg, G. Z. Zhai, D. E. Cane and D. W. Christianson, Science, 1997, 277, 1820 CrossRef CAS PubMed
- D. E. Cane, Compr. Nat. Prod. Chem., 1999, 2, 155–200 CAS
- C. L. Steele, J. Crock, J. Bohlmann and R. Croteau, J. Biol. Chem., 1998, 273, 2078 CrossRef CAS PubMed
- J. Bohlmann, C. L. Steele and R. Croteau, J. Biol. Chem., 1997, 272, 21784–21792 CrossRef CAS PubMed
- X. Y. Chen, Y. Chen, P. Heinstein and V. J. Davisson, Arch. Biochem. Biophys., 1995, 324, 255–266 CrossRef CAS PubMed
- B. Felicetti and D. E. Cane, J. Am. Chem. Soc., 2004, 126, 7212–7221 CrossRef CAS PubMed
- N. L. Brock, S. R. Ravella, S. Schulz and J. S. Dickschat, Angew. Chem., Int. Ed., 2013, 52, 2100–2104 CrossRef CAS PubMed
- S. Lodeiro, Q. Xiong, W. K. Wilson, M. D. Kolesnikova, C. S. Onak and S. P. T. Matsuda, J. Am. Chem. Soc., 2007, 129, 11213–11222 CrossRef CAS PubMed
- T. G. Köllner, C. Schnee, J. Gershenzon and J. Degenhardt, Plant Cell, 2004, 16, 1115–1131 CrossRef PubMed
- T. G. Köllner, P. E. O'Maille, N. Gatto, W. Boland, J. Gershenzon and J. Degenhardt, Arch. Biochem. Biophys., 2006, 448, 83–92 CrossRef PubMed
- J. Rétey, Angew. Chem., Int. Ed. Engl., 1990, 29, 355–361 CrossRef
- S. Garms, T. G. Köllner and W. Boland, J. Org. Chem., 2010, 75, 5590–5600 CrossRef CAS PubMed
- L. S. Vedula, M. J. Rynkiewicz, H.-J. Pyun, R. M. Coates, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 6153–6163 CrossRef CAS PubMed
- L. S. Vedula, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 12719–12727 CrossRef CAS PubMed
- D. W. Christianson, Chem. Rev., 2006, 106, 3412–3442 CrossRef CAS PubMed
- A. Vattekkatte, N. Gatto, E. Schulze, W. Brandt and W. Boland, Org. Biomol. Chem., 2015, 13, 4776–4784 CAS
- A. Warke and C. Momany, Cryst. Growth Des., 2007, 7, 2219–2225 CAS
- J. P. Noel, N. Dellas, J. A. Faraldos, M. Zhao, B. A. Hess, L. Smentek, R. M. Coates and P. E. O'Maille, ACS Chem. Biol., 2010, 5, 377–392 CrossRef CAS PubMed
- C. M. Starks, K. W. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815 CrossRef CAS PubMed
- J. Bohlmann, M. Phillips, V. Ramachandiran, S. Katoh and R. Croteau, Arch. Biochem. Biophys., 1999, 368, 232–243 CrossRef CAS PubMed
- L. Pazouki, H. R. Memari, A. Kännaste, R. Bichele and Ü. Niinemets, Front. Plant Sci., 2015, 6, 111 Search PubMed
- D. A. Nagegowda, M. Gutensohn, C. G. Wilkerson and N. Dudareva, Plant J., 2008, 55, 224–239 CrossRef CAS PubMed
- J. Bohlmann, D. Martin, N. J. Oldham and J. Gershenzon, Arch. Biochem. Biophys., 2000, 375, 261–269 CrossRef CAS PubMed
- F. Chen, D.-K. Ro, J. Petri, J. Gershenzon, J. Bohlmann, E. Pichersky and D. Tholl, Plant Physiol., 2004, 135, 1956–1966 CrossRef CAS PubMed
- J.-X. Ruan, J.-X. Li, X. Fang, L.-J. Wang, W.-L. Hu, X.-Y. Chen and C.-Q. Yang, Front. Plant Sci., 2016, 7, 638 Search PubMed
- P. Mercke, I. F. Kappers, F. W. Verstappen, O. Vorst, M. Dicke and H. J. Bouwmeester, Plant Physiol., 2004, 135, 2012–2024 CrossRef CAS PubMed
- S. Green, E. N. Friel, A. Matich, L. L. Beuning, J. M. Cooney, D. D. Rowan and E. MacRae, Phytochemistry, 2007, 68, 176–188 CrossRef CAS PubMed
- C. Landmann, B. Fink, M. Festner, M. Dregus, K. H. Engel and W. Schwab, Arch. Biochem. Biophys., 2007, 465, 417–429 CrossRef CAS PubMed
- C. C. N. van Schie, M. A. Haring and R. C. Schuurink, Plant Mol. Biol., 2007, 64, 251–263 CrossRef PubMed
- G. Arimura, S. Garms, M. Maffei, S. Bossi, B. Schulze, M. Leitner, A. Mithofer and W. Boland, Planta, 2008, 227, 453–464 CrossRef CAS PubMed
- S. Roeder, A. M. Hartmann, U. Effmert and B. Piechulla, Plant Mol. Biol., 2007, 65, 107–124 CrossRef CAS PubMed
- Y. Iijima, D. R. Gang, E. Fridman, E. Lewinsohn and E. a. Pichersky, Plant Physiol., 2004, 134, 370–379 CrossRef CAS PubMed
- J. S. Yuan, T. G. Köllner, G. Wiggins, J. Grant, J. Degenhardt and F. Chen, Plant J., 2008, 55, 491–503 CrossRef CAS PubMed
- M. Hosoi, M. Ito, T. Yagura, R. P. Adams and G. Honda, Biol. Pharm. Bull., 2004, 27, 1979–1985 CAS
- D. M. Martin, J. Fäldt and J. Bohlmann, Plant Physiol., 2004, 135, 1908–1927 CrossRef CAS PubMed
- S. A. B. McKay, W. L. Hunter, K.-A. Godard, S. X. Wang, D. M. Martin, J. Bohlmann and A. L. Plant, Plant Physiol., 2003, 133, 368–378 CrossRef CAS PubMed
- M. A. Phillips, M. R. Wildung, D. C. Williams, D. C. Hyatt and R. Croteau, Arch. Biochem. Biophys., 2003, 411, 267–276 CrossRef CAS PubMed
- D. P. W. Huber, R. N. Philippe, K. A. Godard, R. N. Sturrock and J. Bohlmann, Phytochemistry, 2005, 66, 1427–1439 CrossRef CAS PubMed
- S. C. Kampranis, D. Ioannidis, A. Purvis, W. Mahrez, E. Ninga, N. A. Katerelos, S. Anssour, J. M. Dunwell, J. Degenhardt, A. M. Makris, P. W. Goodenough and C. B. Johnson, Plant Cell, 2007, 19, 1994–2005 CrossRef CAS PubMed
- M. L. Wise, T. J. Savage, E. Katahira and R. Croteau, J. Biol. Chem., 1998, 273, 14891–14899 CrossRef CAS PubMed
- D. J. Hoelscher, D. C. Williams, M. R. Wildung and R. Croteau, Phytochemistry, 2003, 62, 1081–1086 CrossRef CAS PubMed
- C. G. Jones, C. I. Keeling, E. L. Ghisalberti, E. L. Barbour, J. A. Plummer and J. Bohlmann, Arch. Biochem. Biophys., 2008, 477, 121–130 CrossRef CAS PubMed
- C. G. Jones, J. Moniodis, K. G. Zulak, A. Scaffidi, J. A. Plummer, E. L. Ghisalberti, E. L. Barbour and J. Bohlmann, J. Biol. Chem., 2011, 286, 17445–17454 CrossRef CAS PubMed
- T. Maruyama, M. Ito and G. Honda, Biol. Pharm. Bull., 2001, 24, 1171–1175 CAS
- D. M. Martin and J. Bohlmann, Phytochemistry, 2004, 65, 1223–1229 CrossRef CAS PubMed
- C. F. Lin, Plant Physiol., 2008, 146, 940–951 CrossRef CAS PubMed
- D. Tholl, F. Chen, J. Petri, J. Gershenzon and E. Pichersky, Plant J., 2005, 42, 757 CrossRef CAS PubMed
- V. Portnoy, Plant Mol. Biol., 2008, 66, 647–661 CrossRef CAS PubMed
- A. Sean, L.-G. Fernando and S.-D. Claudia, Mol. Microbiol., 2009, 72, 1181–1195 CrossRef PubMed
- R. S. van der Hoeven, A. J. Monforte, D. Breeden, S. D. Tanksley and J. C. Steffens, Plant Cell, 2000, 12, 2283–2294 CrossRef CAS
- S. M. Colby, J. Crock, B. Dowdle-Rizzo, P. G. Lemaux and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2216–2221 CrossRef CAS
- S. Lee and J. Chappell, Plant Physiol., 2008, 147, 1017–1033 CrossRef CAS PubMed
- I. M. Prosser, R. J. Adams, M. H. Beale, N. D. Hawkins, A. L. Phillips, J. A. Pickett and L. M. Field, Phytochemistry, 2006, 67, 1564–1571 CrossRef CAS PubMed
- Y. Iijima, R. Davidovich-Rikanati, E. Fridman, D. R. Gang, E. Bar, E. Lewinsohn and E. a. Pichersky, Plant Physiol., 2004, 136, 3724–3736 CrossRef CAS PubMed
- A. X. Cheng, C. Y. Xiang, J. X. Li, C. Q. Yang, W. L. Hu, L. J. Wang, Y. G. Lou and X. Y. Chen, Phytochemistry, 2007, 68, 1632–1641 CrossRef CAS PubMed
- F. Deguerry, L. Pastore, S. Q. Wu, A. Clark, J. Chappell and M. Schalk, Arch. Biochem. Biophys., 2006, 454, 123–136 CrossRef CAS PubMed
- C. Schnee, T. G. Köllner, J. Gershenzon and J. Degenhardt, Plant Physiol., 2002, 130, 2049–2060 CrossRef CAS PubMed
- C. Schnee, T. G. Köllner, M. Held, T. C. J. Turlings, J. Gershenzon and J. Degenhardt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1129–1134 CrossRef CAS PubMed
- S. Picaud, M. E. Olsson, M. Brodelius and P. E. Brodelius, Arch. Biochem. Biophys., 2006, 452, 17–28 CrossRef CAS PubMed
- F. Yu, H. Harada, K. Yamasaki, S. Okamoto, S. Hirase, Y. Tanaka, N. Misawa and R. Utsumi, FEBS Lett., 2008, 582, 565–572 CrossRef CAS PubMed
- R. Ito, K. Mori, I. Hashimoto, C. Nakano, T. Sato and T. Hoshino, Org. Lett., 2011, 13, 2678–2681 CrossRef CAS PubMed
- C. D. Poulter, J. C. Argyle and E. A. Mash, J. Am. Chem. Soc., 1977, 99, 957–959 CrossRef CAS PubMed
- C. D. Poulter, P. L. Wiggins and A. T. Le, J. Am. Chem. Soc., 1981, 103, 3926–3927 CrossRef CAS
- C. D. Poulter, D. M. Satterwhite and H. C. Rilling, J. Am. Chem. Soc., 1976, 98, 3376–3377 CrossRef CAS PubMed
- D. E. Cane, J. S. Oliver, P. H. M. Harrison, C. Abell, B. R. Hubbard, C. T. Kane and R. Lattman, J. Am. Chem. Soc., 1990, 112, 4513–4524 CrossRef CAS
- D. N. Brems and H. C. Rilling, J. Am. Chem. Soc., 1977, 99, 8351–8352 CrossRef CAS PubMed
- W. Rittersdorf and F. Cramer, Tetrahedron, 1968, 24, 43–52 CrossRef CAS
-
E. M. Davis and R. Croteau, in Biosynthesis:
Aromatic Polyketides, Isoprenoids, Alkaloids, Springer-Verlag, Berlin, 2000, vol. 209, pp. 53–95 Search PubMed
- R. Croteau, Chem. Rev., 1987, 87, 929–954 CrossRef CAS
- D. M. Satterwhite, C. J. Wheeler and R. Croteau, J. Biol. Chem., 1985, 260, 3901–3908 Search PubMed
- D. E. Cane, P. C. Prabhakaran, J. S. Oliver and D. B. McIlwaine, J. Am. Chem. Soc., 1990, 112, 3209 CrossRef CAS
- A. L. Schilmiller, I. Schauvinhold, M. Larson, R. Xu, A. L. Charbonneau, A. Schmidt, C. Wilkerson, R. L. Last and E. Pichersky, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10865–10870 CrossRef CAS PubMed
- C. Sallaud, D. Rontein, S. Onillon, F. Jabes, P. Duffe, C. Giacalone, S. Thoraval, C. Escoffier, G. Herbette, N. Leonhardt, M. Causse and A. Tissier, Plant Cell, 2009, 21, 301–317 CrossRef CAS PubMed
- M. C. Schulbach, S. Mahapatra, M. Macchia, S. Barontini, C. Papi, F. Minutolo, S. Bertini, P. J. Brennan and D. C. Crick, J. Biol. Chem., 2001, 276, 11624–11630 CrossRef CAS PubMed
- M. C. Schulbach, P. J. Brennan and D. C. Crick, J. Biol. Chem., 2000, 275, 22876–22881 CrossRef CAS PubMed
- F. Beran, P. Rahfeld, K. Luck, R. Nagel, H. Vogel, N. Wielsch, S. Irmisch, S. Ramasamy, J. Gershenzon, D. G. Heckel and T. G. Köllner, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2922–2927 CrossRef CAS PubMed
- F. Lopez-Gallego, S. A. Agger, D. Abate-Pella, M. D. Distefano and C. Schmidt-Dannert, ChemBioChem, 2010, 11, 1093–1106 CrossRef CAS PubMed
- A. Vattekkatte, N. Gatto, T. G. Kollner, J. Degenhardt, J. Gershenzon and W. Boland, Org. Biomol. Chem., 2015, 13, 6021–6030 CAS
- N. Gatto, A. Vattekkatte, T. Kollner, J. Degenhardt, J. Gershenzon and W. Boland, Chem. Commun., 2015, 51, 3797–3800 RSC
- A. Vattekkatte, S. Garms and W. Boland, J. Org. Chem., 2017, 82, 2855–2861 CrossRef PubMed
- L. D. Hurst, C. Pal and M. J. Lercher, Nat. Rev. Genet., 2004, 5, 299–310 CrossRef CAS PubMed
- T. M. Hohn and R. D. Plattner, Arch. Biochem. Biophys., 1989, 272, 137–143 CrossRef CAS PubMed
- S. S. Dehal and R. Croteau, Arch. Biochem. Biophys., 1988, 261, 346–356 CrossRef CAS PubMed
- D. E. Cane, H. J. Ha, C. Pargellis, F. Waldmeier, S. Swanson and P. P. N. Murthy, Bioorg. Chem., 1985, 13, 246–265 CrossRef CAS
- T. M. Hohn and F. Vanmiddlesworth, Arch. Biochem. Biophys., 1986, 251, 756–761 CrossRef CAS PubMed
- D. A. Whittington, M. L. Wise, M. Urbansky, R. M. Coates, R. B. Croteau and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15375–15380 CrossRef CAS PubMed
- M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13543 CrossRef CAS PubMed
- J. A. Aaron, X. Lin, D. E. Cane and D. W. Christianson, Biochemistry, 2010, 49, 1787–1797 CrossRef CAS PubMed
- H. L. King and H. C. Rilling, Biochemistry, 1977, 16, 3815–3819 CrossRef CAS PubMed
- L. C. Tarshis, M. Yan, C. D. Poulter and J. C. Sacchettini, Biochemistry, 1994, 33, 10871–10877 CrossRef CAS PubMed
- S. Frick, R. Nagel, A. Schmidt, R. R. Bodemann, P. Rahfeld, G. Pauls, W. Brandt, J. Gershenzon, W. Boland and A. Burse, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4194–4199 CrossRef CAS PubMed
- C. Rivera-Perez, P. Nyati and F. G. Noriega, Insect Biochem. Mol. Biol., 2015, 64, 44–50 CrossRef CAS PubMed
- D. E. Cane, J. H. Shim, Q. Xue, B. C. Fitzsimons and T. M. Hohn, Biochemistry, 1995, 34, 2480–2488 CrossRef CAS PubMed
- M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Biochemistry, 2002, 41, 1732 CrossRef CAS PubMed
- T. G. Köllner, C. Schnee, S. Li, A. Svatoš, B. Schneider, J. Gershenzon and J. Degenhardt, J. Biol. Chem., 2008, 283, 20779–20788 CrossRef PubMed
- S. Picaud, M. Brodelius and P. E. Brodelius, Phytochemistry, 2005, 66, 961–967 CrossRef CAS PubMed
- S. Green, C. J. Squire, N. J. Nieuwenhuizen, E. N. Baker and W. Laing, J. Biol. Chem., 2009, 284, 8661–8669 CrossRef CAS PubMed
- M. B. Quin, S. N. Michel and C. Schmidt-Dannert, ChemBioChem, 2015, 16, 2191–2199 CrossRef CAS PubMed
- T. G. Köllner, C. Schnee, S. Li, A. Svatos, B. Schneider, J. Gershenzon and J. Degenhardt, J. Biol. Chem., 2008, 283, 20779–20788 CrossRef PubMed
- T. G. Köllner, Plant Cell, 2008, 20, 482–494 CrossRef PubMed
- T. C. J. Turlings, J. H. Tumlinson and W. J. Lewis, Science, 1990, 250, 1251–1253 CAS
- M. E. Hoballah and T. C. Turlings, J. Chem. Ecol., 2005, 31, 2003–2018 CrossRef CAS PubMed
- Z. R. Khan, K. Ampong-Nyarko, P. Chiliswa, A. Hassanali, S. Kimani, W. Lwande, W. A. Overholt, W. A. Overholt, J. A. Picketta, L. E. Smart and C. M. Woodcock, Nature, 1997, 388, 631–632 CrossRef CAS
- J. Degenhardt, Plant Physiol., 2009, 149, 96–102 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |