
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe size-selective interaction of key and lock nanocrystals driven by depletion attraction at the nanoscale†
D.
Hinrichs
,
R.
Himstedt
and
D.
Dorfs
 *
*
Department of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover, Callinstrasse 3A, 30167 Hannover, Germany. E-mail: dirk.dorfs@pci.uni-hannover.de
First published on 3rd April 2018
Abstract
In this article, we study the size-dependent interactions of quasi-spherical nanocrystals with voids of concave nanoparticles of complementary sizes and shapes. Experimental insights into a system with key and lock particles with smaller dimensions than 15 nm are presented, which provide evidence for key–lock specific interaction on this length scale. Using depletion attraction as a driving force, the key–lock interaction is shown to be reversible and independent of the material composition of the key particles. Poly(ethylene glycol) methacrylate was utilized as a depletion agent in toluene, the solvent of the studied key–lock system. For this work, a model system of specifically developed concave manganese oxide nanocrystals, synthesized via a cast–mold approach, in combination with highly monodisperse quasi-spherical gold nanocrystals, was investigated with transmission electron microscopy, optical UV/vis/NIR spectroscopy and powder X-ray diffraction. Size-dependent key–lock interactions are clearly identified to occur. For geometrical reasons, only key particles with smaller particle diameters than the voids of the complementary lock particles are able to enter the void. So the void diameter of the lock particles sets a diameter threshold for the key–lock interaction. Additionally, other key particles like silver, iron oxide and even core–shell structured gold-nickel sulfide nanocrystals show key-in-lock assemblies with concave manganese oxide nanocrystals. This behaviour might open up new routes for size-selective particle sensing.
Introduction
Despite many examples of specific interactions of colloidal particles containing cavities at the mesoscale, such systems with particles at the nanoscale are rarely studied.1,2 This is because the preparation of nanoparticles with specific concave-shaped particle domains is still challenging. Additionally, the commonly used optical microscopy for the analysis of the particle–particle interactions is not applicable at this small dimension any more.Due to these complications, the investigations of feasible key–lock systems at the nanoscale were done mostly theoretically.3–7
Only two experimental studies at the nanoscale have been published so far. George and co-workers prepared concave iron oxide nanoparticles by a cast–mold approach in 2011.1 Mixing these lock particles with complementary gold nanocrystals resulted in the destabilization of the gold colloid. They concluded, based on their data, that some kind of key–lock interaction had taken place. Thereby ligands could be stripped from the gold nanocrystal surface, presumably resulting in particle destabilization. Furthermore, in 2014, Kraus-Ophir and co-workers prepared a polymer film on a conducting tin-doped indium oxide support, with nanometre-sized cavities by nanoparticle-imprinting and subsequent dissolution of these particles.2 Using this macroscopic specimen, they could indirectly analyse the amount of gold nanoparticles taken up by the voids by measuring the charge, which was needed for the complete electro-oxidation. Both experimental studies were conducted only with gold as the material for the “key” particles, and the procedure in both cases inherently destroyed the investigated gold nanocrystals.
In general, at the mesoscale, lock particles are prepared via 2D and 3D lithography,8–11 controlled shell buckling of polymerized silicon oil droplets without templates,12,13 or growth on template particles.14,15 Beside various possible interparticle forces, e.g. van der Waals, magnetic, electrostatic and molecular, mostly the entropic effect of depletion interaction was used as the driving force to initiate the key–lock self-assembly.16 Although theoretical studies predict the key–lock interactions initiated via depletion interaction at the nanoscale, this driving force was never used experimentally for such a system so far.3,4,7
In this paper, we show an experimental study of a novel key–lock system at the nanoscale. We design tailored lock particles consisting of concave-shaped manganese oxide nanocrystals prepared via a cast–mold approach. In addition, highly monodisperse key nanocrystals consisting of various materials are synthesised, e.g. gold, silver, iron oxide and gold-nickel sulfide hybrid nanocrystals. Utilizing depletion attraction as the entropic driving force, the key-in-lock assemblies of these highly crystalline nanoparticles are analysed by transmission electron microscopy. We demonstrate their size-specific but simultaneously material-independent interaction. Furthermore, the assembly is shown to be reversible via dilution of the colloidal solution.
The gathered results of the size-selective nanoparticle interaction are essential for prospective applications, e.g. an analytically used nanoparticle colloid with size-selective nanoparticle recognition properties.
Results and discussion
Concave manganese oxide nanocrystals
Concave nanocrystals or, that means nanoparticles with an accessible void, were synthesized via the so-called cast–mold-approach.1 In general, this procedure consists of two steps: first, a material is grown onto a template of a different material, which has a specific size and shape. Afterwards, this template is removed via an etching process. A void is obtained in the on-grown material with the size and shape of the template. In this work, manganese oxide nanocrystals (MONCs) with concave-shaped regions not yet reported were produced via the nucleation and growth of manganese oxide onto preformed quasi-spherical gold nanocrystals (AuNCs) with a highly monodisperse size distribution. The gold domain of these asymmetric hybrid nanocrystals was removed by etching with iodine afterwards. The etching can only be achieved if the gold domain is in contact with the nanoparticle surface. Only in this case iodine is able to reach and etch the gold. Iodine cannot etch the manganese oxide domain itself. Therefore, the manganese oxide domain stays unchanged in size and shape. Thereby, highly crystalline MONCs, each containing a spheroidal void, could be obtained (for details, see the Experimental section). It should be noted here that manganese oxide was chosen as the material simply and only due to the relatively simple and highly reproducible synthesis procedure for the concave MONCs and not because of any type of specific chemical interaction of this material. Thus, in principle, similar key–lock recognition should be possible with any type of colloidal particles, which can be prepared sufficiently monodisperse with similar spheroidal voids. In Fig. 1A, the schematic reaction pathway is drawn. Starting from AuNCs with an initial diameter of (6.4 ± 0.5) nm (Fig. 1B), an exemplary system of hybrid Au–MONCs was grown with an outer diameter of (12.7 ± 1.3) nm (Fig. 1C). The gold domains were removed to obtain concave MONCs with a spheroidal void (Fig. 1D). With (5.5 ± 0.7) nm, the aperture of these voids is slightly smaller than the pristine AuNC diameter. This difference of 1 nm is a result of the hybrid nanocrystal geometry. The gold nanocrystal is less than half overgrown by the manganese oxide. Therefore, the imprint in the manganese oxide is smaller than the diameter of the gold.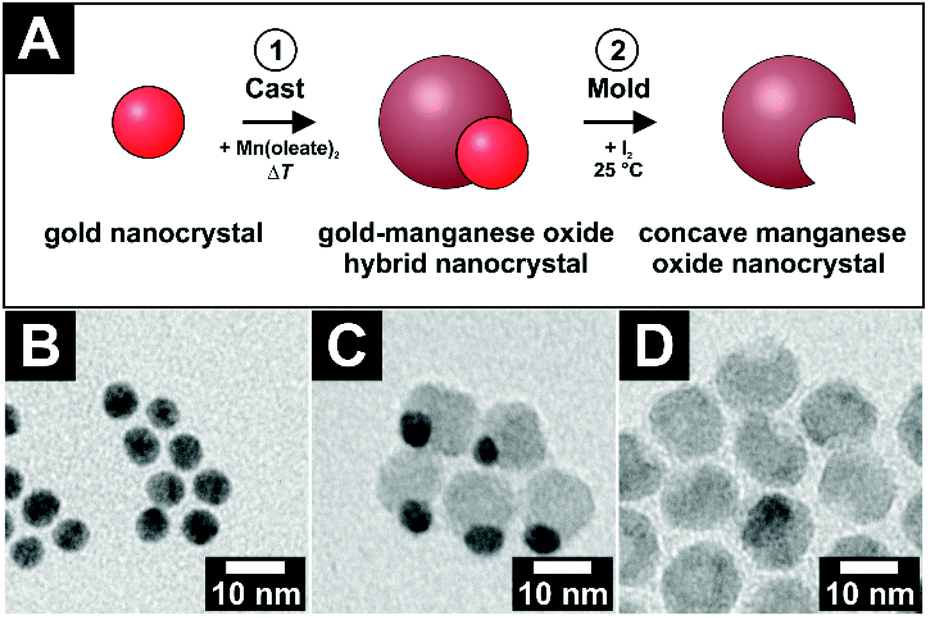 | ||
| Fig. 1 (A) Panel A shows a scheme of the cast–mold approach, which is used to obtain concave manganese oxide nanocrystals. In the first step, manganese oxide is grown onto quasi-spherical gold nanocrystals via the thermal decomposition of manganese oleate. In the second step, the gold domain of the hybrid nanocrystals is etched by iodine. Thereby a void complementary to the shape and size of the gold nanocrystal is formed inside the manganese oxide nanocrystals. (B–D) TEM bright field images of (B) gold nanocrystals with diameters of (6.4 ± 0.5) nm, (C) gold-manganese oxide hybrid nanocrystals with an outer diameter of (12.7 ± 1.3) nm, and (D) the respective etched particles with a spheroidal void with an aperture of around (5.5 ± 0.7) nm. | ||
The exact geometry of the Au–MONC hybrid particles can be tuned by varying the number of manganese oxide nucleation sides per gold particle. To obtain asymmetric Janus-like hybrid particles with one manganese oxide domain per particle, a preliminary ligand exchange for the AuNCs from oleylamine to octadecanethiol is necessary.17 The much higher binding affinity of the thiol group to gold in comparison with an amine group suppresses multiple nucleations of manganese oxide on the thiol-protected surface of one gold particle. So, only one manganese oxide domain per AuNC will nucleate and Janus-like hybrids will be obtained. Without such a ligand exchange, multiple nucleations of manganese oxide per AuNC initiate the growth of flower-like hybrid particles.
Interestingly, multiple nucleations of manganese oxide on large gold nanocrystals open up the possibility of creating large voids, which are formed by up to four fused manganese oxide domains (see Fig. S1 and S2†). Most frequently, particles with two or three fused domains can be found. In Fig. 2A and B such Au–MONCs and concave MONCs with an outer diameter of roughly 25 nm to 30 nm and an inner void diameter of (9.3 ± 0.9) nm are shown. Because approximately half of each AuNC is overgrown with manganese oxide, the aperture of the voids has nearly the same diameter as the inner void in this case.
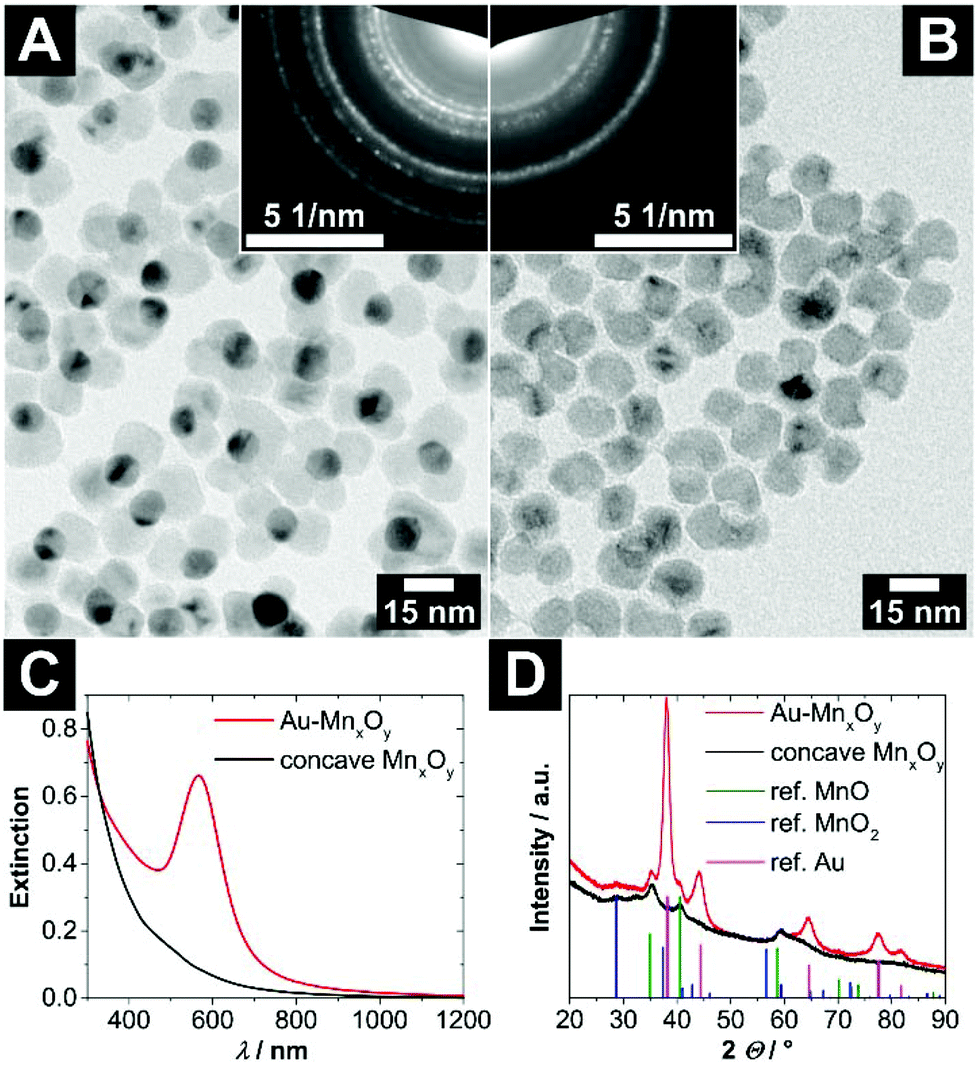 | ||
| Fig. 2 (A) TEM bright field image of hybrid Au–MONCs with a (10.8 ± 0.8) nm large gold domain. The upper right inlay shows the selected area electron diffraction of these hybrid nanocrystals. (B) TEM bright field image of the same particles after etching of the gold domains via iodine. In the upper left inlay, the selected area electron diffraction of these concave MONCs is depicted. The assignment of the ring patterns is given in Fig. S4 (ESI†). (C) UV/vis/NIR extinction spectra of the Au–MONCs and concave MONCs. (D) Powder X-ray diffraction patterns of the Au–MONCs and the respective concave MONCs. The reference structural data show the reflection positions of manganosite (MnO, PDF card #: 01-075-6876), pyrolusite (MnO2, PDF card #: 01-071-0071), and gold (Au, PDF card #: 00-004-0784) displayed as vertical lines. | ||
The UV/vis/NIR extinction spectrum of the hybrid Au–MONCs in Fig. 2C shows an absorption band at around 566 nm, which originates from the localized surface plasmon resonance (LSPR) of the gold domain. In comparison with the position of the resonance frequency of the pristine AuNCs, which is located at 531 nm (see Fig. S3†), the LSPR band of the hybrid Au–MONCs is shifted bathochromically. The reason for this is the higher dielectric permittivity of manganese oxide in contrast to toluene. After the treatment of the hybrid Au–MONCs with iodine to dissolve the gold domains, the concave MONCs show a non-characteristic absorption, which increases towards shorter wavelengths. No LSPR-induced absorption can be found any more. This is a strong indicator for the complete absence of all gold domains within the whole sample.
The powder XRD patterns in Fig. 2D are in line with the optical measurements. The reflections of gold in the hybrid Au–MONCs at 38.2, 44.4, 64.6, 77.5 and 81.7° 2Θ disappear after iodine treatment. Furthermore, also in the TEM investigation (Fig. 2B), no gold can be found after iodine treatment. All in all, these findings prove the quantitative absence of gold after the etching procedure. However, the powder XRD patterns show that the manganese oxide domains of the hybrid nanocrystals as well as of the concave nanocrystals are not a pure phase, but rather a mixture of mainly manganosite and pyrolusite.
The synthesis of concave manganese oxide nanocrystals via the described cast–mold approach is highly versatile. Using AuNCs with different diameters makes it possible to obtain concave nanocrystals with differently sized void apertures; see the ESI for details (Fig. S5†).
Besides gold etching with iodine, which yields well-shaped and quantitative gold-free concave MONCs, there is also the possibility of using cyanide to dissolve all gold domains. The iodine procedure has one drawback: already small amounts of residual iodine on the surface of the concave MONCs can interfere with the consecutive key–lock experiments by inducing AuNC coalescence (see the section ‘Key and lock interaction’). Therefore, it is essential to thoroughly clean the etched product. However, etching the gold via a two-phase reaction using an aqueous potassium cyanide solution also yields concave MONCs. To obtain a colloidal stable and quantitatively gold-free product, the addition of a phase-transfer catalyst is necessary (here: 18-crown-6-ether). This fact is inherently connected to a drawback of this method. The cyanide ions in the organic phase are so strongly activated18,19 that they do not only etch the gold domains but also significantly dissolve parts of the manganese oxide phase. Especially the inner region is affected, which was the pristine surrounding of the seed (see Fig. S6† for details). Interestingly, the broadening of the void diameters can be used as some kind of post-synthetic fine-tuning for the void diameters of a given Au–MONC batch. All in all, the iodine-based etching procedure in combination with a thorough cleaning of the product results in more defined voids than those from the cyanide procedure.
Independent of the type of etching procedure used, whether iodine or potassium cyanide in combination with 18-crown-6-ether, we would like to emphasise that it is extremely important to thoroughly check the concave product whether the etching procedure was quantitative for all particles (e.g. TEM, UV/vis, SAED). Otherwise the remaining gold could lead to misinterpretation of the later key–lock experiments.
Quasi-spherical gold nanocrystals
For the investigation of size-specific interparticle interactions of concave and convex nanocrystals, not only monodisperse voids are necessary. Also, highly monodisperse convex particles with an almost spherical shape are needed, to keep the system as simple as possible and to gain information about the interaction of the specific combination of just one particle size with a specific void size. Therefore, gold nanocrystals provided the best opportunities for this purpose, due to their well-developed synthesis procedure.Quasi-spherical gold nanocrystals (AuNCs), which are soluble in organic solvents, could be synthesized in various diameters with very narrow size distributions by varying the reaction temperature and utilising a seed-mediated growth approach. The diameters of the AuNCs increase with decreasing reaction temperature. AuNCs with larger diameters than approx. 6–7 nm can only be obtained via seed-mediated growth. For details, see the Experimental section. In Fig. 3A–D, a selection of AuNCs is depicted, which were used for further investigation of the key and lock interaction later on. The UV/vis extinction spectra of these AuNCs in Fig. 3E show absorption bands between 500 nm and 600 nm, which are typical for gold nanoparticles. The origin of these absorption bands is the localized surface plasmon resonance (LSPR) of the conduction band electrons of the gold. In the short-wavelength regime at and below 400 nm, the absorption is mostly caused by interband transitions. Thereby it is proportional to the gold mass concentration and independent of the nanocrystal size.20 Hence, the spectra were normalised at 400 nm for a better comparability of the LSPR-induced absorption bands. In general, three tendencies can be gathered comparing the normalised UV/vis extinction spectra of the gold nanocrystals, which are expected for monodisperse gold nanocrystals. First, the oscillator strength of the LPSR band increases with increasing particle size. Second, a narrowing of the LSPR absorption band occurs with increasing particle size in the intrinsic size regime. Third, the maximum of the LSPR absorption band shifts bathochromically with increasing particle size.
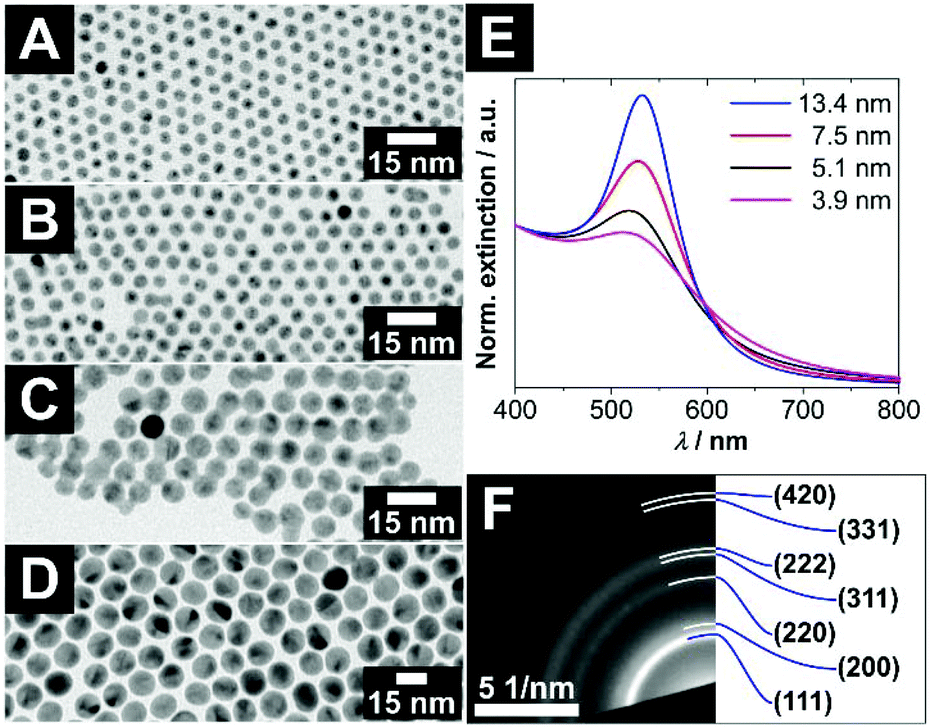 | ||
| Fig. 3 (A–D) TEM bright field images of highly monodisperse, oleylamine stabilized, quasi-spherical gold nanocrystals with various diameters of (A) (3.9 ± 0.3) nm, (B) (5.1 ± 0.6) nm, (C) (7.5 ± 0.6) nm and (D) (13.4 ± 0.9) nm. Additional overview images and size distributions can be found in Fig. S7 (ESI†). (E) Normalised UV/vis extinction spectra of the differently sized gold nanocrystals. (F) The panel shows a selected area electron diffraction of ≈5 nm large gold nanocrystals with assigned lattice planes of fcc gold. | ||
Key and lock interaction
Nanocrystals with a concave-shaped region will from now on be called lock particles. In addition, nanocrystals with a complementary convex shape will be called key particles; see Fig. S8 (ESI†). To investigate the interactions of the colloidal key and lock particles, the particle concentrations of the nanocrystal stock solutions were determined, and approximately, the same particle amounts were mixed with each other in toluene.Mixing concave manganese oxide lock particles (MONCs) with voids of (9.3 ± 0.9) nm diameter with smaller gold key particles (AuNCs) of (7.5 ± 0.6) nm diameter results in a stable colloidal solution. Even though the keys could geometrically fit into the voids, almost no key-in-lock assemblies can be observed in the TEM micrograph, see Fig. S9 (ESI†). The rare key-in-lock structures could be stochastically overlain particles. The colloidal solution is stable for days without any changes. This experiment is in contradiction with a published work on a gold–iron oxide key–lock system.1 Up to now, this is the only published experimental work, which outlined the key and lock interaction of colloidal nanocrystals smaller than 15 nm in size. In that regard, George et al. synthesized concave iron oxide nanocrystals via the cast–mold approach. They used these lock particles in combination with key nanoparticles consisting of gold of two different sizes. Mixing the pure lock and key particle solutions in pure toluene led to the destabilization of the system when the diameter of the keys was smaller than diameter of the void. The AuNCs formed large undefined structures, which were observed via dynamic light scattering and TEM. The authors concluded that the AuNCs were destabilized by the void due to the ligands being stripped off while the key dips into the void. The thermodynamic driving force of this process remained unstated.
We have performed several control experiments, which clearly reveal that the findings by George et al. are likely not explainable by key–lock interactions but rather by the residual iodine contamination from the etching step of the concave nanocrystals. These experiments are summarized in detail in the ESI (Fig. S10 and S11† and the corresponding discussion).
Many theoretical and also experimental investigations of the key–lock systems at the mesoscale utilized depletion interaction as a thermodynamic driving force to push the keys into the complementary locks.9–14 Briefly, adding a non-adsorbing polymer to a solution of colloidal hard spheres induces an effective depletion layer on the surface of the particles. The reason for this is the loss of configurational entropy of the polymer in the region near the surface. The respective volume is called excluded volume. If the hard spheres come close enough to each other, the excluded volumes of them will overlap and inherently increase the overall accessible volume for the polymer in the system. This induces an entropic driving force for the system to agglomerate (this means increased accessible volume for the polymer).21 The depletion interaction is stronger for particles with complementary shapes because in that case the overlap of the excluded volumes is larger. In a system with a mixture of lock and key particles, the entropy of the system will considerably increase if the keys fit into the voids of the locks.
Indeed, adding poly(ethylene glycol) methacrylate (PEGMA) to a mixture of AuNCs and concave MONCs initiates an interaction in the previously stable colloidal solution. The mixture gets turbid after minutes with a PEGMA volume fraction of φ = 0.3. In Fig. 4, the product of four combinations of keys with different sizes and locks with the same void diameter are shown. For the three samples with key particles smaller than the void diameter, key-in-lock assemblies can be seen; see Fig. 4A–C. This proves that depletion interaction takes place and induces not only an agglomeration of the particles but also a specific interaction of the differently shaped particles. In contrast, for the experiment with larger key particles in comparison with the void diameter, no key-in-lock assemblies can be observed at all; see Fig. 4D. (However, if the multiple domains of one concave MONC become somehow fragmented, nanocrystals with a very wide void aperture are formed. These rare particles have such a large concave region that they can also interact with very large gold nanocrystals and key-in-lock combinations can be found; see Fig. S12.†) For geometrical reasons, no key of a larger diameter can enter a smaller void. This means, the void diameter sets an upper threshold for keys that can enter the void. Nevertheless, also this larger-key-smaller-void-solution gets turbid due to agglomeration because the depletion attraction always leads to some kind of unspecific background agglomeration of particles, even in the case of quasi-spherical hard spheres. Especially this unspecific agglomeration of all particles in the samples makes a reliable statistical analysis of the experiments almost impossible, e.g. the ratio of the key-in-lock assemblies to separated non-interacted particles. Furthermore, very large agglomerates in the samples could contain different particle amounts of key and lock particles, which would direct the other parts of the system to a non-equilibrium of key and lock particles. Nevertheless, we here clearly report for the first time the depletion attraction driven size-selective recognition of nanoparticles by concave nanoparticles.
 | ||
| Fig. 4 TEM bright field images of key–lock experiments with quasi-spherical gold nanocrystals (AuNCs) of different sizes and concave manganese oxide nanocrystals (MONCs). While the lock particles with inner void diameters of (9.3 ± 0.9) nm are the same in all experiments, the diameter of the key particles varies from (A) (3.9 ± 0.3) nm to (B) (5.1 ± 0.6) nm, (C) (7.5 ± 0.6) nm and (D) (13.4 ± 0.9) nm. Poly(ethylene glycol) methacrylate was added in a volume fraction of 0.3 to all mixtures to induce depletion attraction. | ||
This key–lock interaction is a reversible process. All particles of the system are continuously stabilised by a ligand shell and only pushed into assemblies by the pseudo-attractive force of the depletion interaction. If the key–lock system with an initial volume fraction of φ(PEGMA) = 0.3 is diluted with toluene by a factor of roughly 2, the key-in-lock assemblies, as well as all other agglomerates start to dissolve again; see Fig. 5. The product is a stable colloidal solution of key and lock particles. The particles are not changed in size and shape in comparison with the pristine key and lock particles.
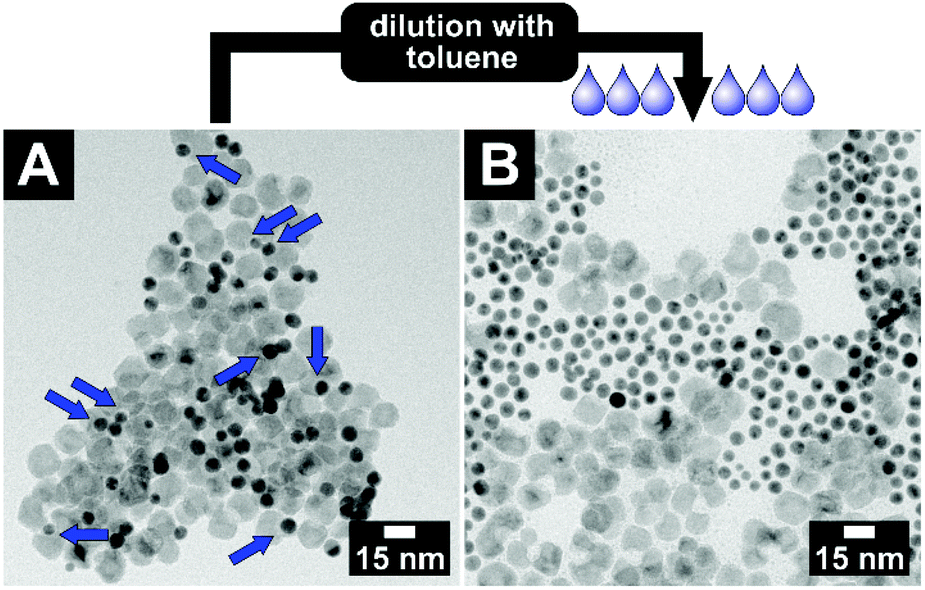 | ||
| Fig. 5 The reversibility of the key–lock interaction: depletion attraction, the entropic driving force of the system, can easily be reduced or switched off by dilution. TEM bright field images of (A) a key and lock particle mixture with a poly(ethylene glycol) methacrylate volume fraction of φ = 0.3. To guide the eye, the blue arrows point towards key-in-lock assemblies. (B) After dilution of this sample by a factor of roughly 2, no key-in-lock assemblies can be observed anymore. | ||
The interaction of the key and lock particles should exclusively depend on the key and void diameters and not on the kind of material they consist of. To confirm this hypothesis, key particles with appropriate diameters consisting of silver, iron oxide and a core–shell structure of gold-nickel sulfide (and thus particles with very different surface characters) were synthesised. For the structural analysis of the pure nanocrystals, see Fig. S13–S15 (ESI†). In the key–lock experiments, these key particles were mixed with manganese oxide lock particles and the interaction was initiated via the addition of PEGMA. For all three material compositions, key-in-lock assemblies can be found; see Fig. 6. The findings for these materials are not as clear as for the gold particles, simply because the well-known and well-established gold particle samples have much narrower size distributions. Nevertheless, they clearly demonstrate that as expected, the depletion attraction driven process at the nanoscale shown in this paper is not a material-related property, but instead only a shape-driven property, which hence should be easily transferrable to other material combinations.
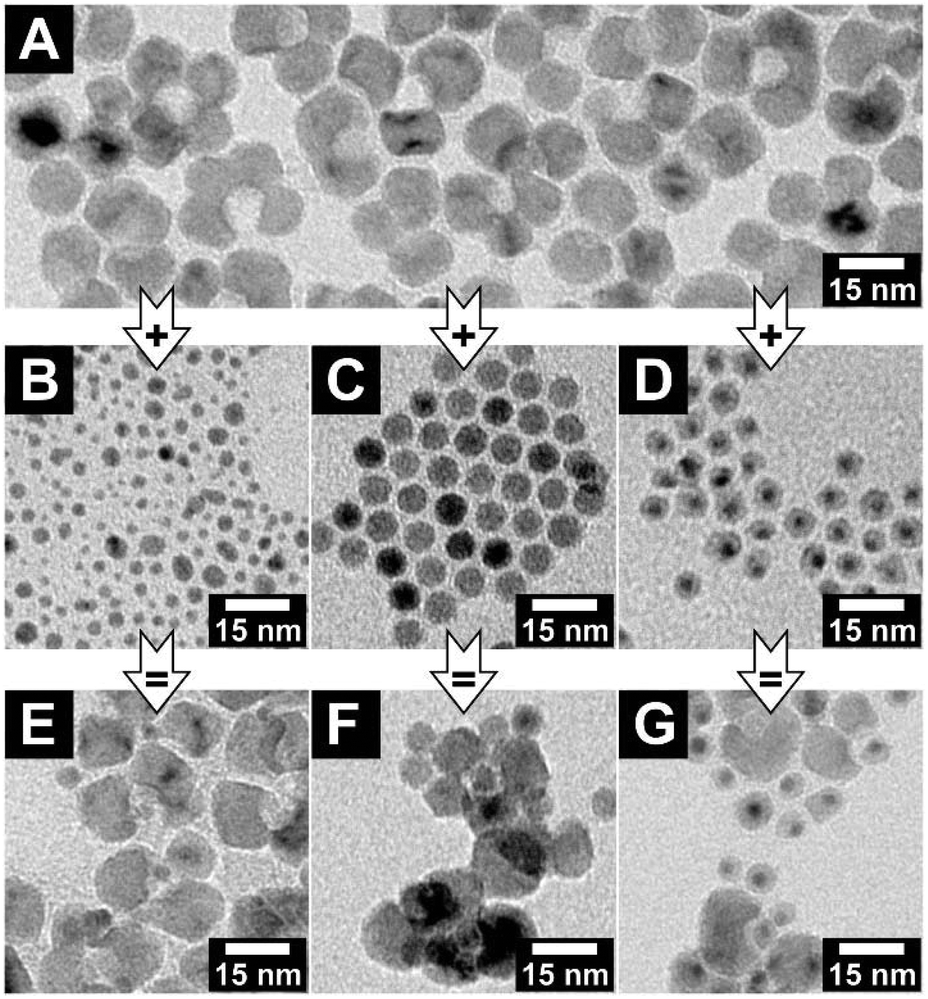 | ||
| Fig. 6 TEM bright field images of (A) concave manganese oxide nanocrystals with void diameters of (9.3 ± 0.9) nm. (B–D) Pure quasi-spherical silver, iron oxide and gold-nickel sulfide core–shell nanocrystals, with diameters of (3.1 ± 1.2) nm, (7.6 ± 0.4) nm and (6.5 ± 1.0) nm, respectively. (E–G) Key-in-lock assemblies of manganese oxide lock nanocrystals (E) with silver key nanocrystals, (F) with iron oxide key nanocrystals, and (G) with gold-nickel sulfide key nanocrystals. | ||
Conclusions
Concave manganese oxide nanocrystals (MONCs), with an accessible void, can interact with convex-shaped nanocrystals of complementary or smaller size and shape. The interaction is analogous to a key–lock system. Only if the convex key particle is smaller than the void of the concave lock particle, the key can enter the void. Additionally, the system requires a driving force to make the nanocrystals interact with each other. Here, depletion attraction was utilized as an entropic driving force, which was enabled by the addition of poly(ethylene glycol) methacrylate to the pristine stable colloidal nanocrystal mixtures. The interaction of key and lock nanocrystals was proven to be dependent on the size but independent of the material composition of the key particles. Quasi-spherical key nanocrystals consisting of gold, silver, iron oxide and gold-nickel sulfide were examined in combination with manganese oxide lock particles. The fundamental experimental findings are the proof of reversible key–lock interaction at the length scale of a few nanometres. This opens up possibilities for the further development of sensory applications to detect nanoparticles with specific sizes utilizing key–lock interactions. In principle, our findings should be extendable towards all types of chemically inert matching key and lock particles, which are mixed with an also chemically inert depletion attraction agent. Further improvements for the quantification of key-in-lock assemblies will have to be done, e.g. by the immobilization of lock particles onto macroscopic surfaces like glass slides. Additionally, the shape of the voids will be an interesting parameter for key–lock interactions at the nanoscale. For this purpose, cubic or cylindrical voids can be formed via the cast–mold approach using cubic or cylindrical seed particles. Besides the gain of fundamental knowledge, this could open up possibilities for the cheap and fast investigations of nanocrystal shapes without the need for expensive TEM.Experimental section
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100g), and dispersed in toluene (0.3 mL).
100g), and dispersed in toluene (0.3 mL).
Methods
Abbreviations
| NCs | Nanocrystals |
| AuNCs | Gold nanocrystals |
| MONCs | Manganese oxide nanocrystals |
| LSPR | Localized surface plasmon resonance |
| PEGMA | Poly(ethylene glycol) methacrylate |
| SAED | Selected area electron diffraction |
| TEM | Transmission electron microscopy |
| XRD | X-ray diffraction |
| NIR | Near infra-red |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. D. and D. H. acknowledge financial support from DFG Research Grant DO 1580/5-1. R. H. is grateful to the Hannover School for Nanotechnology for financial support. Additionally, the authors would like to thank the Laboratory of Nano and Quantum Engineering for the possibility to use the TEM. The authors wish to thank Armin Feldhoff and Jürgen Caro for providing access to powder XRD measurement facilities.References
- C. George, D. Dorfs, G. Bertoni, A. Falqui, A. Genovese, T. Pellegrino, A. Roig, A. Quarta, R. Comparelli, M. L. Curri, R. Cingolani and L. Manna, J. Am. Chem. Soc., 2011, 133, 2205–2217 CrossRef PubMed.
- S. Kraus-Ophir, J. Witt, G. Wittstock and D. Mandler, Angew. Chem., 2014, 53, 294–298 CrossRef PubMed.
- H. Wu, Y. Li, D. Kadirov, S. Zhao, X. Lu and H. Liu, Langmuir, 2017, 33, 11817–11824 CrossRef PubMed.
- H. Y. Chang, C. W. Huang, Y. F. Chen, S. Y. Chen, Y. J. Sheng and H. K. Tsao, Langmuir, 2015, 31, 13085–13093 CrossRef PubMed.
- G. van Anders, D. Klotsa, N. K. Ahmed, M. Engel and S. C. Glotzer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4812–E4821 CrossRef PubMed.
- G. van Anders, N. K. Ahmed, R. Smith, M. Engel and S. C. Glotzer, ACS Nano, 2014, 8, 931–940 CrossRef PubMed.
- Z. Jin and J. Wu, J. Phys. Chem. B, 2011, 115, 1450–1460 CrossRef PubMed.
- P. Y. Wang and T. G. Mason, J. Am. Chem. Soc., 2015, 137, 15308–15314 CrossRef PubMed.
- U. Okabe, T. Okano and H. Suzuki, Sens. Actuators, A, 2017, 254, 43–53 CrossRef.
- S. Badaire, C. Cottin-Bizonne and A. D. Stroock, Langmuir, 2008, 24, 11451–11463 CrossRef PubMed.
- T. Tigges and A. Walther, Angew. Chem., 2016, 55, 11261–11265 CrossRef PubMed.
- S. Sacanna, W. T. Irvine, P. M. Chaikin and D. J. Pine, Nature, 2010, 464, 575–578 CrossRef PubMed.
- S. Sacanna, W. T. M. Irvine, L. Rossi and D. J. Pine, Soft Matter, 2011, 7, 1631–1634 RSC.
- Y. Wang, Y. Wang, X. Zheng, G. R. Yi, S. Sacanna, D. J. Pine and M. Weck, J. Am. Chem. Soc., 2014, 136, 6866–6869 CrossRef PubMed.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51–55 CrossRef PubMed.
- K. J. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef PubMed.
- I. Schick, S. Lorenz, D. Gehrig, A. M. Schilmann, H. Bauer, M. Panthofer, K. Fischer, D. Strand, F. Laquai and W. Tremel, J. Am. Chem. Soc., 2014, 136, 2473–2483 CrossRef PubMed.
- J. H. Zubrick, B. I. Dunbar and H. D. Durst, Tetrahedron Lett., 1975, 16, 71–74 CrossRef.
- C. J. Pedersen and H. K. Frensdorff, Angew. Chem., 1972, 11, 16–25 CrossRef.
- T. Hendel, M. Wuithschick, F. Kettemann, A. Birnbaum, K. Rademann and J. Polte, Anal. Chem., 2014, 86, 11115–11124 CrossRef PubMed.
- H. N. W. Lekkerkerker and R. Tuinier, Colloids and the Depletion Interaction, Springer, 2011, vol. 833 Search PubMed.
- S. Peng, Y. Lee, C. Wang, H. Yin, S. Dai and S. Sun, Nano Res., 2008, 1, 229–234 CrossRef.
- W. Zhu, R. Michalsky, O. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef PubMed.
- T. D. Schladt, T. Graf and W. Tremel, Chem. Mater., 2009, 21, 3183–3190 CrossRef.
- R. Hufschmid, H. Arami, R. M. Ferguson, M. Gonzales, E. Teeman, L. N. Brush, N. D. Browning and K. M. Krishnan, Nanoscale, 2015, 7, 11142–11154 RSC.
- R. Himstedt, P. Rusch, D. Hinrichs, T. Kodanek, J. Lauth, S. Kinge, L. D. A. Siebbeles and D. Dorfs, Chem. Mater., 2017, 29, 7371–7377 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional UV/vis/NIR extinction spectra, TEM overview images, data of the cyanide etching, size and the phase determination of key particles. See DOI: 10.1039/c8nr01893f |
| This journal is © The Royal Society of Chemistry 2018 |