
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferential effects of graphene materials on the metabolism and function of human skin cells†
Javier
Frontiñán-Rubio
 a,
M. Victoria
Gómez
a,
Cristina
Martín
a,
Jose M.
González-Domínguez
a,
Mario
Durán-Prado
*b and
Ester
Vázquez
*ac
a,
M. Victoria
Gómez
a,
Cristina
Martín
a,
Jose M.
González-Domínguez
a,
Mario
Durán-Prado
*b and
Ester
Vázquez
*ac
aInstituto Regional de Investigación Científica Aplicada (IRICA), University of Castilla-La Mancha, 13071, Ciudad Real, Spain. E-mail: ester.vazquez@uclm.es
bCell Biology area, Faculty of Medicine, University of Castilla-La Mancha, 13071, Ciudad Real, Spain. E-mail: mario.duran@uclm.es
cFaculty of Chemical Science and Technology, University of Castilla-La Mancha, 13071, Ciudad Real, Spain
First published on 12th June 2018
Abstract
Graphene-related materials (GRMs) such as graphene oxide (GO) and few-layer graphene (FLG) are used in multiple biomedical applications; however, there is still insufficient information available regarding their interactions with the main biological barriers such as skin. In this study, we explored the effects of GO and FLG on HaCaTs human skin keratinocytes, using NMR-based metabolomics and fluorescence microscopy to evaluate the global impact of each GRM on cell fate and damage. GO and FLG at low concentrations (5 μg mL−1) induced a differential remodeling of the metabolome, preceded by an increase in the level of radical oxygen species (ROS) and free cytosolic Ca2+. These changes are linked to a concentration-dependent increase in cell death by triggering apoptosis and necrosis, the latter being predominant at higher concentrations of the nanostructures. In addition, both compounds reduce the ability of HaCaT cells to heal wounds. Our results demonstrate that the GO and FLG used in this study, which mainly differ in their oxidation state, slightly trigger differential effects on HaCaTs cells, but with evident outcomes at the cellular and molecular levels. Their behavior as pro-apoptotic/necrotic substances and their ability to inhibit cell migration, even at low doses, should be considered in the development of future applications.
1. Introduction
GRMs (graphene-related materials) have emerged as promising tools in the field of biomedicine, and have found applications in bio-imaging,1 drug delivery,2,3 diagnosis4 and many other areas.5–9 However, despite the rapid progress in the development of new applications, there is still not enough information about how GRMs could affect human health, particularly regarding such highly exposed barriers as skin.The toxic effects of GRMs seem to be influenced by concentration, lateral dimension, surface structure, functionalization and dispersion method.10–16 As such, it is very important to perform studies in which GRMs are thoroughly characterized, defining the relationships between physicochemical characteristics and biological responses.9
Among the GRMs, graphene oxide (GO) has become the most used because of its solubility in water.17 Some studies have already described how GO induces cytotoxicity through an increase in reactive oxygen species (ROS), which drives the activation of the intrinsic (mitochondrial) apoptotic pathway but also provokes necrosis.18–20 FLG also seems to interact with HaCaT keratinocytes by altering the plasma membrane and inducing significant mitochondrial damage at high concentrations.21 However, to date there has been no in-depth comparison of the effects of both GRMs (GO and FLG) on human cells going from the molecular level (small scale), to the cell process level (large scale). Regarding information at the molecular level, metabolomics, the study of small molecules in living organisms, provides useful information for understanding the physiological state of living systems. Nuclear magnetic resonance (NMR) spectroscopy is one of the leading analytical techniques for performing metabolomics studies. NMR spectroscopy provides unique structural information among the most abundant compounds present in a mixture without the need for sample fractionation or derivatization.22 Most importantly, NMR spectroscopy allows accurate quantification over a wide dynamic range, it is highly reproducible and is non-destructive. To our knowledge, our work is the first study on the effects of FLG on non-transformed human cell metabolomics, and the first to compare those effects with GO-induced metabolomics alterations, and also the first that correlates those effects with other cellular alterations such as cytotoxicity, ROS alterations or motility alterations. There has been only one previous study reporting metabolomics changes in human tumor cells treated with reduced GO (rGO).23 Previous works have studied the effects of GO in zebrafish,24Chlorella vulgaris (bacteria),25,26 and plant cells.27
Herein, we report a systematic study involving the analysis of two GRMs (GO and FLG), which mainly differ in their oxidation state. We evaluated the effects of these two nanostructures on metabolome remodeling, cytotoxicity and cell motility, dissecting the cellular processes governing these responses, as the alteration of mitochondrial ROS and free cytosolic Ca2+ levels. We used HaCaT cells as a model of the skin barrier. HaCaTs are non-transformed keratinocytes, which are the most suitable model for skin cytotoxicity studies28 and also the most used to assess the toxicity of new nanomaterials at the skin level.29–31 NMR spectroscopy was chosen to identify and quantify the most abundant metabolites present on exposed HaCaT cells. Statistical analysis of the NMR data revealed the metabolites that were altered by GO and FLG in our assays.
2. Results and discussion
Skin is the main human barrier and the first likely to interact with GRMs by direct contact. Using human HaCaT keratinocytes, we have analyzed the effects of two different, yet related GRMs (GO and FLG) at the molecular level on metabolomic, ROS, Ca2+ and cell processes, cell death and motility. Special attention has been paid to the physicochemical characterization of the GRMs.2.1 Characterization of FLG and GO
From the different characterization techniques applied to both nanostructures, we obtained the following results (Fig. 1). The composition of GO and FLG was assessed by TGA and elemental analysis. Thermogravimetric plots (Fig. 1A) show very different thermal profiles for both nanostructures, in agreement with their distinct functional group contents. GO showed a weight loss of 42.7%, while FLG only displayed a 6.1 wt% loss. Both results are consistent with their respective elemental analyses (Fig. 1B) where the oxygen content was drastically different, as expected. GO has abundant functional groups, while FLG is mostly composed of carbon, with minimal amounts of heteroatoms. From the atomic content of nitrogen in FLG (Fig. 1B), a residual melamine content of 0.65 wt% was estimated. This value has been used to calculate melamine concentration in control samples for FLG.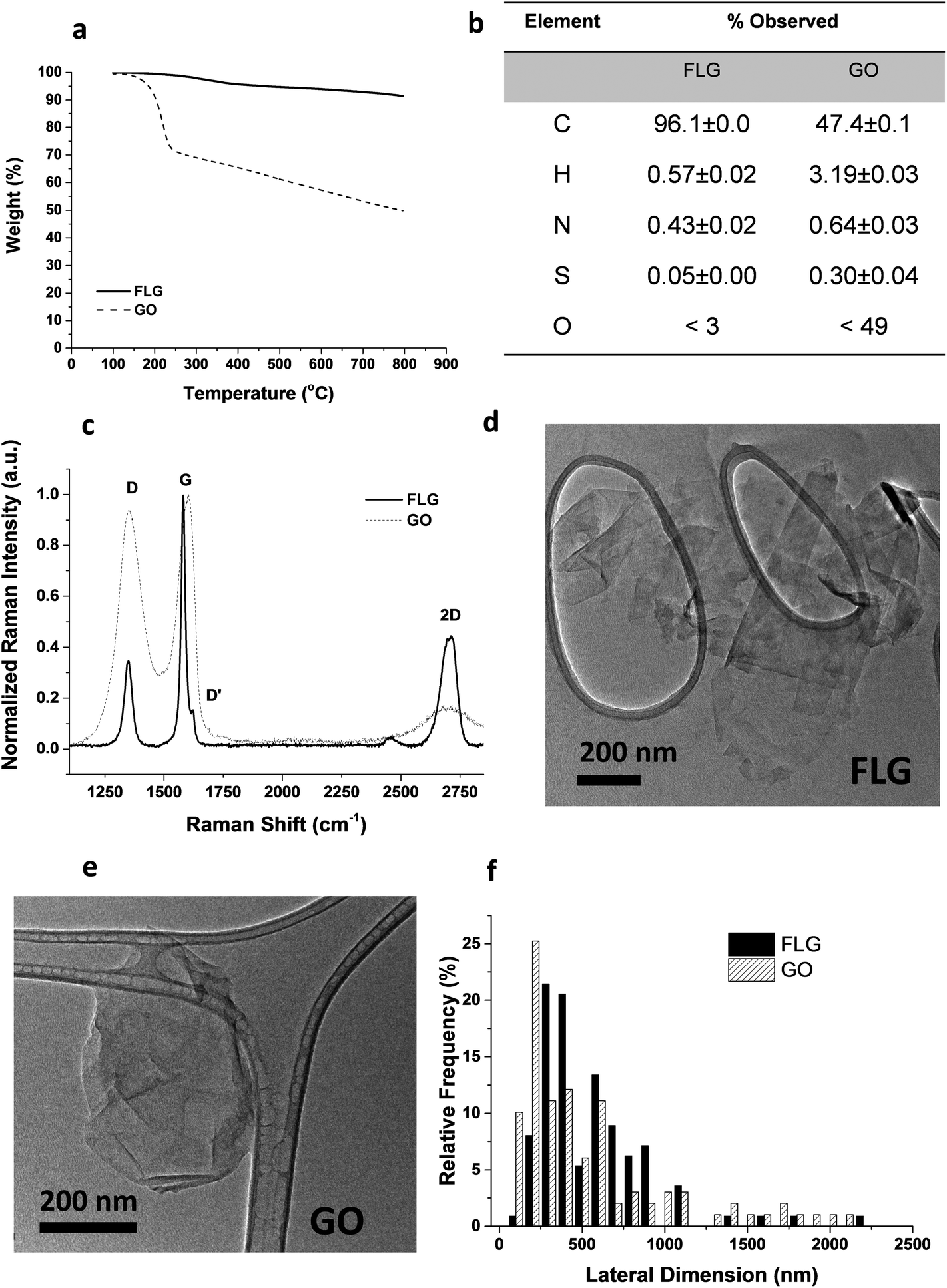 | ||
| Fig. 1 Physicochemical characterization of the GO and FLG used in this study. (a) Thermogravimetric analysis, (b) elemental analysis, (c) Raman spectroscopy, (d–e) TEM images, (f) lateral size distribution from TEM observations. | ||
Structural features in both nanostructures were assessed using Raman spectroscopy (Fig. 1C). GO exhibits prominent G and D bands (ID/IG = 0.94), ascribed to its highly defective structure.32 In contrast, FLG has a Raman profile consistent with its high crystallinity. In this case, ID/IG = 0.35, and the full-width at half maximum (FWHM) of the 2D band (66.2 cm−1) confirmed the few-layered nature.33 Additional insight into the number of layers in FLG was acquired via an established formula,34 which relates the different intensities of G and 2D bands of FLG and graphite, respectively. According to this calculation, our FLG was composed of ∼4 layers.35 For GO, the characterization performed by the supplier suggests that its thickness was close to the monolayer.36
In addition, since a stainless-steel flask was used during the ball-milling process, TXRF was performed to ensure the absence of metals (especially Fe) in the FLG sample, revealing a Fe content of only 0.19 ppm. TXRF results from both GO and FLG are available in the ESI (Table S1†).
TEM characterization (Fig. 1D–E) provided a direct visualization of the sheet morphology in GO and FLG. The lateral size distribution (Fig. 1F) sampled in both nanostructures confirmed their similarity in size and shape.
2.2 Metabolomic cell profile
Metabolomics can generate much information about the relationship between nanomaterials and human cells.27,37,38 Fig. S1† shows the typical 1H NMR spectrum of an aqueous extract from untreated HaCaT cells, our control experiment. Focusing on the analysis of the 1H NMR spectrum of the aqueous extract from untreated cells, we identified NMR peaks to elucidate which metabolites could be detected in our experimental conditions. The NMR peak identification was carried out by matching spectral data to reference spectra from the Human Metabolome Database (HMDB) and from Birmingham Metabolite Library (BML-NMR) (see Fig. S2† as an example) as well as by assistance from the software Chenomx Profiler. In addition, 2D NMR experiments, J-resolved, 1H–1H-TOCSY and 1H–13C HSQC, were conducted to assist with spectral assignment (Fig. S3–S5†). The 2D NMR experiments were carried out using a Bruker 800 MHz spectrometer equipped with a cryoprobe for gaining signal sensitivity, thus enabling the detection of the 13C NMR peaks for certain metabolites that were present at very low concentrations, as observed for succinate, methionine, phosphocholine, glycerophosphocholine, among others. For selected metabolites, i.e. phosphocreatine, ATP, UTP, UDP, the addition of standard samples further assisted in peak identification. A total of 27 metabolites (Fig. S1†) could be identified in our experimental conditions. A list is shown in Fig. 2A. Comparison with previous NMR metabolomics studies on HaCaT cells indicated a correct assignment of NMR peaks.37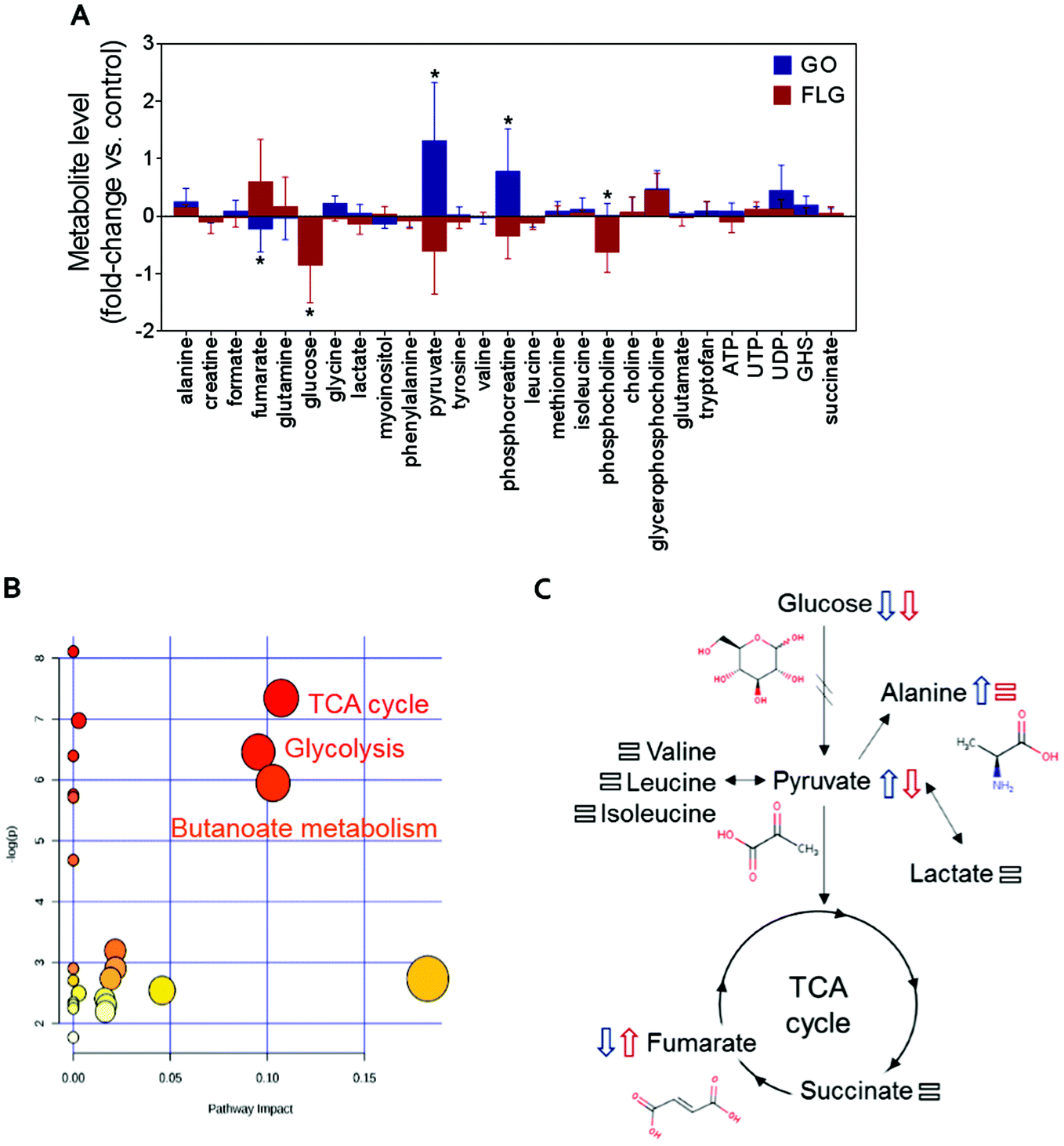 | ||
| Fig. 2 The impact of GO and FLG on HaCat cells: (a) relative changes in the identified metabolites induced by GO (blue) and FLG (red) (n = 6). (b), (c) Representative altered cellular pathways. | ||
Focused on investigating the influence of GO and FLG on the metabolomics cell profile, HaCaT cells were treated with 5 μg mL−1 of GO or FLG for 7 days. We considered this concentration because it is a dose that triggers cell death in a low percentage. This fact allows the analysis of the main metabolic changes in the viable, but probably damaged, remaining cells. 1H-NMR experiments for both the control samples and the cells treated with GO or FLG were registered at 298 K with a 500 MHz NMR spectrometer. The control sample for the cell extract treated with GO corresponded to a cell extract that was not exposed to any material. Since FLG contains trace amounts of melamine, the latter was used in exactly the same concentration in control experiments with the cell extracts.33 Comparison of the treated and untreated 1H NMR spectra did not show any new metabolites for the treated sample, as was expected. The variations in concentration induced in the metabolites were investigated through quantification of all NMR signals in the 1H-NMR spectra for all samples. Focused on avoiding the integration of overlapped signals and thus achieving higher accuracy in the absolute amount of every metabolite, the quantification was carried out by manual peak picking using Global Spectral Deconvolution (GSD) from Mnova (MestreLab Research). The selected signals for all metabolites detected in the 1H-NMR spectrum were quantified using the module from Mnova for a targeted spectral analysis, Simple Mixture Analysis (SMA), which uses the integral of a standard solution of known concentration (TSP, 0.1 mM) for extracting absolute concentration amounts of each metabolite. For higher accuracy in the quantification, a separated sample containing only TSP at pH 7.4 was prepared and its 1H NMR spectrum was registered at 298 K and with similar acquisition parameters to the study sample. The extracted values for the absolute amount (mM) for every metabolite were treated statistically by differential analysis and classification, which was performed with MEV4, and pathway enrichment with MetaboAnalyst.
The addition of GO to HaCaT cells resulted in an increase in alanine, pyruvate, phosphocreatine, glycerophosphocholine and UDP levels by 0.3, 1.2, 0.8, 0.6 and 0.5-fold vs. control, respectively (Fig. 2A).
Treatment of HaCaT cells with FLG resulted in 0.7 and 0.6-fold increases in fumarate and glycerophosphocholine levels vs. control, respectively, and a decrease in pyruvate, phosphocreatine and phosphocholine levels of −0.5, −0.3 and −0.6-fold vs. control, respectively. Treatment with GO or FLG also reduced glucose levels by −0.5 and −0.9-fold vs. control, respectively, and was correlated with the slight decrease in leucine, acetate and phenylalanine levels. Enrichment analysis revealed the alteration in major metabolic pathways including those related to protein biosynthesis, the urea cycle, ammonia recycling and methionine metabolism, among others (Fig. 2B). All these changes are related to critical cell processes (Fig. 2B and C) and could probably directly impact cell viability and motility.
The down-shift in the glucose level is especially relevant as it is usually associated with increased cell death. Different studies, including ours, have shown that a 5 μg mL−1 dose of GRMs slightly increases apoptosis and cell toxicity.21,39,40 The observed decrease in glucose denotes that GO and FLG remodel cellular energetic pathways, probably by interaction with mitochondria and the overproduction of ROS.20 Moreover, the levels of some of the metabolites located in mitochondria, such as phosphocholine, are altered by both FLG and GO treatment, which could be associated with mitochondrial damage.
Interestingly, cells treated with GO exhibited an increase of pyruvate levels, while the opposite trend was observed for FLG. This fact could account for a differential effect of GO and FLG in glycolysis and the tricarboxylic acid cycle (TCA) or Krebs cycle. Pyruvate plays a critical role as an intermediate compound in the metabolism of carbohydrates, proteins, and fats. In our study, we did not notice significant alterations in valine, leucine, isoleucine and lactate levels, which are directly related to pyruvate. In fact, pyruvate also acts as an intermediate in alanine metabolism, and in our study, we observed an increase in alanine levels in cells treated with GO (Fig. 2A).
Significant and differential changes in fumarate levels were found in cells treated with GO (decrease) or FLG (increase). Fumarate is a precursor of L-malate in the TCA cycle. It is formed by the oxidation of succinic acid by succinate dehydrogenase. It is also considered an oncometabolite because of its high levels observed in tumors or biofluids surrounding tumors and because of its stabilization of HIF1α, promoting tumor growth and development.41,42 Fumarate also plays a pivotal role in the TCA cycle,43 which is altered in cells treated with GO or FLG (Fig. 2B). Therefore, these findings suggest that the TCA cycle may be compromised in cells treated with FLG. Recently, Oliveira et al. observed a decrease in creatine/creatine phosphate in HaCaT cells treated with cobalt ferrite nanoparticles44 and also an increase in fumarate levels in cells treated with silver nanoparticles.37 Decreased levels of phosphocreatine can be linked to diverse consequences for the cells, such as increased permeability of the plasma membrane.45 In our study, increased levels of phosphocreatine induced by GO could probably be due to a cytoprotective mechanism triggered by cells to overcome the damage.
We also noticed significant changes in phosphocholine levels. Phosphocholine is the precursor metabolite of choline in the glycine, serine and threonine metabolism pathways and an intermediate between choline and cytidine-diphosphate choline in the glycerophospholipid metabolism pathway. Phosphocholine together with glycerophosphocholine (GPC) are the two major forms of choline storage in the cytosol. We also observed a non-significant increase in GPC levels in cells treated with GO and FLG. Alterations observed in phosphocholine could be directly related to an increase in 1-alkyl-2-acetyl GPC levels, a GPC-derived lipid mediator involved in numerous inflammatory processes and stress responses in keratinocytes, which could compromise processes as would healing.46 However, it has recently been reported that the immunomodulatory effect of GO functionalized with NH2 groups avoids this problem, shifting the immune response to T helper-1/M1.10
In summary, in this study, GO and FLG treatment of HaCaT cells caused altered concentrations of metabolites related to glycolysis, the TCA cycle and butanoate metabolism (Fig. 2B), and there are some interesting alterations that should be studied more deeply, such as fumarate increase induced by FLG. Some of the alterations induced by GRMs imply the remodeling of cellular energetic pathways, probably by interaction with mitochondria and overproduction of ROS.
2.3 The effects of GO and FLG on ROS and free cytosolic Ca2+ levels
ROS and Ca2+ are essential and interconnected signaling molecules in homeostatic/normal cell conditions.47 However, the overproduction of ROS and increased levels of Ca2+ are linked to diverse cellular processes, from metabolic alterations to cell death.48–52Our results show that GO and FLG increase the levels of ROS and Ca2+ in a concentration- and time-dependent manner for up to 7 days. Specifically, GO induced a significant increase of 64.8% in mitochondrial ROS after 24 h in cells treated with 100 μg mL−1 (***p < 0.001). After 7 days of GO exposure, mitochondrial ROS was 39.1% (*p < 0.05), while cells treated with FLG elicited an increase of 61.1% at 100 μg mL−1 for 7 days (###p < 0.001). Thus, GO showed a stronger effect compared to FLG (a < 0.05) even at short incubation times (Fig. 3A).
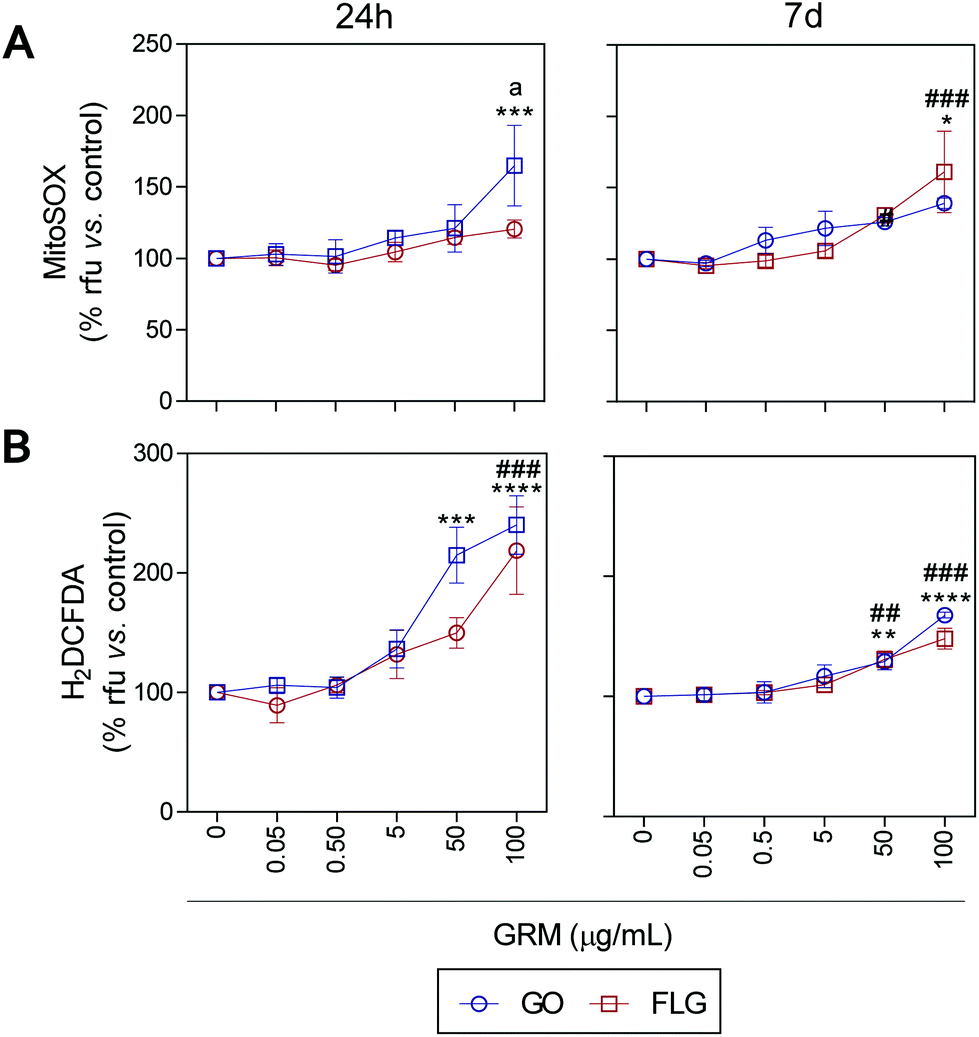 | ||
| Fig. 3 The impact of GO and FLG on O2˙− and H2O2 levels: (a), (b) MitoSOX-AM (O2˙−) and H2DCF-DA (H2O2) levels in cells treated with GO (blue) or FLG (red) for 24 h or 7 d (n = 4) (ESI Fig. 6†). | ||
The significant difference could be related to different levels of oxidation in the GRMs used. GO also induced a significant increase in cytosolic ROS of 114.8% and 140% in cells treated for 24 h with 50 μg mL−1 and 100 μg mL−1, respectively (***p < 0.001). Cells incubated for 7 days with 50 μg mL−1 and 100 μg mL−1 showed an increase of 28% (**p < 0.001) and 67.7% (****p < 0.0001), respectively (Fig. 3B). Meanwhile, in cells incubated with 100 μg mL−1 of FLG for 24 h we observed a significant increase of 118.5% (####p < 0.0001). In cells incubated for 7 days with 100 μg mL−1, O2˙− increased by 48% (###p < 0.001) (Fig. 3B). To sum up, GO is more potent at increasing ROS levels, specifically at short incubation times, but levels tend to recover at longer incubation times, leading to the activation of a protective cellular antioxidant response.
It is well known that overproduction of ROS can trigger the calcium-signaling pathway.47 Moreover, Ca2+ plays an important role in numerous cellular processes related to metabolism, cell death, signaling processes, etc.49,50 Nevertheless, elevated free cytosolic Ca2+ levels can damage the mitochondrial electron transport chain, leading to mitochondrial damage and overproduction of ROS.53 This fact motivated us to check the effect of GO and FLG on the free cytosolic Ca2+ levels. Our results show that the addition of 100 μg mL−1 of GO to HaCaTs elicited a 47% increase (#p < 0.05) in Ca2+ levels over a very short time of 2 h (Fig. 4). At an intermediate time of 24 h, 5, 50 and 100 μg mL−1 GO increased Ca2+ levels by 40% (*p < 0.05), 70% and 82% (****p < 0.0001), respectively (Fig. 4). Over the long-term (7 days), GO induced a significant increase in Ca2+ levels of 118.2% in cells treated with only higher doses (100 μg mL−1) (****p < 0.0001) (Fig. 4). Similar results were obtained for FLG at short (2 h) and moderate (24 h) incubation times (Fig. 4). Remarkably, over long incubation times (7 days), FLG was more potent than GO at increasing the free cytosolic Ca2+ levels. Specifically, 50 and 100 μg mL−1 of FLG increased Ca2+ levels to 124% (####p < 0.0001) and 174.2% (####p < 0.0001), respectively, compared to 78% (aap < 0.01) and 56% (ap < 0.05) for the same doses of GO (Fig. 4).
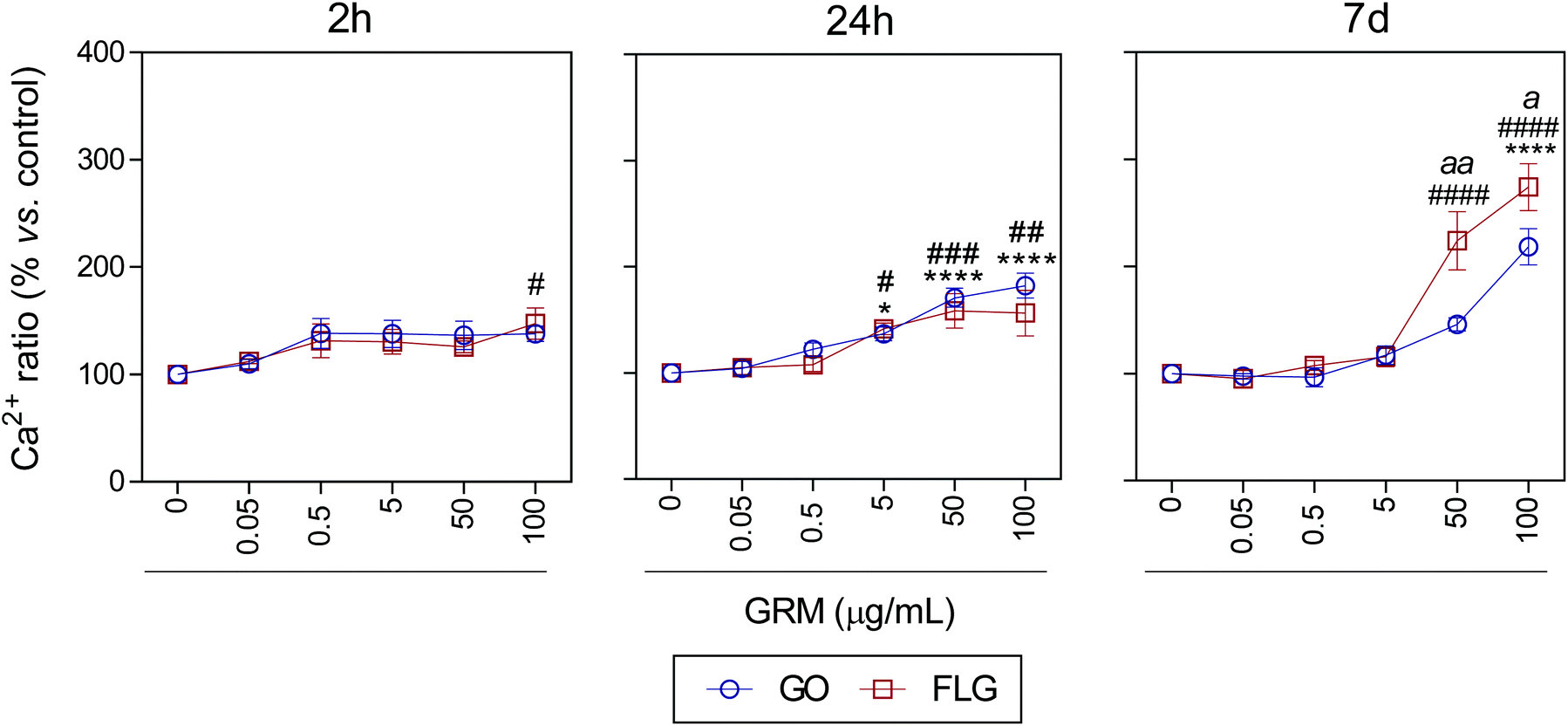 | ||
| Fig. 4 The effects of GO and FLG on free cytosolic Ca2+ levels. Fluo4-AM levels in cells treated with GO (blue) or FLG (red) over 2 h, 24 h and 7 d (n = 4) (ESI images Fig. 7†). | ||
These results are in line with that reported previously for GO and FLG in other cell lines. Indeed, GO and FLG were previously shown to increase ROS generation,54,55 which was paralleled by an increase in the free cytosolic Ca2+ levels in a concentration-dependent way. Whereas increased ROS levels had been postulated as a primary cytotoxicity mechanism of GO,55,56 cytosolic Ca2+ is strongly related to several key glycolytic and Krebs cycle enzymes that generate or metabolize pyruvate.51,57,58 Moreover, several nanomaterials induced Ca2+ homeostasis changes and apoptosis59–61 through direct damage to the mitochondria.40,53 The decrease in glucose observed in our metabolomics study in cells treated for 7 days with 5 μg mL−1 of GO and FLG denotes changes in cellular energetic pathways, probably by interaction with mitochondria and overproduction of ROS.20 With this dose, we observed a non-significant increase in ROS levels (Fig. 3) so we tested the total antioxidant capacity in these cells and noticed a significant decrease of 18% (**p < 0.01) and 10% (#p < 0.05) in cells treated with GO and FLG, respectively (ESI Fig. 9†).
In summary, although our results obtained for GO and FLG are quite similar in terms of the elevation of ROS and free cytosolic Ca2+ levels, the effect of FLG is greater at regulating both O2˙− and Ca2+ at high concentrations and long incubation times.
2.4 The influence of GO and FLG on cell necrosis and apoptosis
It has been previously reported that different GRMs could provoke cell toxicity, inducing apoptosis, necrosis and autophagy, and all these processes are related to cellular stress.21,54,62,63 Indeed, a recent study describes significant cellular damage induced by FLG and GO (1–100 μg mL−1) in HaCaT cells after 72 h,21 similar to that reported for skin fibroblasts exposed to 50 μg mL−1 GO.64We assessed the effect of GO and FLG on cell viability, necrosis and apoptosis at different times from 24 h to 7 d. First, there was a dose- and time-dependent reduction in the number of cells per field, which was significant with doses higher than 100 μg mL−1 for both compounds, with a stronger effect from GO compared to FLG (Fig. 5A and ESI Fig. 8†). In particular, the incubation of HaCaTs with 100 and 200 μg mL−1 of GO for 24 h exhibited a reduction of 47.5% and 46.3%, respectively (***p < 0.001; **p < 0.01, Fig. 5A). This was mainly due to the induction of necrosis, reaching values of 13% and 23% (****p < 0.001, Fig. 5B) and apoptosis to a minor extent, reaching low but significantly increased values of 5% and 6.5% (****p < 0.0001, Fig. 5C). At this same time, FLG showed similar cytotoxic effects, being slightly less potent than GO (Fig. 5A–C).
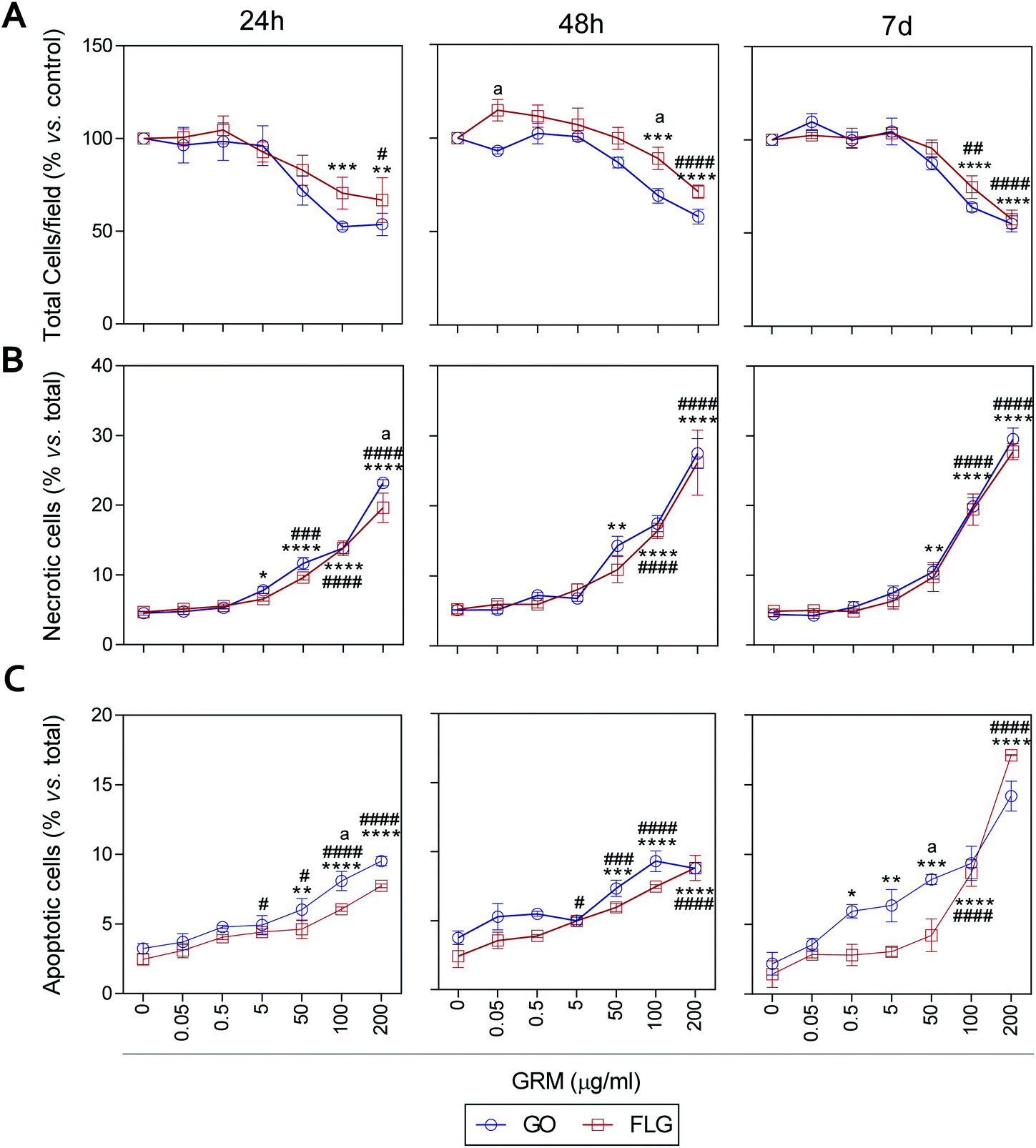 | ||
| Fig. 5 The effect of GO and FLG on HaCaTs viability, necrosis and apoptosis. (a) The total number of cells per field. (b) The percentage of necrotic cells, (c) percentage of apoptotic cells in HaCaTs treated with GO (blue) or FLG (red) during 24 h, 48 h and 7 d (n = 4). | ||
Results for both compounds were similar after 48 h of incubation, with GO being slightly more potent than FLG (Fig. 5A–C and ESI Fig. 4†) at reducing the number of total cells per field, probably by its pro-apoptotic effect but also by the inhibition of cell adhesion, as described before.65 A 7-day incubation of HaCaTs with 100 and 200 μg mL−1 GO provoked a reduction of 36.2% (***p < 0.001) and 55% (****p < 0.0001), respectively (Fig. 5A and ESI Fig. 4†), which was mainly due to an induction of necrosis, yielding values of 14.8% and 25.2% (****p < 0.0001) (Fig. 5B), respectively, and to apoptosis to a minor extent, reaching low but significantly increased values of 9.36% and 14.18% (****p < 0.0001, Fig. 5C). Lower doses such as 0.5, 5 and 50 μg mL−1 also increased apoptosis by 5.9% (*p < 0.05), 6.3% (**p < 0.01) and 8.22% (***p < 0.001), respectively. For this same incubation time, FLG showed similar cytotoxic effects, being slightly less potent than GO (Fig. 5A–C).
Our results support the ability of GO and FLG to increase the levels of mitochondrial and cytosolic ROS as well as the levels of free cytosolic calcium,54–56 which are known to affect metabolism51,57,58 and lead to apoptosis.59–61 Surprisingly, the cytotoxicity induced by both compounds is due to cell necrosis, preferentially from apoptosis, which is indicative of the induction of physical cell damage. This may be caused by the interaction of the GRMs with the plasma membrane presenting damage-associated molecular patterns (DMAPs), as previously suggested by Pelin and colleagues for the same cell model.21 Interestingly, Li et al.66 observed and modeled the interaction between FLG and keratinocyte plasma membranes, associating it with cell damage possibly activated by DAMPs.67–69
2.5 GO and FLG inhibit cell motility but regulate different structures of the actin cytoskeleton
Actin polymerization can be altered by an increase in Ca2+ levels, which impairs the dynamics of the actin cytoskeleton and reduces cell motility.70 The physical interaction of GRMs with cell membranes also alters the dynamics of actin and may lead to cytoskeletal disruption.66,71,72 Furthermore, GRMs can make contact with actin filaments, interfering with their dynamics.73 Since cell motility is a critical step in wound closure after injury, we wondered if motility and actin structures related to cell movement and adhesion to the substrate were affected by GO and FLG in HaCaTs human keratinocytes.Increasing concentrations of GO and FLG resulted in a concentration-dependent inhibition of HaCaT cells (Fig. 6), which were reduced by 80% at 200 μg mL−1 GRMs (*** & ###p < 0.001). Since cell motility depends on actin specialized structures such as lamellipodia, filopodia and stress fibers (related to cell polarization, anchorage and contraction, respectively), we used confocal microscopy to analyze the impact of GRMs on these structures. This strategy revealed that treatment with GO reduced the number of lamellipodia positive cells at 50, 100 and 200 μg mL−1 by 17.6 (*p < 0.05), 17.5 (*p < 0.05) and 23.7% (***p < 0.001), respectively (Fig. 6B). At 100 and 200 μg mL−1, the number of invadopodia positive cells (21.9% and 33.5%) (Fig. 6C) and stress fiber positive cells (18.6% and 22.9%) (Fig. 6D) were also reduced. Moreover, GO at 50 and 200 μg mL−1 doses increased the number of filopodia positive cells (Fig. 6E). FLG decreased the number of lamellipodia positive cells (Fig. 6B) at concentrations from 5 μg mL−1 (11.9%; ##p < 0.01) to 200 μg mL−1 (27%; ####p < 0.0001). At higher doses of 100 and 200 μg mL−1, it also reduced the number of invadopodia positive cells (15.9% and 15% respectively; #p < 0.05) (Fig. 6C) without altering stress fibers (Fig. 6D). Moreover, filopodia were increased (Fig. 6E) at 50 μg mL−1 (#p < 0.05).
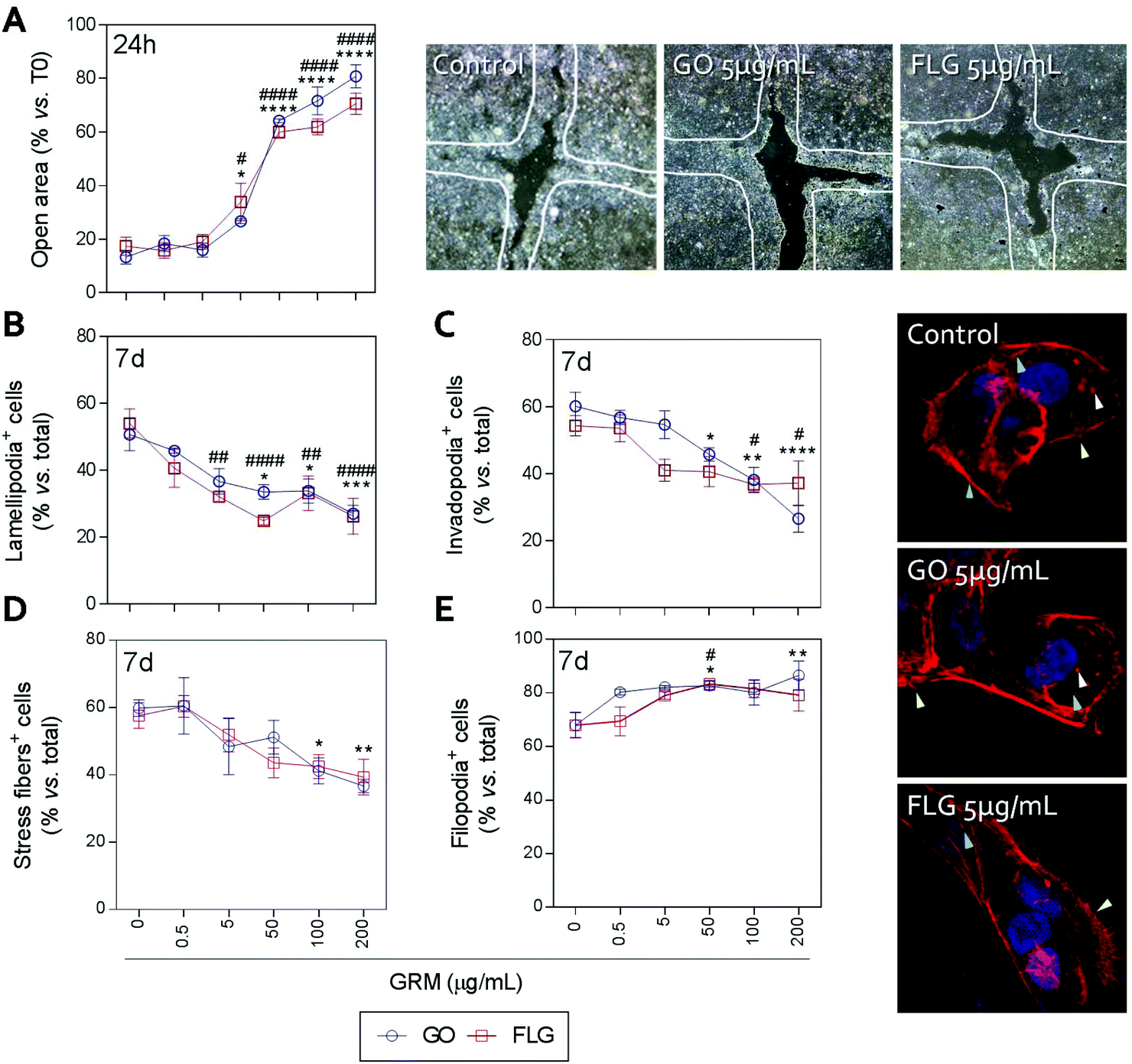 | ||
| Fig. 6 The effect of GO and FLG on HaCaTs motility and F-actin organization. (a) The percentage of open areas in cells treated with GO (blue) or FLG (red) over a 24 h period. (b) The percentage of lamellipodia positive cells (green arrow), (c) the percentage of invadopodia positive cells (white arrow), (d) the percentage of stress fiber positive cells (blue arrow), (e) the percentage of filopodia positive cells (yellow arrow) in HaCaTs treated with GO (blue) or FLG (red) over 7 days (n = 4). | ||
Overall, the data indicate that FLG and GO, besides inhibiting cell migration, affect the dynamics of the actin cytoskeleton in human non-transformed epithelial cells in a differential manner, probably by their effect on ROS and free cytosolic Ca2+ levels, though we cannot discard the involvement of the regulation of genes related to the actin cytoskeleton as described before.74
These results indicate that both GO and FLG could significantly affect biological processes like wound healing, where the proliferation and migration of skin cells are mandatory. This fact is clearly in contrast to different recent publications that show that graphene-containing scaffolds induce wound healing, at a faster rate in comparison to other compounds, in mice, rat and rabbit models.75–77 A recent study indicated that skin grafts, based on hybrid graphene oxide-natural biopolymer nanofibers, are effective in wound healing in vivo.78 These polymers stimulate the proliferation of fibroblasts and the infiltration of immune cells as polymorphonuclear cells. Another study showed that graphene foams loaded with bone marrow-derived mesenchymal stem cells can be used in wound healing processes.79 However, in all these studies, GRMs are immobilized in other materials. As a whole, our study shows that GO or FLG reduces wound healing and actin-motility structures, probably by the direct effect of GRMs on the cell, altering redox homeostasis, metabolism, etc. For future biomedical applications, it is essential to characterize the impact of GRMs according to their functionalization, size, lateral dimensions, incubation time and dose, etc. It is also critical to determine whether GRMs could be released from the different scaffolds and interact directly with cells, inducing irreversible damage.
3. Conclusions
NMR-based metabolomics revealed the differential effects of GO and FLG on HaCaT cells, altering the levels of different metabolites involved in biochemical processes such as glycolysis, TCA cycle and butanoate metabolism. These alterations could be associated with the modulation of the global cell-bioenergetics status, suggesting that mitochondria function could be compromised. However long-term studies must be performed to ascertain whether cells can recover from this initial damage. The different oxidation states of GO and FLG may be responsible for their subtle but differential effects on HaCaT cells at cellular levels. Our results showed that GO and FLG induced a significant dose- and concentration-dependent increase in the levels of the free cytosolic Ca2+, with both compounds being indistinguishable in short time periods (up to 24 h), but FLG was more potent at 7 days and at high concentrations (>50 μg mL−1). FLG was also more potent than GO at increasing the level of ROS at 24 h. Interestingly, the levels of H2O2 were higher at shorter times, but tended to recover the basal level at 7 days. The differential regulation of Ca2+ and ROS was reflected in a differential remodeling of the metabolome related to processes like protein biosynthesis, the urea cycle, the glucose and alanine cycle. After 7 days of incubation, GO and FLG had different effects on cell death, with FLG being more potent at triggering apoptosis by exerting similar effects to provoking necrosis. Moreover, both compounds reduced cell motility to a similar magnitude and in a dose-dependent manner, although GO preferentially inhibited the formation of lamellipodia and invadopodia, which are structures involved in directional cell migration and substrate degradation. Since actin remodeling and cell migration are impaired by treatment with free GRMs, our results suggest that processes like wound healing could be compromised. Graphene foam is a promising candidate for novel scaffolds in skin tissue regeneration and bioengineering and it is therefore essential to understand and dissect the mechanisms governing its toxic effects and to determine the exact toxicity range for each GRM. In summary, our results demonstrate that GO and FLG, compounds with different oxidation states, trigger differential effects on HaCaTs cells, which are evident at cellular levels and are governed at the molecular level.4. Experimental section
4.1 FLG and GO production and characterization
FLG was prepared by a ball milling treatment following a protocol described elsewhere,33 using a Retsch PM 100 planetary mill under air atmosphere. Graphite (7.5 mg SP-1 graphite powder, purchased from Bay Carbon, Inc.) and melamine (22.5 mg Sigma Aldrich ref. M2659) were ball milled at 100 rpm for 30 min, dispersed in 20 mL of water and sonicated for 1 min to produce a black suspension. Melamine was removed by dialysis against hot water and the resulting dispersion was left to settle for 5 days. The precipitate, consisting of poorly exfoliated graphite, was discarded and the liquid fraction was carefully extracted, freeze-dried and used as a fine powder.GO was obtained from Grupo Antolin Ingeniería (Burgos, Spain); it was produced by the oxidation of carbon fibers (GANF Helical-Ribbon Carbon Nanofibres, GANF®) with a KMnO4/H2SO4 mixture and sodium nitrate at 0 °C.36 The concomitant carbon debris and other possible acid traces were removed by washing with Milli-Q water, with sequential cycles of re-dispersion/centrifugation (4000 rpm, 30 min), discarding the supernatant liquid in each cycle until the pH of the GO aqueous suspension was ∼5. The GO suspension was then freeze-dried for further use.
Thermogravimetric analysis (TGA) was carried out, from 100 °C to 800 °C, using a TGA Q50 (TA Instruments) at 10 °C min−1 under nitrogen flow. For comparative purposes, GO and FLG maximum weight losses were taken at 600 °C.
At least 20 individual Raman spectra, at different random locations of the sample, were collected using an inVia Raman microspectrophotometer (Renishaw plc, United Kingdom). The laser wavelength was 532 nm, together with a 100× objective (N.A. 0.85) and an incident power density below 1 mW μm−2 for FLG, or below 0.1 mW μm−2 in the case of GO.
Elemental analysis was performed with a LECO CHNS-932 analyzer, completely burning the sample with four doses of oxygen and quantifying the released gases by thermal conductivity.
The morphologies and lateral sizes of both nanostructures were analyzed using a Jeol JEM 1011 transmission electron microscope (TEM) (Jeol, Japan). Lateral dimension distribution was calculated by using Fiji® software and probing at least 100 flakes in each case.
In addition, total reflection X-ray fluorescence (TXRF) of FLG and GO were performed using a Bruker-S2 PicoFox TXRF spectrometer.
4.2 Cell culture
HaCaT cells were maintained in DMEM high glucose medium (Sigma-Aldrich) containing 10% FBS (Sigma-Aldrich) and 1% antibiotic/antimycotic (Sigma-Aldrich), at 37 °C and 5% CO2. Experiments were performed in DMEM containing 10% FBS. All cells used in this study were up to the 15th passage.4.3 Sample preparation for NMR experiments
Cells were grown in 3 × 175 cm2 flasks and treated for 7 days with 5 μg mL−1 GO or FLG. Cells were detached, rinsed twice in PBS and homogenized by sonication in deuterated D2O (99.98 atom% D) (Sigma-Aldrich) (n = 6).4.4 NMR measurements
The NMR experiments were carried out using a Varian Inova 500 spectrometer operating at 499.77 MHz for 1H and at 125.678 MHz for 13C. The spectrometer was equipped with a four-nucleus 5 mm 1H {15N–31P}PFG high-field indirect detection probe. Standard 1D spectra with water suppression (presat) were recorded as previously reported by us80 with a spectral width of 8 kHz, 32k data points, a 90° pulse width of 11.5 μs, a 4 s relaxation delay and 400 scans, at 298 K and using pulse sequences from the Varian library. Manual phase and baseline corrections were applied to all 1D spectra for processing. J-Resolved 2D homo- and heteronuclear correlation experiments, 1H–1H-TOCSY, and 1H–13C HSQC studies were carried out at 298 K for NMR peak assignment in an 800 MHz (1H) Avance NMR spectrometer (Bruker, Billerica, MA, USA) equipped with 1H, 13C, 15N cryoprobe and Z-gradients, following the Bruker library. In addition, matching 1D and 2D data to reference spectra in both the Human Metabolome Database (HMDB) and the Birmingham Metabolite Library (BML-NMR), and the use of Chenomx NMR Suite 8.2 were used to assist in peak identification. The addition of standard samples to the mixture of metabolites was carried out to confirm peak identification for selected metabolites. The NMR peak quantification was assessed by Simple Mixtures Analysis (SMA, Mnova 11.0) by means of a known concentration of TSP (0.1 mM) as standard, and by manual peak picking using Global Spectral Deconvolution (GSD) from Mnova. A separated sample tube containing only TSP at pH 7.4 was prepared and its 1H NMR spectrum was registered at 298 K and with similar acquisition parameters to the study sample.4.5 Statistical analysis
Data are expressed as mean ± S.E.M., obtained from a minimum of three independent experiments. Statistical analysis was carried out with GraphPad Prism 6, using Two-way ANOVA (Bonferroni test). Differences were considered significant at p < 0.05.4.6 Determination of apoptosis, necrosis and viability
HaCaT were seeded in 96 well plates and incubated for 24 h, 48 h and 7 d with increasing concentrations of GO and FLG. After each treatment, cells were incubated with 10 μg mL−1 EtBr and 1 μM Calcein-AM. Viable (green) and necrotic cells (red) were determined using Cytation 5 (BioTek). Immediately after image acquisition, the cells were fixed and permeabilized for 2 min in ice-cold methanol and stained with 1 μg mL−1 Hoescht. Apoptotic nuclei were determined according to morphological criteria.80 For viability, necrosis and apoptosis, the results are expressed as percentage vs. total cells (n = 4).4.7 Determination of total antioxidant capacity
Cells were seeded in 96 well plates and incubated for 24 h or 7 days with 5 μg mL−1 of GO or FLG. Total antioxidant capacity was determined in culture media using a commercial kit, following the manufacturer's instructions (MAK187, Sigma-Aldrich). Results are expressed as ratios (vs. control) of nmol (n = 4).4.8 Determination of O2˙−, H2O2 and Ca2+ in single cells
Mitochondrial O2˙−, total H2O2 and free cytosolic Ca2+ levels were determined using the fluorescent probes MitoSox, H2DCF-DA and Fluo-4, respectively (Thermo Fischer). HaCaT cells were seeded in 96 well plates and incubated for 24 h, 48 h or 7 days with increasing concentrations of GO and FLG. After each treatment, the cells were loaded for 30 min with 1 μM of the fluorescent probe (one independent probe per assay), washed in fresh medium and imaged using a Nikon TiU microscope (20× objective). Images were analyzed and processed with ImageJ. Results show the percentage of cell signal vs. control (n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 4).
= 4).
4.9 Wound healing assay
HaCaT were plated in 12-well plates, cultured to confluence and then serum-starved for 12 h in media. A cross was scratched into the monolayer with a 10 mL pipet tip and the medium was replaced with fresh medium containing GO or FLG. The percentage of wound closure was calculated by measuring the open area free of cells for each image, using ImageJ, immediately after making the scratch and 24 h after treatment. Results shown are an average of n = 4.4.10 Remodeling of the actin cytoskeleton
To assess the effect of the compounds on the remodeling of the actin cytoskeleton, cells were incubated for 24 h with GO and FLG ranging from 0.5 to 200 μg mL−1, fixed for 10 min in 4% paraformaldehyde at room temperature, then washed three times with PBS and incubated with ActinRed™ 555 Ready Probes® (Thermo-Fisher). Images were obtained using a Zeiss LSM-600 confocal microscope (63× objective). To quantify the effects of the treatments on actin reorganization and therefore on cell mobility, cells with a static (filopodia positive) and a migratory (lamellipodia, invadopodia or stress fibers positive) phenotype were counted in a double-blind procedure. At least 100 cells in 4 independent experiments were counted for each group, considering positive cells with migratory phenotype. Results are expressed as percentages of positive cells normalized vs. control.Conflicts of interest
The authors declare no conflict of interest.Funding sources
This work has been supported from the EU Graphene-based disruptive technologies, Flagship project (no. 696656), and from Spanish Ministry of Economy and Competitiveness (MINECO) under project grant CTQ2014-53600-R.References
- J. M. Yoo, J. H. Kang and B. H. Hong, Chem. Soc. Rev., 2015, 44, 4835–4852 RSC.
- S. Gurunathan and J. H. Kim, Int. J. Nanomed., 2016, 11, 1927–1945 CrossRef PubMed.
- H. Dong, C. Dong, T. Ren, Y. Li and D. Shi, J. Biomed. Nanotechnol., 2014, 10, 2086–2106 CrossRef PubMed.
- L. Feng, L. Wu and X. Qu, Adv. Mater., 2013, 25, 168–186 CrossRef PubMed.
- A. Gulzar, P. Yang, F. He, J. Xu, D. Yang, L. Xu and M. O. Jan, Chem.–Biol. Interact., 2017, 262, 69–89 CrossRef PubMed.
- J. Byun, J. Microbiol. Biotechnol., 2015, 25, 145–151 CrossRef PubMed.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef PubMed.
- G. Lalwani, A. M. Henslee, B. Farshid, L. Lin, F. K. Kasper, Y. X. Qin, A. G. Mikos and B. Sitharaman, Biomacromolecules, 2013, 14, 900–909 CrossRef PubMed.
- G. Reina, J. M. Gonzalez-Dominguez, A. Criado, E. Vazquez, A. Bianco and M. Prato, Chem. Soc. Rev., 2017, 46, 4400–4416 RSC.
- M. Orecchioni, D. Bedognetti, L. Newman, C. Fuoco, F. Spada, W. Hendrickx, F. M. Marincola, F. Sgarrella, A. F. Rodrigues, C. Menard-Moyon, G. Cesareni, K. Kostarelos, A. Bianco and L. G. Delogu, Nat. Commun., 2017, 8, 1109 CrossRef PubMed.
- A. Bianco, Angew. Chem., Int. Ed., 2013, 52, 4986–4997 CrossRef PubMed.
- D. Bitounis, H. Ali-Boucetta, B. H. Hong, D. H. Min and K. Kostarelos, Adv. Mater., 2013, 25, 2258–2268 CrossRef PubMed.
- A. B. Seabra, A. J. Paula, R. de Lima, O. L. Alves and N. Duran, Chem. Res. Toxicol., 2014, 27, 159–168 CrossRef PubMed.
- X. Hu and Q. Zhou, Chem. Rev., 2013, 113, 3815–3835 CrossRef PubMed.
- P. Wick, A. E. Louw-Gaume, M. Kucki, H. F. Krug, K. Kostarelos, B. Fadeel, K. A. Dawson, A. Salvati, E. Vazquez, L. Ballerini, M. Tretiach, F. Benfenati, E. Flahaut, L. Gauthier, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2014, 53, 7714–7718 CrossRef PubMed.
- M. Orecchioni, D. A. Jasim, M. Pescatori, R. Manetti, C. Fozza, F. Sgarrella, D. Bedognetti, A. Bianco, K. Kostarelos and L. G. Delogu, Adv. Healthcare Mater., 2016, 5, 276–287 CrossRef PubMed.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef PubMed.
- M. C. Duch, G. R. Budinger, Y. T. Liang, S. Soberanes, D. Urich, S. E. Chiarella, L. A. Campochiaro, A. Gonzalez, N. S. Chandel, M. C. Hersam and G. M. Mutlu, Nano Lett., 2011, 11, 5201–5207 CrossRef PubMed.
- N. Chatterjee, H. J. Eom and J. Choi, Biomaterials, 2014, 35, 1109–1127 CrossRef PubMed.
- L. Ou, B. Song, H. Liang, J. Liu, X. Feng, B. Deng, T. Sun and L. Shao, Part. Fibre Toxicol., 2016, 13, 57 CrossRef PubMed.
- M. Pelin, L. Fusco, V. Leon, C. Martin, A. Criado, S. Sosa, E. Vazquez, A. Tubaro and M. Prato, Sci. Rep., 2017, 7, 40572 CrossRef PubMed.
- J. L. Markley, R. Bruschweiler, A. S. Edison, H. R. Eghbalnia, R. Powers, D. Raftery and D. S. Wishart, Curr. Opin. Biotechnol, 2017, 43, 34–40 CrossRef PubMed.
- G. Jiao, X. Li, N. Zhang, J. Qiu, H. Xu and S. Liu, RSC Adv., 2014, 4, 44712–44717 RSC.
- C. Ren, X. Hu, X. Li and Q. Zhou, Biomaterials, 2016, 93, 83–94 CrossRef PubMed.
- S. Ouyang, X. Hu and Q. Zhou, ACS Appl. Mater. Interfaces, 2015, 7, 18104–18112 Search PubMed.
- X. Hu, Y. Gao and Z. Fang, Carbon, 2016, 109, 65–73 CrossRef.
- X. Hu, S. Ouyang, L. Mu, J. An and Q. Zhou, Environ. Sci. Technol., 2015, 49, 10825–10833 CrossRef PubMed.
- S. Gibbs, Skin Pharmacol. Physiol., 2009, 22, 103–113 CrossRef PubMed.
- A. A. Shvedova, V. Castranova, E. R. Kisin, D. Schwegler-Berry, A. R. Murray, V. Z. Gandelsman, A. Maynard and P. Baron, J. Toxicol. Environ. Health, Part A, 2003, 66, 1909–1926 CrossRef PubMed.
- C. Gong, G. Tao, L. Yang, J. Liu, H. He and Z. Zhuang, Mol. Biol. Rep., 2012, 39, 4915–4925 CrossRef PubMed.
- H. Liang, C. Jin, Y. Tang, F. Wang, C. Ma and Y. Yang, J. Appl. Toxicol., 2014, 34, 367–372 CrossRef PubMed.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467–4472 CrossRef PubMed.
- V. Leon, J. M. Gonzalez-Dominguez, J. L. Fierro, M. Prato and E. Vazquez, Nanoscale, 2016, 8, 14548–14555 RSC.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef PubMed.
- J. Russier, V. León, M. Orecchioni, E. Hirata, P. Virdis, C. Fozza, F. Sgarrella, G. Cuniberti, M. Prato, E. Vázquez, A. Bianco and L. G. Delogu, Angew. Chem., Int. Ed., 2017, 56, 3014–3019 CrossRef PubMed.
- H. Varela-Rizo, I. Rodriguez-Pastor, C. Merino and I. Martin-Gullon, Carbon, 2010, 48, 3640–3643 CrossRef.
- J. Carrola, V. Bastos, J. M. Ferreira de Oliveira, H. Oliveira, C. Santos, A. M. Gil and I. F. Duarte, Arch. Biochem. Biophys., 2016, 589, 53–61 CrossRef PubMed.
- C. Jin, Y. Liu, L. Sun, T. Chen, Y. Zhang, A. Zhao, X. Wang, M. Cristau, K. Wang and W. Jia, J. Appl. Toxicol., 2013, 33, 1442–1450 CrossRef PubMed.
- A. Sasidharan, S. Swaroop, P. Chandran, S. Nair and M. Koyakutty, Nanomedicine, 2016, 12, 1347–1355 CrossRef PubMed.
- M. H. Lim, I. C. Jeung, J. Jeong, S. J. Yoon, S. H. Lee, J. Park, Y. S. Kang, H. Lee, Y. J. Park, H. G. Lee, S. J. Lee, B. S. Han, N. W. Song, S. C. Lee, J. S. Kim, K. H. Bae and J. K. Min, Acta Biomater., 2016, 46, 191–203 CrossRef PubMed.
- H. Lu, R. A. Forbes and A. Verma, J. Biol. Chem., 2002, 277, 23111–23115 CrossRef PubMed.
- R. R. J. Collins, K. Patel, W. C. Putnam, P. Kapur and D. Rakheja, Clin. Chem., 2017, 63, 1812–1820 Search PubMed.
- M. Sciacovelli and C. Frezza, Free Radicals Biol. Med., 2016, 100, 175–181 CrossRef PubMed.
- A. B. Oliveira, F. R. de Moraes, N. M. Candido, I. Sampaio, A. S. Paula, A. de Vasconcellos, T. C. Silva, A. H. Miller, P. Rahal, J. G. Nery and M. F. Calmon, J. Proteome Res., 2016, 15, 4337–4348 CrossRef PubMed.
- M. Tokarska-Schlattner, R. F. Epand, F. Meiler, G. Zandomeneghi, D. Neumann, H. R. Widmer, B. H. Meier, R. M. Epand, V. Saks, T. Wallimann and U. Schlattner, PLoS One, 2012, 7, e43178 Search PubMed.
- S. M. Prescott, G. A. Zimmerman, D. M. Stafforini and T. M. McIntyre, Annu. Rev. Biochem., 2000, 69, 419–445 CrossRef PubMed.
- G. Y. Li, B. Fan and Y. C. Zheng, Biomed. Environ. Sci., 2010, 23, 371–377 CrossRef PubMed.
- B. Halliwell, Br. J. Exp. Pathol., 1989, 70, 737–757 Search PubMed.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef PubMed.
- J. Kim and R. P. Sharma, Toxicol. Sci., 2004, 81, 518–527 CrossRef PubMed.
- J. G. McCormack, A. P. Halestrap and R. M. Denton, Physiol. Rev., 1990, 70, 391–425 CrossRef PubMed.
- D. Bakowski and A. B. Parekh, Curr. Biol., 2007, 17, 1076–1081 CrossRef PubMed.
- E. J. Park, G. H. Lee, B. S. Han, B. S. Lee, S. Lee, M. H. Cho, J. H. Kim and D. W. Kim, Arch. Toxicol., 2015, 89, 1557–1568 CrossRef PubMed.
- Y. Chang, S. T. Yang, J. H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef PubMed.
- A. Sasidharan, L. S. Panchakarla, A. R. Sadanandan, A. Ashokan, P. Chandran, C. M. Girish, D. Menon, S. V. Nair, C. N. Rao and M. Koyakutty, Small, 2012, 8, 1251–1263 CrossRef PubMed.
- G. Lalwani, M. D'Agati, A. M. Khan and B. Sitharaman, Adv. Drug Delivery Rev., 2016, 105, 109–144 CrossRef PubMed.
- S. R. Martin, R. R. Biekofsky, M. A. Skinner, R. Guerrini, S. Salvadori, J. Feeney and P. M. Bayley, FEBS Lett., 2004, 577, 284–288 CrossRef PubMed.
- S. Muallem, Curr. Biol., 2007, 17, R549–R551 CrossRef PubMed.
- H. Afeseh Ngwa, A. Kanthasamy, Y. Gu, N. Fang, V. Anantharam and A. G. Kanthasamy, Toxicol. Appl. Pharmacol., 2011, 256, 227–240 CrossRef PubMed.
- F. T. Andon and B. Fadeel, Acc. Chem. Res., 2013, 46, 733–742 CrossRef PubMed.
- S. Hussain and S. Garantziotis, Autophagy, 2013, 9, 101–103 CrossRef PubMed.
- E. Mari, S. Mardente, E. Morgante, M. Tafani, E. Lococo, F. Fico, F. Valentini and A. Zicari, Int. J. Mol. Sci., 2016, 17, 1995 CrossRef PubMed.
- Y. Li, Y. Liu, Y. Fu, T. Wei, L. Le Guyader, G. Gao, R. S. Liu, Y. Z. Chang and C. Chen, Biomaterials, 2012, 33, 402–411 CrossRef PubMed.
- K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Nanoscale Res. Lett., 2011, 6, 8 Search PubMed.
- N. V. Vallabani, S. Mittal, R. K. Shukla, A. K. Pandey, S. R. Dhakate, R. Pasricha and A. Dhawan, J. Biomed. Nanotechnol., 2011, 7, 106–107 CrossRef PubMed.
- Y. Li, H. Yuan, A. von dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef PubMed.
- S. Y. Seong and P. Matzinger, Nat. Rev. Immunol., 2004, 4, 469–478 CrossRef PubMed.
- E. Hirata, E. Miyako, N. Hanagata, N. Ushijima, N. Sakaguchi, J. Russier, M. Yudasaka, S. Iijima, A. Bianco and A. Yokoyama, Nanoscale, 2016, 8, 14514–14522 RSC.
- S. Bengtson, K. B. Knudsen, Z. O. Kyjovska, T. Berthing, V. Skaug, M. Levin, I. K. Koponen, A. Shivayogimath, T. J. Booth, B. Alonso, A. Pesquera, A. Zurutuza, B. L. Thomsen, J. T. Troelsen, N. R. Jacobsen and U. Vogel, PLoS One, 2017, 12, e0178355 Search PubMed.
- G. Bellomo and F. Mirabelli, Ann. N. Y. Acad. Sci., 1992, 663, 97–109 CrossRef PubMed.
- S. Gurunathan, J. Han, J. H. Park and J. H. Kim, Int. J. Nanomed., 2014, 9, 1783–1797 CrossRef PubMed.
- J. Yuan, H. Gao and C. B. Ching, Toxicol. Lett., 2011, 207, 213–221 CrossRef PubMed.
- M. C. Matesanz, M. Vila, M. J. Feito, J. Linares, G. Goncalves, M. Vallet-Regi, P. A. Marques and M. T. Portoles, Biomaterials, 2013, 34, 1562–1569 CrossRef PubMed.
- M. Xu, J. Zhu, F. Wang, Y. Xiong, Y. Wu, Q. Wang, J. Weng, Z. Zhang, W. Chen and S. Liu, ACS Nano, 2016, 10, 3267–3281 CrossRef PubMed.
- B. Lu, T. Li, H. Zhao, X. Li, C. Gao, S. Zhang and E. Xie, Nanoscale, 2012, 4, 2978–2982 RSC.
- P. Thangavel, R. Kannan, B. Ramachandran, G. Moorthy, L. Suguna and V. Muthuvijayan, J. Colloid Interface Sci., 2018, 517, 251–264 CrossRef PubMed.
- R. Deepachitra, V. Ramnath and T. P. Sastry, RSC Adv., 2014, 4, 62717–62727 RSC.
- N. Mahmoudi, N. Eslahi, A. Mehdipour, M. Mohammadi, M. Akbari, A. Samadikuchaksaraei and A. Simchi, J. Mater. Sci. Mater. Med., 2017, 28, 73 CrossRef PubMed.
- Z. Li, H. Wang, B. Yang, Y. Sun and R. Huo, Mater. Sci. Eng., C, 2015, 57, 181–188 CrossRef PubMed.
- M. Duran-Prado, J. Frontinan, R. Santiago-Mora, J. R. Peinado, C. Parrado-Fernandez, M. V. Gomez-Almagro, M. Moreno, J. A. Lopez-Dominguez, J. M. Villalba and F. J. Alcain, PLoS One, 2014, 9, e109223 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr00897c |
| This journal is © The Royal Society of Chemistry 2018 |