
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monodisperse CoSn2 and FeSn2 nanocrystals as high-performance anode materials for lithium-ion batteries†
Shutao
Wang‡
 ab,
Meng
He‡
ab,
Marc
Walter
ab,
Frank
Krumeich
a,
Kostiantyn V.
Kravchyk
ab and
Maksym V.
Kovalenko
*ab
ab,
Meng
He‡
ab,
Marc
Walter
ab,
Frank
Krumeich
a,
Kostiantyn V.
Kravchyk
ab and
Maksym V.
Kovalenko
*ab
aETH Zürich, Department of Chemistry and Applied Biosciences, Vladimir Prelog Weg 1, 8093 Zürich, Switzerland
bEmpa-Swiss Federal Laboratories for Materials Science and Technology, Laboratory for thin films and photovoltaics, Überlandstrasse 129, 8600 Dübendorf, Switzerland. E-mail: mvkovalenko@ethz.ch
First published on 29th March 2018
Abstract
Nanostructured metal alloys show great promise as replacement materials for graphite anodes, and could improve the energy and power density of present-day lithium-ion batteries (LIBs). Herein, we report a facile colloidal synthesis of CoSn2 and FeSn2 nanocrystals (NCs) via the reaction of Co or Fe NCs and SnCl2 in oleylamine under reducing conditions. Among our pure Sn NCs and mixtures of Co or Fe and Sn NCs, monodisperse CoSn2 nanoalloys showed considerably improved cycling stability. In particular, CoSn2 delivered a stable average capacity of 650 mA h g−1 for 5000 cycles at a high current density of 1984 mA g−1, which is among the highest reported cycling stabilities for Sn-based anode materials.
Rechargeable lithium-ion batteries (LIBs) are the main electricity storage technology for portable electronic devices and electric vehicles owing to their high energy densities, long cycling lifetimes, and high power performance.1 Improvements to present-day LIBs might be achieved by partial or complete replacement of graphite – the major commercial anode material with a charge storage capacity of 372 mA h g−1 – by alloying or application of conversion-type materials with higher capacities.2–4 Tin (Sn) is a promising candidate for replacing graphite owing to its high charge storage capacity (992 mA h g−1 for Li22Sn5 formation).5–9 However, harnessing the storage ability of Sn is complicated by its poor capacity retention owing to the large volume changes (up to 300%) during lithiation/delithiation. This effect leads to mechanical disintegration of the electrodes and thus loss of electrical connectivity. These difficulties can be mitigated through nanostructuring, in particular, when the active material is embedded into an elastic and conductive network.9–20 For example, the cycling performance of 10 nm Sn nanocrystals (NCs), synthesized through colloidal methods, was demonstrated to be better than that of 150–50 nm Sn NCs.9 As an alternative to scaling the active material, alloying with other metals (such as M–Sn alloys) has also been demonstrated to be an effective strategy; where M is electroactive (Sb,15,21 Ge,22 Bi,23 Mg24) or inactive (Fe,25,26 Co,27,28–54 Ni,55 Cu56) with regard to Li. The superior performance of such alloys can be attributed to the additional metal acting as a matrix, buffering volume changes and preventing electrochemical aggregation of Sn particles.30,57,58 Herein, we attempt to tailor the electrochemical properties of Sn by combining nanostructuring, and alloying with an electrochemically non-active metal. In particular, we combine Sn with Co or Fe, which are two metals that do not form alloys with Li. On the basis of bulk phase diagrams and earlier studies on microcrystalline materials,29,30,32,59 we targeted the synthesis of CoSn2 and FeSn2 NCs. We then aimed to determine whether uniform CoSn2 and FeSn2 nanoalloys, composed of mixed elements with atomic homogeneity, exhibit advantageous electrochemical performances compared with those of elemental Sn NCs and their mixtures with Co (Fe) NCs (obtained by simple mechanical mixing). Although there have been many studies on Co–Sn27,28,36–38,41,45 and Fe–Sn25,26 nanoscopic alloys for LIBs, our comparative study requires Co, Fe, Sn, CoSn2, and FeSn2 to be obtained in the form of uniform colloidal NCs of very similar sizes. After accomplishing these syntheses, we find that both CoSn2 and FeSn2 NCs are indeed unlike the mixtures of their elemental NCs. Higher capacities and extended cyclabilities were achieved, highlighting the importance of the nanoscopic homogeneity of alloyed NCs.
CoSn2 NCs were synthesized via successive reduction of Co and Sn precursors. First, dried oleylamine was mixed with CoCl2, and further dried under vacuum followed by injection of a LiN(iPr)2 (lithium diisopropylamide, LDA) solution at 270 °C, and, 30 s later at 235 °C, injection of SnCl2 into the solution. In this reaction, the LDA served as a mild base for the partial deprotonation of oleylamine, thereby promoting the formation of highly reactive metal-oleylamido complexes.9,60 The latter species almost instantly decomposed, causing a burst of nucleation and NC formation. The reaction mixture was maintained at ca. 235 °C for an additional 4 h to form uniform Co–Sn alloy NCs.
Transmission electron microscopy (TEM) images and X-ray diffraction (XRD) patterns (Fig. 1b) confirmed the formation of uniform and highly crystalline tetragonal CoSn2 NCs (space group I4/mcm, a = b = 6.363 Å, c = 5.456 Å, JCPDS no. 25-0256) with sizes on the order of 10 nm. Crystalline FeSn2 NCs (JCPDS no. 25-0415, space group I4/mcm, a = b = 6.539 Å, c = 5.325 Å) of the same average size could be synthesized in a similar fashion by replacing CoCl2 with FeCl2 (Fig. 1c, for experimental details see ESI†). Notably, the size of the CoSn2 NCs could be tuned in the range of 5–13 nm with a narrow size-dispersion (Table S1, Fig. S1†). To compare the characteristics of the alloys and pure metals, we also synthesized elemental Sn and Co NCs, using previously published procedures (see examples on Fig. S2†).9,60 The galvanostatic cycling measurements for CoSn2 NCs are summarized in Fig. 2. The as-synthesized NCs were surface-functionalized with long-chain capping molecules (namely, oleylamine and oleic acid), rendering the NCs colloidally stable in non-polar solvents. For electrochemical testing, these electrically insulating molecular layers were removed by treatment with a 1 M solution of hydrazine in acetonitrile (ACN) for 2 h, a treatment typically used for colloidal quantum dots.61 Untreated NCs yielded no operational electrodes.
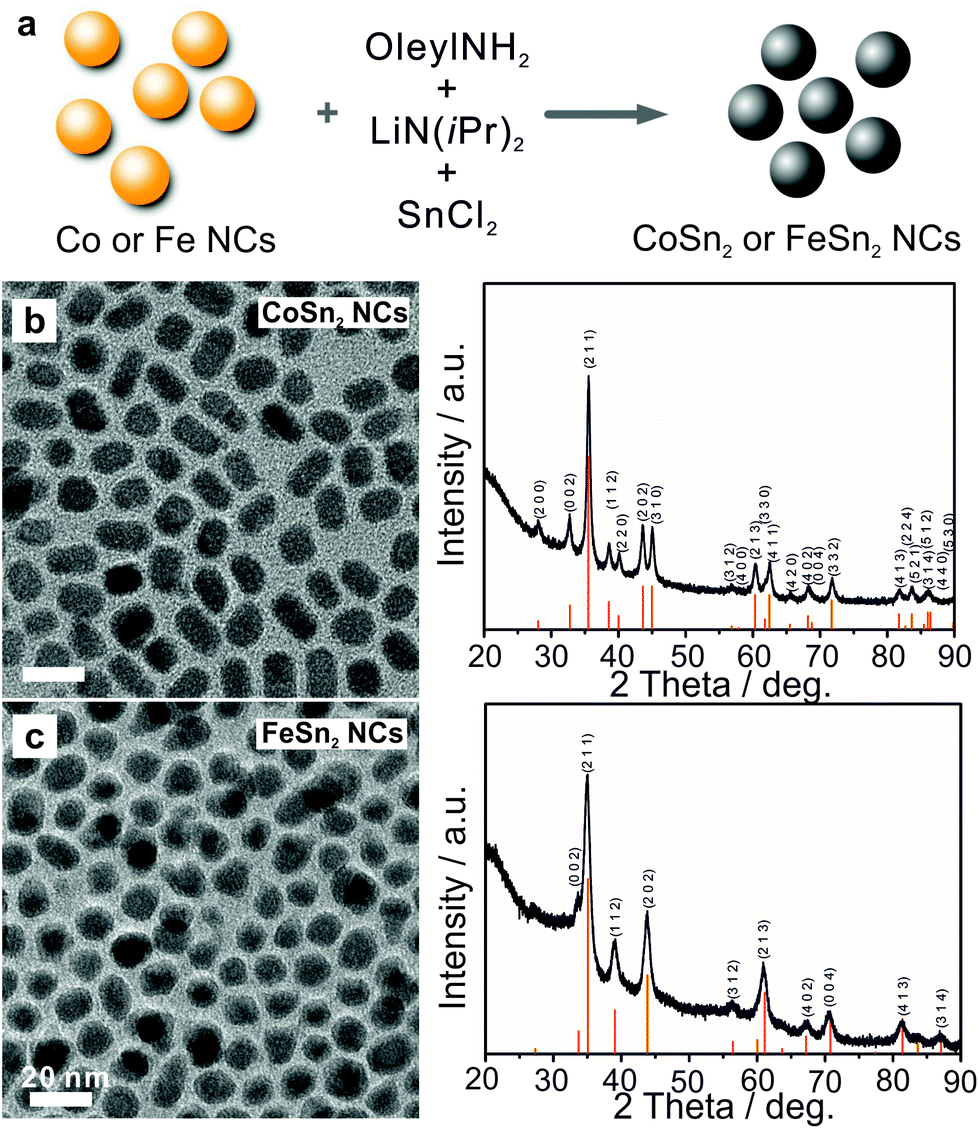 | ||
| Fig. 1 (a) Schematic illustration of one-pot synthesis, TEM and corresponding XRD patterns of (b) CoSn2 and (c) FeSn2 NCs. | ||
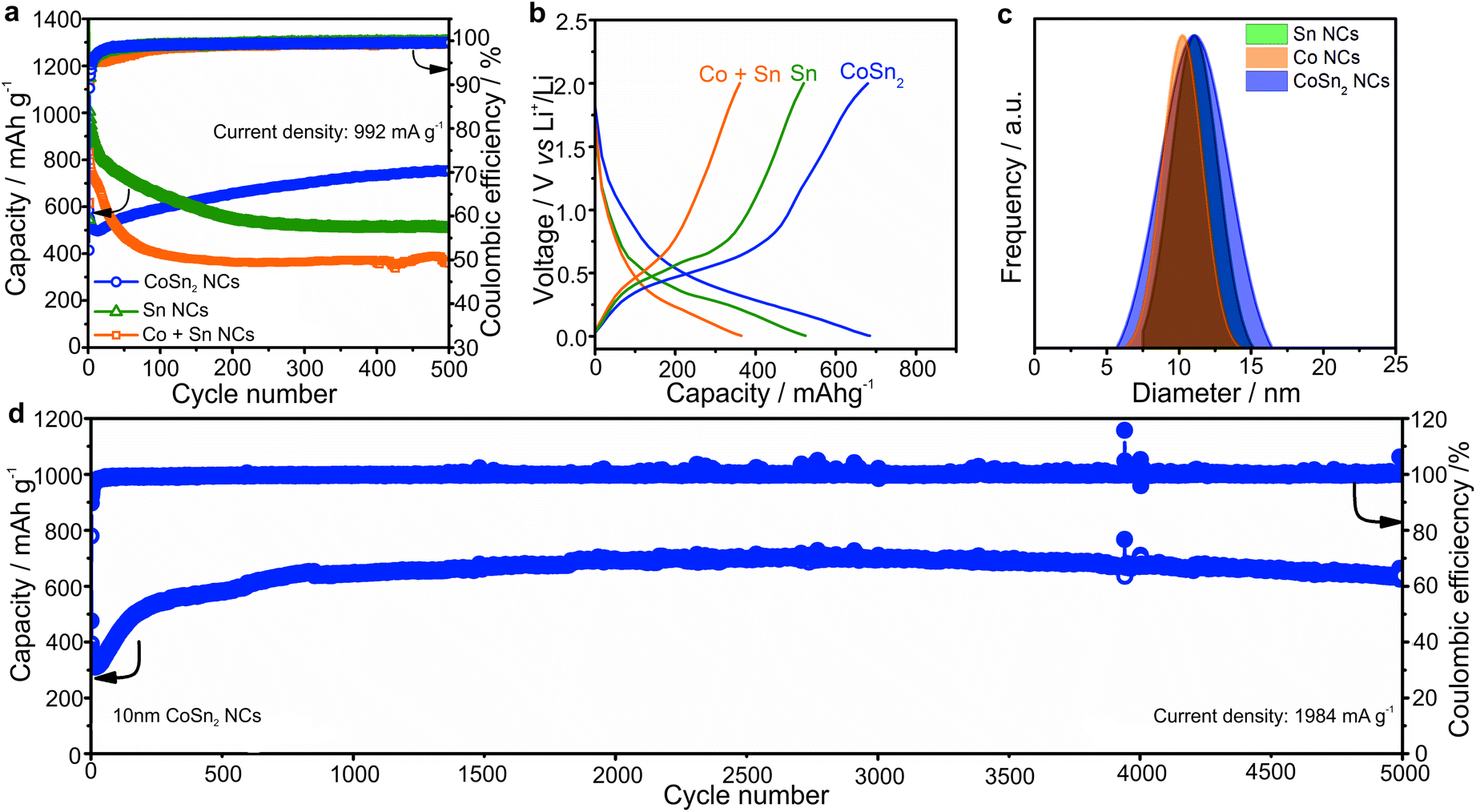 | ||
| Fig. 2 (a) Comparison of the cycling stability for CoSn2 NCs, Sn NCs and mixtures of Co NCs with two equivalents of Sn NCs, all of ca. 10–12 nm mean particle size, in Li-ion half-cells at a current density of 992 mA g−1. Initial coulombic efficiencies of CoSn2 NCs, Sn NCs and mixture of Co and Sn NCs were 52%, 59% and 63%. (b) Corresponding galvanostatic charge/discharge curves for the 250th cycle. (c) Size distributions of the tested NCs (obtained with Nano Measurer software). (d) Long-term cycling stability of ca. 10 nm CoSn2 NCs at current density of 1984 mA g−1. All cells were cycled at room temperature in the potential range of 0.005–2.000 V vs. Li+/Li. | ||
All electrodes contained 64 wt% of active material, 15 wt% carboxymethylcellulose (CMC) as binder and 21 wt% carbon black as a conductive additive. Electrochemical tests were performed in Li-ion half-cells with elemental lithium acting as both counter and reference electrode.
A comparison of the capacity retention of CoSn2, Sn, and mixtures of Co NCs with two equivalents of Sn NCs for cycling at a current density of 992 mA g−1 (rate of 1C for Sn) is shown in Fig. 2a (all NCs were ca. 10 nm large).
In agreement with the higher theoretical capacity of Sn (992 mA h g−1vs. 795 mA h g−1 for CoSn2), the Sn NCs delivered the highest capacities in the first cycles. However, upon prolonged cycling, the capacity of the Sn NCs, and mechanical mixtures of Sn and Co NCs, gradually decreased. Conversely, the capacities of the CoSn2 NCs increased to ∼750 mA h g−1. In all cases, the coulombic efficiency was initially low, but increased to more than 99%. However, the most distinct feature of the CoSn2 NCs was their high long-term cycling stability, even at high cycling rates (see Fig. 2d). In particular, at a current density of 1984 mA g−1, very stable capacities, on average 650 mA h g−1, were obtained up to 5000 cycles.
As follows from ex situ XRD measurements of CoSn2 NCs before and after electrochemical cycling (Fig. 3), CoSn2 phase can be fully recovered after delithiation reaction suggesting that the overall lithiation and delithiation mechanism of CoSn2 NCs can be described by the equation:
| CoSn2 + 8.8 Li+ + 8.8e− ↔ 2Li4.4Sn + Co. | (1) |
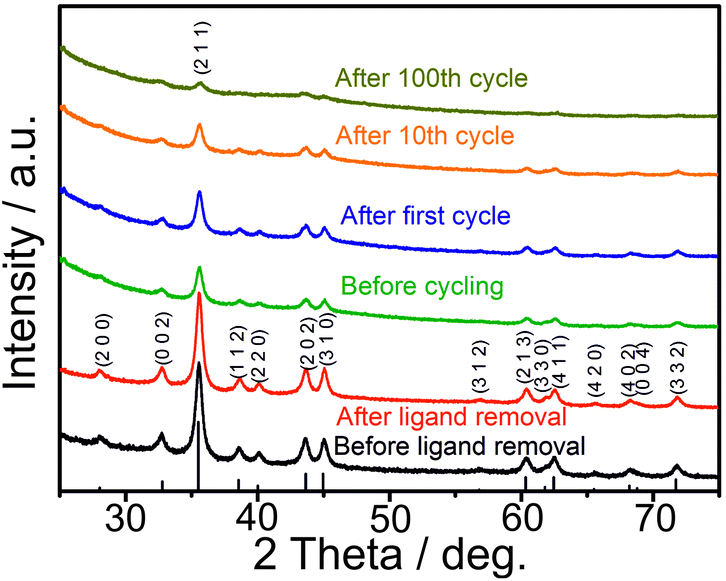 | ||
| Fig. 3 Ex situ XRD patterns of CoSn2 NCs before and after (charged CoSn2 NCs) electrochemical cycling. | ||
Ex situ XRD measurements of the CoSn2 NC electrodes show continuous amorphization of the material during cycling as indicated by the steady decrease of the signal intensity and broadening of the peaks. The anisotropic expansion of such amorphous phases can lead to lower mechanical stress during cycling, compared with that of crystalline elemental Sn NCs, which may explain the observed high cycling stability of CoSn2 NCs. In addition, the amorphization of CoSn2 NCs during cycling might facilitate the lithation/delithiation reaction and therefore lead to a higher utilization of the capacity as indicated by the increasing values during initial cycling. Notably, the ex situ XRD measurements indicate that Co-domains are not fully inactive after the first lithiation, but take a part in full CoSn2 recovery.
Similar electrochemical cycling behavior was obtained with FeSn2 NCs (Fig. 4). Namely, stable capacities in the range of 500–600 mA h g−1 were retained for at least 500 cycles, clearly outperforming mixtures of Sn and Fe NCs of a similar size. We attribute this effect to the closer contact of Fe and Sn domains due to the full recovery of FeSn2 phase after charge (see Fig. S3†), which can lead to a more effective buffering of the volume changes and prevent aggregation of Sn-domains.
 | ||
| Fig. 4 (a) Comparison of the cycling stability for FeSn2 NCs and mixtures of Sn and Fe NCs in Li-ion half-cells at a current density of 992 mA g−1 (see Fig. S4 and S5† for comparison of cycling stability and rate capability of FeSn2 and CoSn2 NCs) with (b) the corresponding size distributions (obtained with Nano Measurer software) of all tested NCs. Initial coulombic efficiencies of FeSn2 NCs and mixture of Fe and Sn NCs were 56% and 54%. All cells were cycled at room temperature in the potential range 0.005–2.0 V vs. Li+/Li with sodium alginate as binder. | ||
Conclusions
In conclusion, we have developed a novel synthesis of highly uniform colloidal CoSn2 and FeSn2 NCs, all with mean particle sizes in the range of 10–12 nm. Both CoSn2 and FeSn2 NCs showed electrochemical performance superior to that of Sn NCs and mixtures of individual Sn and Co or Fe NCs. In particular, CoSn2 NCs exhibited high reversible specific charge-storage capacities and cycling stability. Specifically, CoSn2 NCs delivered a stable average capacity of 650 mA h g−1 for 5000 cycles at a high density of 1984 mA g−1 (rate of 2C for Sn). This high-performance cycling stability can be attributed to the presence of Co domains, which acted as a matrix that buffers the volumetric changes occurring during lithiation/delithiation, and prevents the aggregation of Sn NCs and amorphization of the electrode material. Such high-rate performance illustrates the potential for use of nanostructured Co–Sn materials, or their mixtures with graphite, as alternative anode materials for high-power Li-ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by ETH Zürich (Grant No. ETH-56 12-2), the Competence Center for Energy and Mobility (CCEM, project SLIB), the Swiss Federal Commission for Technology and Innovation (CTI-Project No. 14698.2 PFIW-IW) and the CTI Swiss Competence Centers for Energy Research (SCCER, Heat and Electricity Storage).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
.
- Y. Yao, N. Xu, D. Guan, J. Li, Z. Zhuang, L. Zhou, C. Shi, X. Liu and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 39425–39431 CAS
- C. Tang, Y. Liu, C. Xu, J. Zhu, X. Wei, L. Zhou, L. He, W. Yang and L. Mai, Adv. Funct. Mater., 2018, 28, 1704561 CrossRef
- C. Tang, J. Zhu, X. Wei, L. He, K. Zhao, C. Xu, L. Zhou, B. Wang, J. Sheng and L. Mai, Energy Storage Mater., 2017, 7, 152–156 CrossRef
- W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 1160–1165 CrossRef CAS
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336–2340 CrossRef CAS
- M. Wachtler, J. O. Besenhard and M. Winter, J. Power Sources, 2001, 94, 189–193 CrossRef CAS
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470–474 CrossRef CAS PubMed
- K. Kravchyk, L. Protesescu, M. I. Bodnarchuk, F. Krumeich, M. Yarema, M. Walter, C. Guntlin and M. V. Kovalenko, J. Am. Chem. Soc., 2013, 135, 4199–4202 CrossRef CAS PubMed
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed
- C. K. Chan, R. N. Patel, M. J. O'Connell, B. A. Korgel and Y. Cui, ACS Nano, 2010, 4, 1443–1450 CrossRef CAS PubMed
- M. F. Oszajca, M. I. Bodnarchuk and M. V. Kovalenko, Chem. Mater., 2014, 26, 5422–5432 CrossRef CAS
- M. He, M. Walter, K. V. Kravchyk, R. Erni, R. Widmer and M. V. Kovalenko, Nanoscale, 2015, 7, 455–459 RSC
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255–1262 CrossRef CAS PubMed
- J. B. Cook, E. Detsi, Y. Liu, Y.-L. Liang, H.-S. Kim, X. Petrissans, B. Dunn and S. H. Tolbert, ACS Appl. Mater. Interfaces, 2017, 9, 293–303 CAS
- Y. Jin, Y. Tan, X. Hu, B. Zhu, Q. Zheng, Z. Zhang, G. Zhu, Q. Yu, Z. Jin and J. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 15388–15393 CAS
- H. Zhang, X. Huang, O. Noonan, L. Zhou and C. Yu, Adv. Funct. Mater., 2017, 27, 1606023 CrossRef
- M. Walter, M. I. Bodnarchuk, K. V. Kravchyk and M. V. Kovalenko, Chimia, 2015, 69, 724–728 CrossRef CAS PubMed
- M. Walter, S. Doswald and M. V. Kovalenko, J. Mater. Chem. A, 2016, 4, 7053–7059 CAS
- M. I. Bodnarchuk, K. V. Kravchyk, F. Krumeich, S. Wang and M. V. Kovalenko, ACS Nano, 2014, 8, 2360–2368 CrossRef CAS PubMed
- A. Trifonova, M. Wachtler, M. Winter and J. O. Besenhard, Ionics, 2002, 8, 321–328 CrossRef CAS
- D. Larcher, A. S. Prakash, J. Saint, M. Morcrette and J. M. Tarascon, Chem. Mater., 2004, 16, 5502–5511 CrossRef CAS
- Y. Ye, P. Wu, X. Zhang, T. G. Zhou, Y. W. Tang, Y. M. Zhou and T. H. Lu, RSC Adv., 2014, 4, 17401–17404 RSC
- C. Q. Zhang, J. P. Tu, X. H. Huang, Y. F. Yuan, S. F. Wang and F. Mao, J. Alloys Compd., 2008, 457, 81–85 CrossRef CAS
- X. Y. Fan, F. S. Ke, G. Z. Wei, L. Huang and S. G. Sun, J. Alloys Compd., 2009, 476, 70–73 CrossRef CAS
- B. Feng, J. Xie, G. S. Cao, T. J. Zhu and X. B. Zhao, New J. Chem., 2013, 37, 474–480 RSC
- A. D. W. Todd, R. E. Mar and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A597–A604 CrossRef CAS
- S.-I. Lee, S. Yoon, C.-M. Park, J.-M. Lee, H. Kim, D. Im, S.-G. Doo and H.-J. Sohn, Electrochim. Acta, 2008, 54, 364–369 CrossRef CAS
- P. P. Ferguson, A. D. W. Todd and J. R. Dahn, Electrochem. Commun., 2008, 10, 25–31 CrossRef CAS
- P. P. Ferguson, M. L. Martine, A. E. George and J. R. Dahn, J. Power Sources, 2009, 194, 794–800 CrossRef CAS
- Z. Chen, J. Qian, X. Ai, Y. Cao and H. Yang, J. Power Sources, 2009, 189, 730–732 CrossRef CAS
- X.-Y. Fan, F.-S. Ke, G.-Z. Wei, L. Huang and S.-G. Sun, J. Alloys Compd., 2009, 476, 70–73 CrossRef CAS
- L. Huang, J.-S. Cai, Y. He, F.-S. Ke and S.-G. Sun, Electrochem. Commun., 2009, 11, 950–953 CrossRef CAS
- F. Nacimiento, R. Alcántara and J. L. Tirado, J. Electrochem. Soc., 2010, 157, A666–A671 CrossRef CAS
- J. He, H. Zhao, J. Wang, J. Wang and J. Chen, J. Alloys Compd., 2010, 508, 629–635 CrossRef CAS
- X.-L. Wang, W.-Q. Han, J. Chen and J. Graetz, ACS Appl. Mater. Interfaces, 2010, 2, 1548–1551 CAS
- F. Nacimiento, P. Lavela, J. Tirado and J. Jiménez-Mateos, J. Solid State Electrochem., 2012, 16, 953–962 CrossRef CAS
- G. Ferrara, L. Damen, C. Arbizzani, R. Inguanta, S. Piazza, C. Sunseri and M. Mastragostino, J. Power Sources, 2011, 196, 1469–1473 CrossRef CAS
- Z. Du and S. Zhang, J. Phys. Chem. C, 2011, 115, 23603–23609 CAS
- L.-J. Xue, Y.-F. Xu, L. Huang, F.-S. Ke, Y. He, Y.-X. Wang, G.-Z. Wei, J.-T. Li and S.-G. Sun, Electrochim. Acta, 2011, 56, 5979–5987 CrossRef CAS
- M.-Y. Li, C.-L. Liu, M.-R. Shi and W.-S. Dong, Electrochim. Acta, 2011, 56, 3023–3028 CrossRef CAS
- D.-H. Nam, R.-H. Kim, C.-L. Lee and H.-S. Kwon, J. Electrochem. Soc., 2012, 159, A1822–A1826 CrossRef CAS
- G. Ferrara, C. Arbizzani, L. Damen, M. Guidotti, M. Lazzari, F. G. Vergottini, R. Inguanta, S. Piazza, C. Sunseri and M. Mastragostino, J. Power Sources, 2012, 211, 103–107 CrossRef CAS
- R. M. Gnanamuthu, Y. N. Jo and C. W. Lee, J. Alloys Compd., 2013, 564, 95–99 CrossRef CAS
- J. R. Gonzalez, F. Nacimiento, R. Alcantara, G. F. Ortiz and J. L. Tirado, CrystEngComm, 2013, 15, 9196–9202 RSC
- X. Liu, J. Xie, H. Zhao, P. Lv, K. Wang, Z. Feng and K. Świerczek, Solid State Ionics, 2015, 269, 86–92 CrossRef CAS
- B.-O. Jang, S.-H. Park and W.-J. Lee, J. Alloys Compd., 2013, 574, 325–330 CrossRef CAS
- N. Mahmood, C. Zhang, F. Liu, J. Zhu and Y. Hou, ACS Nano, 2013, 7, 10307–10318 CrossRef CAS PubMed
- N. Mahmood, J. Zhu, S. Rehman, Q. Li and Y. Hou, Nano Lett., 2015, 15, 755–765 CrossRef CAS PubMed
- G. O. Park, J. Yoon, J. K. Shon, Y. S. Choi, J. G. Won, S. B. Park, K. H. Kim, H. Kim, W.-S. Yoon and J. M. Kim, Adv. Funct. Mater., 2016, 26, 2800–2808 CrossRef CAS
- X. Shi, H. Song, A. Li, X. Chen, J. Zhou and Z. Ma, J. Mater. Chem. A, 2017, 5, 5873–5879 CAS
- J. Zhu, D. Wang, T. Liu and C. Guo, Electrochim. Acta, 2014, 125, 347–353 CrossRef CAS
- J. Liu, Y. R. Wen, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 6387–6392 CrossRef CAS PubMed
- W. X. Lei, Y. Pan, Y. C. Zhou, W. Zhou, M. L. Peng and Z. S. Ma, RSC Adv., 2014, 4, 3233–3237 RSC
- N. Tamura, Y. Kato, A. Mikami, M. Kamino, S. Matsuta and S. Fujitani, J. Electrochem. Soc., 2006, 153, A1626–A1632 CrossRef CAS
- C. M. Ionica-Bousquet, P. E. Lippens, L. Aldon, J. Olivier-Fourcade and J. C. Jumas, Chem. Mater., 2006, 18, 6442–6447 CrossRef CAS
- L. O. Vogt and C. Villevieille, J. Mater. Chem. A, 2017, 5, 3865–3874 CAS
- M. He, L. Protesescu, R. Caputo, F. Krumeich and M. V. Kovalenko, Chem. Mater., 2015, 27, 635–647 CrossRef CAS
- D. V. Talapin and C. B. Murray, Science, 2005, 310, 86–89 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr08261d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |