
White light emission in Bi3+/Mn2+ ion co-doped CsPbCl3 perovskite nanocrystals†

Abstract
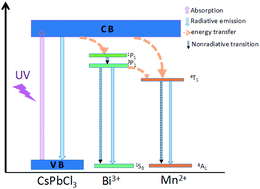
Colloidal perovskite nanocrystals (NCs), especially the fully inorganic cesium lead halide (CsPbX3, X = Cl, Br, I) NCs, have been considered as promising candidates for lighting and display applications due to their narrow band emission, tunable band gap and high photoluminescence quantum yields (QYs). However, owing to the anion exchange in the CsPbX3 NCs, stable multi-color and white light emissions are difficult to achieve, thus limiting their practical optoelectronic applications. In this work, dual ion Bi3+/Mn2+ codoped CsPbCl3 perovskite NCs were prepared through the hot injection method for the first time to the best of our knowledge. Through simply adjusting the doping ion concentrations, the codoped NCs exhibited tunable emissions spanning the wide range of correlated color temperature (CCT) from 19 000 K to 4250 K under UV excitation. This interesting spectroscopic behaviour benefits from efficient energy transfer from the perovskite NC host to the intrinsic energy levels of Bi3+ or Mn2+ doping ions. Finally, taking advantage of the cooperation between the excitonic transition of the CsPbCl3 perovskite NC host and the intrinsic emissions from Bi3+ and Mn2+ ions, white light emission with the Commission Internationale de l'Eclairage (CIE) color coordinates of (0.33, 0.29) was developed in the codoped CsPbCl3 NCs.
- This article is part of the themed collections: Nanoscale 10th Anniversary: Top Authors, Nanoscale Most Popular Articles and Editor’s Choice: Perovskite Nanomaterials and Devices
 Please wait while we load your content...
Please wait while we load your content...

