
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidized Co–Sn nanoparticles as long-lasting anode materials for lithium-ion batteries†
Marc
Walter
ab,
Simon
Doswald
ab,
Frank
Krumeich
 a,
Meng
He
ab,
Roland
Widmer
c,
Nicholas P.
Stadie‡
ab and
Maksym V.
Kovalenko
*ab
a,
Meng
He
ab,
Roland
Widmer
c,
Nicholas P.
Stadie‡
ab and
Maksym V.
Kovalenko
*ab
aDepartment of Chemistry and Applied Biosciences, ETH Zürich – Swiss Federal Institute of Technology Zürich, Vladimir Prelog Weg 1, 8093 Zürich, Switzerland. E-mail: mvkovalenko@ethz.ch
bEmpa-Swiss Federal Laboratories for Materials Science and Technology, Laboratory for thin films and photovoltaics, Überlandstrasse 129, 8600 Dübendorf, Switzerland
cEmpa-Swiss Federal Laboratories for Materials Science and Technology, Nanotech@surfaces Laboratory, Überlandstrasse 129, 8600 Dübendorf, Switzerland
First published on 1st February 2018
Abstract
Herein, we present the synthesis and systematic comparison of Sn- and Co–Sn-based nanoparticles (NPs) as anode materials for lithium-ion batteries. These nanomaterials were produced via inexpensive routes combining wet chemical synthesis and dry mechanochemical methods (ball milling). We demonstrate that oxidized, nearly amorphous CoSn2Ox NPs, in contrast to highly crystalline Sn and CoSn2 NPs, exhibit high cycling stability over 1500 cycles, retaining a capacity of 525 mA h g−1 (92% of the initial capacity) at a high current density of 1982 mA g−1. Moreover, when cycled in full-cell configuration with LiCoO2 as the cathode, such CoSn2Ox NPs deliver an average anodic capacity of 576 mA h g−1 over 100 cycles at a current of 500 mA g−1, with an average discharge voltage of 3.14 V.
Introduction
Despite an intensive research effort to develop new materials for rechargeable lithium-ion batteries (LIBs) over the past four decades, graphite remains the most common anode material in commercial devices. This is a surprising circumstance considering the fact that graphite possesses relatively low gravimetric and volumetric capacities (372 mA h g−1 and 820 mA h cm−3, respectively) compared to a large number of both alloying- (e.g., Si, Ge, and Sn) and conversion-type materials (e.g., Fe3O4, MoS2, and SnSb).1,2 The primary reasons for this are the rapid decline in capacity of many of these alternative anode materials due to massive volume changes during cycling (e.g., ΔV = 100–300%) causing mechanical disintegration of the electrodes, and/or due to irreversible reactions during charge and discharge. It has been demonstrated in a variety of case studies that these issues can be mitigated by using nanostructuring strategies in the synthesis of the active anode materials.3–26 Nevertheless, the commercialization of such high-capacity alloying- and conversion-type anodes has remained hampered for several reasons. In conversion-type anodes, a major fraction of the capacity is often obtained at potentials above 1.0 V vs. Li+/Li, reducing the voltage of the full-cell and thus the energy density of the corresponding battery. Secondly, the synthesis of nanostructured electrodes is often elaborate and therefore costly. Tin remains among the few materials which are realistic candidates to replace graphite in commercial LIBs, due to its high gravimetric and volumetric capacities (992 mA h g−1 and ∼7300 mA h cm−3), low delithiation potential (0.2–1 V), high electrical conductivity, low cost and natural abundance. In fact, anodes comprising amorphous Sn–Co–C nanocomposites have indeed been employed in commercial rechargeable batteries (e.g., Nexelion™, Sony Corp., Japan) since at least 2005. The success of this product has triggered further intensive research on Co–Sn-based anodes for LIBs.27–54 The superior cycling stability of many Co–Sn-based materials over elemental Sn has been attributed to two factors: (1) the effective buffering of volume changes occurring during the lithiation of Sn by the inactive Co matrix and (2) the prevention of aggregation of Sn particles during delithiation due to the preferred formation of intermetallic Co–Sn phases.28,55–58 Notable recent examples of nanostructured Co–Sn-based materials include, for instance, CoSnO3 NPs in a graphene network47 and Sn–Co NPs encapsulated in carbon spheres,59 which both exhibit stable capacities over ≥100 cycles in a wide potential range of up to 3.0 V vs. Li+/Li.In this report, we present a systematic comparison of well-defined Co–Sn nanoparticle (NP) based materials as anodes for LIBs. We introduce a simple synthetic procedure which can be used to prepare either amorphous Co NPs or crystalline Sn NPs, and subsequently demonstrate their conversion into crystalline non-oxidized CoSn2 NPs or oxidized, nearly amorphous CoSn2Ox NPs by simple mechanochemical methods. Half-cell electrochemical experiments were carried out to compare pure Sn NPs to both Co–Sn NP systems, indicating superior characteristics of the CoSn2Ox NPs. Finally, full-cell experiments were carried out on this most promising system using LiCoO2 as the cathode; stable cycling with anodic capacities of 576 mA h g−1 for 100 cycles could be demonstrated at a current of 500 mA g−1, with an average discharge voltage of 3.14 V. Note: In the work described herein, “CoSn2 NPs” is used to refer to the highly crystalline, non-oxidized CoSn2 material (seen by X-ray diffraction to be phase-pure), while the term “CoSn2Ox NPs” is used for oxidized and mostly amorphous NPs (with <5 nm crystal domain sizes).
Experimental
Colloidal synthesis of Sn and Co NPs
To prepare Sn NPs, a solution of NaBH4 (96 mmol, 98%, ACBR) in anhydrous 1-methyl-2-pyrrolidone (NMP, 85 mL, 99.5%, Fisher BioReagents) was heated to 60 °C under flowing N2. Then, a solution of SnCl2 (1 mmol, ≥99%, Alfa Aesar) in anhydrous NMP (3 mL) was injected and the reaction mixture was immediately cooled to room temperature using a water-ice bath. To prepare Co NPs, a NaBH4 solution in anhydrous NMP (32 mmol in 15 mL) was heated to 150 °C under flowing N2, followed by the injection of CoCl2 (8 mmol, ≥98%, Sigma-Aldrich) and immediate cooling to room temperature using a water-ice bath. The obtained Co NPs and Sn NPs were purified by washing once with dimethyl sulfoxide and then two times with water after separation from the supernatant by centrifugation. Finally, the reaction product was dried under vacuum at room temperature.Synthesis of crystalline CoSn2 NPs and oxidized CoSn2Ox NPs
For the preparation of Co–Sn based NPs, Sn NPs (1.4 mmol) were ball-milled for 4 hours with Co NPs (0.7 mmol) at a frequency of 30 s−1 using a Fritsch Pulverisette 23 mill (10 mL ZrO2 vessel, loaded with two 10 mm ZrO2 balls). In order to prepare crystalline CoSn2 NPs, the vessel was loaded and sealed under nitrogen atmosphere. Oxidized CoSn2Ox NPs were obtained when the starting Co and Sn NPs were loaded in air.Electrode fabrication, cell assembly and electrochemical measurements
The following battery components were used: carbon black (CB, Super C65, TIMCAL), carboxymethylcellulose (CMC, Grade: 2200, Daicel Fine Chem Ltd), fluoroethylene carbonate (FEC, Solvay, battery grade), 1 M LiPF6 in ethylene carbonate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethyl carbonate (EC:DMC, 1:1 by volume, Merck, battery grade), glass microfiber separator (GF/D, Whatman), and Cu foil (9 μm, MTI Corporation). For electrode preparation, aqueous slurries were prepared by mixing the respective NPs (64 wt%) with CB (21 wt%), CMC (15 wt%) and water using a planetary ball-mill (Fritsch Pulverisette 7 mill), and then coated onto Cu current collectors. The electrodes were then dried at 80 °C under vacuum for 12 h prior to assembling the cells. Material loadings were 0.5 mg cm−2 for half-cell and 1 mg cm−2 for full-cell experiments. Electrochemical measurements were conducted in air-tight coin-type cells assembled in an Ar-filled glove box (O2 < 0.1 ppm, H2O < 0.1 ppm) using elemental lithium as the counter electrode for half-cells and LiCoO2 on Al foil (MTI Corporation, ∼20 mg cm−2) as the cathode in full-cells. Glass microfiber was used as the separator in all cases. A standard solution of 1 M LiPF6 in EC:DMC with 3% FEC was used as the electrolyte. Galvanostatic cycling tests were performed at room temperature on a MPG2 multi-channel workstation (BioLogic). Anodic capacities were determined corresponding to the mass of the Co–Sn material in both half- and full-cell experiments.
:dimethyl carbonate (EC:DMC, 1:1 by volume, Merck, battery grade), glass microfiber separator (GF/D, Whatman), and Cu foil (9 μm, MTI Corporation). For electrode preparation, aqueous slurries were prepared by mixing the respective NPs (64 wt%) with CB (21 wt%), CMC (15 wt%) and water using a planetary ball-mill (Fritsch Pulverisette 7 mill), and then coated onto Cu current collectors. The electrodes were then dried at 80 °C under vacuum for 12 h prior to assembling the cells. Material loadings were 0.5 mg cm−2 for half-cell and 1 mg cm−2 for full-cell experiments. Electrochemical measurements were conducted in air-tight coin-type cells assembled in an Ar-filled glove box (O2 < 0.1 ppm, H2O < 0.1 ppm) using elemental lithium as the counter electrode for half-cells and LiCoO2 on Al foil (MTI Corporation, ∼20 mg cm−2) as the cathode in full-cells. Glass microfiber was used as the separator in all cases. A standard solution of 1 M LiPF6 in EC:DMC with 3% FEC was used as the electrolyte. Galvanostatic cycling tests were performed at room temperature on a MPG2 multi-channel workstation (BioLogic). Anodic capacities were determined corresponding to the mass of the Co–Sn material in both half- and full-cell experiments.
Materials characterization
Transmission electron microscopy (TEM) images were obtained with a Philips CM30 microscope operated at 300 kV, using carbon-coated Cu grids as substrates (Ted-Pella). Energy-dispersive X-ray spectroscopy (EDX) measurements were carried out using a NanoSEM 230 scanning electron microscope. Scanning transmission electron microscopy (STEM) images and EDX spectroscopy maps were collected on a FEI Talos F200X microscope operated at 200 kV. Powder X-ray diffraction (XRD) was measured on a STOE STADI P diffractometer (Cu-Kα1 irradiation, λ = 1.540598 Å). X-ray photoelectron spectroscopy (XPS) measurements were carried out in normal emission using a monochromatized Al Kα X-ray radiation source and a Scienta R3000 display analyzer.Results and discussion
Inexpensive routes to Co–Sn-based NPs were derived by combining the wet colloidal synthesis of unary metallic Co and Sn NPs with dry mechanochemical reactions between them (Fig. 1a), conducted in or without the presence of air.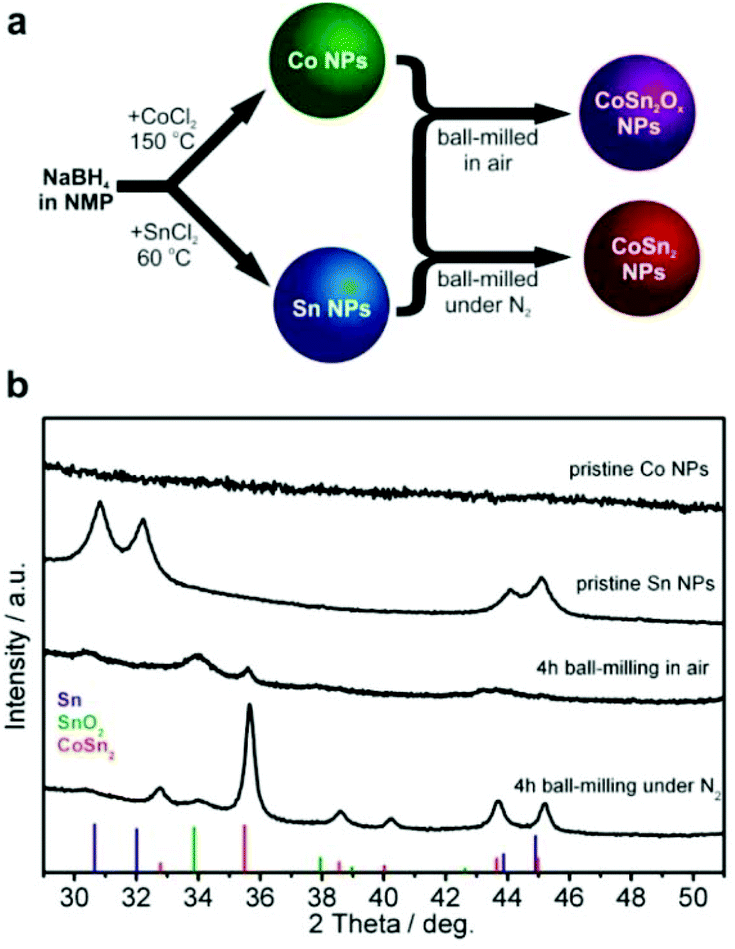 | ||
| Fig. 1 (a) Schematic of the preparation of Co–Sn NPs. (b) X-ray diffraction (XRD) patterns of colloidally synthesized Co and Sn NPs and mechanochemically prepared CoSn2Ox and crystalline CoSn2 NPs. Reference patterns: tetragonal SnO2, space group P42/mnm (136), a = 4.7391 Å, c = 3.1869 Å, ICDD PDF entry 00-077-0448; tetragonal CoSn2, space group I4/mcm (140), a = 6.363 Å, c = 5.456 Å, ICDD PDF entry 00-025-0256. | ||
Unary metallic NPs of Co (amorphous) and Sn (crystalline) were synthesized by an extension of the method that we previously reported for Sb NPs60 based on the borohydride reduction of metal chlorides in NMP. In contrast to the reduction reactions of SbCl360 and SnCl2 which exhibited suitable kinetics at 60 °C, a higher temperature of 150 °C was required for CoCl2. The resulting Co NPs were amorphous (Fig. 1b, see also Fig. S1 and S2a and b†), whereas the Sn NPs were highly crystalline in the form of β-Sn (Fig. 1b, indexed as phase-pure tetragonal Sn, space group I41/amd (141), a = 5.831 Å, c = 3.182 Å, ICDD PDF entry no.: 00-004-0673). High-resolution transmission electron microscopy (HR-TEM) image of Sn NP along with selected area electron diffraction (SAED) and d-spacing is shown on the Fig. S2c and d.† Mixtures of Co and Sn NPs (molar ratio 1:2) were then ball-milled either in air or under a nitrogen atmosphere with the intention of intimately mixing and alloying these materials and to study the effects of oxidation. Synthesis in air yielded a largely amorphous/poorly crystalline product, with broad and weak XRD reflections corresponding to SnO2 and CoSn2. For simplicity, the resulting product is denoted as CoSn2Ox NPs throughout this work. In agreement with the XRD results (Fig. 1b), d-spacing values deduced from HR-TEM measurements of CoSn2Ox NPs indicate the presence of small crystalline domains composed of CoSn2 and SnO2 (Fig. S3†). Elemental mapping with energy-dispersive X-ray spectroscopy in scanning TEM (EDX-STEM) studies confirms oxidation throughout the material, with a rather homogeneous distribution of Co, Sn, and O (Fig. S4†). As follows from XPS spectra (see Fig. S5†), both cobalt and tin are completely oxidized on the surface, most probably forming Co(OH)2 and SnO2. In addition, minor amount of metallic tin was observed in agreement with XRD results. In comparison with tin, cobalt is hardly detectable by XPS. Ball-milling under inert conditions, however, resulted in the formation of highly crystalline CoSn2 NPs (Fig. 1, see also Fig. S2e and f†), with only a minor content of SnO2. In all cases, the NPs obtained were small: 4–7 nm for Co NPs, 5–10 nm for Sn NPs and 6–20 nm for CoSn2Ox NPs and CoSn2 NPs (Fig. 2 and see also Fig. S6†). Importantly, as control experiments, attempts to synthesize Co–Sn nanomaterials with commercial microcrystalline powders of Co and Sn did not result in the formation of the CoSn2 phase (Fig. S7†).
 | ||
| Fig. 2 Transmission electron microscopy (TEM) images of colloidally synthesized Co and Sn NPs and mechanochemically prepared CoSn2Ox and CoSn2 NPs. | ||
Lithium-ion half-cell experiments
In order to compare the electrochemical properties of Sn, CoSn2Ox NPs and CoSn2 NPs, Li-ion half-cells were assembled. Electrodes were composed of NPs, carbon black (CB), and CMC binder (in a ratio of 64:21:15 by weight), and were tested against elemental lithium using 1 M LiPF6 in EC:DMC. FEC was added to the electrolyte to improve cycling stability by forming a stable SEI layer.61–65 All specific capacities and currents presented herein correspond to the combined mass of Sn and Co, excluding CB and CMC. Fig. 3 shows the capacity retention performance of CoSn2Ox NPs compared to Sn NPs over 1500 cycles at a high current density of 1984 mA g−1 in the potential range of 0.005–1.0 V vs. Li+/Li.
 | ||
| Fig. 3 (a) Cycling stability measurements of Sn NPs, CoSn2 NPs and CoSn2Ox NPs in lithium-ion half-cells at a current of 1984 mA g−1 within the potential range of 0.005–1.0 V. Galvanostatic charge/discharge curves for (b) Sn NPs, (c) CoSn2 NPs, and (d) CoSn2Ox NPs after a number of cycles corresponding to the half-cells in (a) (1st discharge curves with full capacity range are shown on Fig. S8a–c†). | ||
Since lithium is reversibly stored in all of the systems studied herein primarily via the Sn → Li4.4Sn conversion reaction (theoretical capacity: 992 mA h g−1), a current density of 992 mA g−1 was designated as the 1C rate.66 Assuming that Co does not contribute to any Li-ion capacity, the theoretical capacity of CoSn2 is 795 mA h g−1. During galvanostatic cycling of the half-cells, the upper cut-off potential was limited to 1.0 V in order to include only the region relevant to subsequent full-cell experiments. As can be seen in Fig. 3, at a high current density of 1984 mA g−1 (“2C”), Sn NPs, CoSn2Ox NPs, and CoSn2 NPs initially show similar capacities of 550–600 mA h g−1, with the highest values typically reached after 50–100 cycles. The increase of the capacities in the initial cycles might be attributed to restructuring processes in the electrode, which lower the resistivity and hence lead to a higher utilization of the capacity. Upon extended cycling, Sn NPs exhibit significant capacity fading within 400 cycles, while Co–Sn-based NPs show a much better retention of their capacity. Specifically, CoSn2 NPs retain a capacity of 462 mA h g−1 after 1500 cycles (corresponding to a reduction of 18%). CoSn2Ox NPs exhibit even better cyclability, retaining a capacity of 525 mA h g−1 (a reduction of 8%) after 1500 cycles. It should be noted that for all three systems, the average coulombic efficiency (CE) is 99.6% during cycling. First-cycle CE is, however, as low as ∼30%, which can be attributed to the formation of solid electrolyte interface (SEI) and the reduction of surface oxides to their corresponding metals with Li2O as a byproduct.
With such a high cycling stability, the CoSn2Ox NPs in this work compare favorably to the majority of recently investigated Sn-based Li-ion anode materials (see Table S1† for a detailed comparison), especially when considering that the cycling performed herein was restricted to potentials below 1.0 V vs. Li+/Li in order to obtain values that are truly relevant for full-cells.8,47–54,67–72 This superior cycling stability of NPs incorporating Co and Sn compared to pure Sn NCs might be attributable to two effects. Firstly, due to the fact that it does not form lithium alloys, Co can serve as an inactive matrix during cycling and therefore buffer the volume changes caused by the lithiation/delithiation of Sn. Secondly, the presence of Co can prevent Sn NCs from aggregating upon delithiation and therefore further improve the retention of the starting structure of the active anode material.28,55,56 Further, cyclic voltammetry (CV) measurements indicate a third possible effect: lithiation/delithiation occurs through fewer (most likely amorphous) phase transitions for Co–Sn-based NPs compared to Sn NCs (Fig. S9–11†). It is well known that pure Sn forms a multitude of intermediate crystalline phases during lithiation, leading to increased anisotropic strain in the particles during cycling and therefore lower cycling stability.
The difference between CoSn2 NPs and CoSn2Ox NPs in terms of cycling stability might be attributed to the fact that CoSn2Ox NPs are highly oxidized. The surface oxides are likely converted into Li2O during the first cycle(s), which is also known to inhibit the sintering of Sn domains during cycling.66 It should be noted that the average delithiation potentials for Sn NCs, CoSn2 NPs, and CoSn2Ox NPs are equally low, with an average value of ∼0.5 V vs. Li+/Li (Fig. 3b–d).
Apart from by the mechanical ball-milling of Co and Sn NPs, crystalline CoSn2 NPs can also be synthesized by the same colloidal method as used herein to synthesize the pure Co and Sn NPs by simultaneous injection of SnCl2/CoCl2 into a solution of NaBH4 in NMP. However, such a synthesis strategy suffers from an imbalance between the reactivities of the two metal chlorides. A high temperature of 150 °C is necessary to reduce Co2+, and the resulting CoSn2 NPs are larger than 20 nm, which is significantly larger than the Co–Sn based NPs prepared by ball-milling (see Fig. S12†). As a result of the larger size of colloidally synthesized CoSn2 NPs, much poorer capacity retention was observed: the capacity reduced to <200 mA h g−1 after 100 cycles (see Fig. S13†).
To evaluate the rate capability of the Co–Sn based NPs developed in this work, galvanostatic cycling was performed at current rates between 0.2C to 10C (Fig. 4, 1C = 992 mA g−1). Due to the similar particle sizes of these systems, and therefore similar reaction kinetics in all cases, comparable rate capabilities were observed. The only exception was at currents of 0.5C–2C where Sn NPs showed ∼50 mA h g−1 lower capacities compared to CoSn2 NPs. Even at rates as high as 10C, all three materials still retained a capacity of ∼350 mA h g−1. Interestingly, it was observed that at such high currents, Li-ion capacities increased during cycling, resulting in the same or even higher capacities during the stepwise decrease of the rate back to 0.2C. Especially for CoSn2Ox NPs, the slight difference in capacity compared to CoSn2 NPs initially observed at rates of 0.5C–2C becomes fully diminished during cycling. For comparison, graphite is known to exhibit much poorer rate capability,73,74 which was confirmed in this work by control experiments using graphite anodes (TIMCAL, KS6) tested under identical conditions (Fig. S14†).
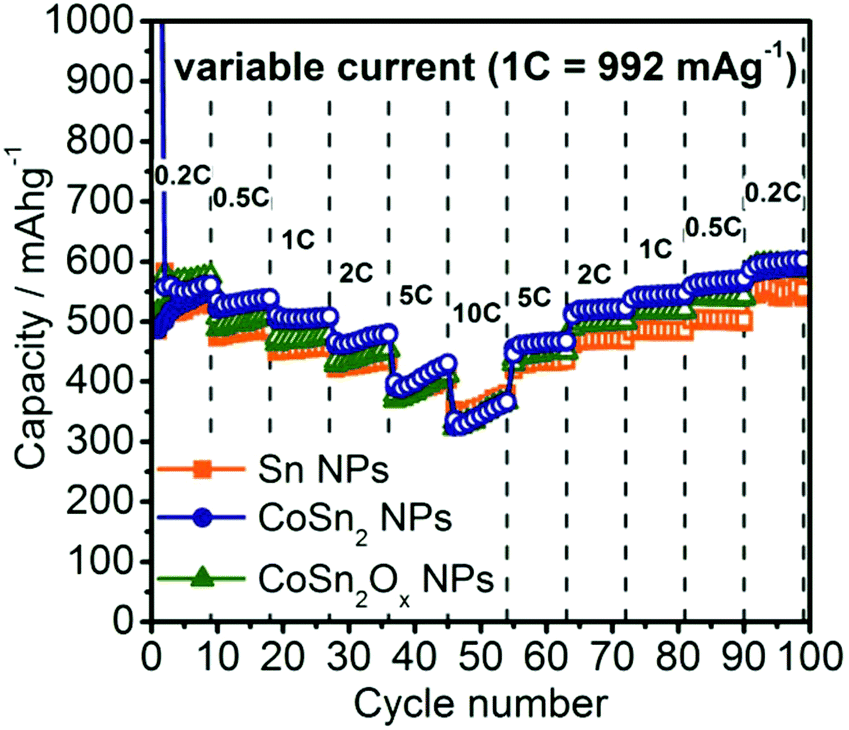 | ||
| Fig. 4 Rate capability tests for Sn, CoSn2, and CoSn2Ox NPs in lithium-ion half-cells within the potential range of 0.005–1.0 V. | ||
Lithium-ion full-cell experiments
In order to investigate the applicability of Co–Sn-based NPs as anode materials for commercial batteries, anode-limited full-cells using LiCoO2 as the cathode were assembled (Fig. 5). Based on the results of the half-cell experiments, CoSn2Ox NPs were selected for primary investigation in full-cell experiments. Herein, all specific capacities and currents correspond to the mass of CoSn2Ox NPs. Full-cells of CoSn2Ox/LiCoO2 were cycled galvanostatically at a current of 500 mA g−1 in the potential range of 2.0–3.9 V. Under such conditions, CoSn2Ox NPs exhibit an average capacity of 576 mA h g−1 upon extended cycling, similar to the values observed in half-cell experiments. Neglecting the excess of cathode material used in this work, the theoretical charge storage capacity of the cell is estimated as 112.6 mA h g−1 based on: Ccell = CanodeCcathode/(Canode + Ccathode). Taking the average discharge voltage of 3.14 V into account, the resulting average specific energy density for the CoSn2Ox/LiCoO2 cell is 353 W h kg−1 and, most importantly, it remains stable at this value for 100 cycles (see Fig. 5c). This specific energy density is comparable to that of the state-of-the-art graphite/LiCoO2 system (∼360 W h kg−1 based on 140 mA h g−1/3.7 V vs. Li+/Li for LiCoO2 and 372 mA h g−1/0.15 V vs. Li+/Li for graphite).75 However, considering the much higher density of bulk β-Sn (7.3 g cm−3) and Co (8.9 g cm−3) compared to graphite (2.2 g cm−3), CoSn2Ox NPs can theoretically exhibit improved volumetric energy densities by up to ∼40% (see also Table S2†). For a large variety of portable electronic devices, the importance of volumetric energy density is greater than that of gravimetric energy density.76 For comparison, the improvement of the volumetric energy density of the Sony Nexelion™ battery is 20% over identical cells using graphite anodes.77 | ||
| Fig. 5 Electrochemical performance of a lithium-ion full-cell battery with CoSn2Ox NPs as the anode and LiCoO2 as the cathode material. (a) Galvanostatic charge/discharge curves during the 50th cycle (see also Fig. S15†). (b) Capacity retention and coulombic efficiency. (c) Specific energy density and discharge voltage. Cells were cycled at a current of 500 mA g−1 in the potential range of 2.0–3.9 V. Specific capacities and currents correspond to the mass of CoSn2Ox NPs. | ||
Conclusions
Co and Sn NPs with diameters of ≤10 nm were synthesized via the simple reduction of their respective chlorides using NaBH4 in NMP, and were subsequently converted into intermetallic Co–Sn NPs by ball-milling. The resulting nanostructured materials can be seen as well-defined model systems, suited for investigating the effects of crystallinity and composition on electrochemical properties upon lithiation/delithiation cycling. Despite the fact that Sn and CoSn2 NPs show good cycling stability for several hundred cycles, CoSn2Ox NPs show the most outstanding retention of capacity, losing only 8% of their initial capacity over 1500 cycles at 1984 mA g−1. In addition, in lithium-ion full-cell experiments with LiCoO2 as the cathode material, CoSn2Ox NPs provide capacities of on average 576 mA h g−1 with an average discharge voltage of 3.14 V. This system, therefore, exhibits stable specific energy densities comparable to state-of-the-art LIBs based on graphite, and potentially much higher volumetric energy densities due to the higher density of CoSn2Ox. Considering that high rates were used in both half- and full-cell experiments, the CoSn2Ox NPs presented herein also offer a potential improvement in power density over cells assembled using conventional graphite anodes and also over Sony's commercialized Nexelion™ battery which has been designed specifically for low power (e.g., camcorder) devices.77Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by ETH Zürich (Grant No. ETH-56 12-2), the Competence Center for Energy and Mobility (CCEM, project SLIB), the Swiss Federal Commission for Technology and Innovation (CTI-Project No. 14698.2 PFIW-IW) and the CTI Swiss Competence Centers for Energy Research (SCCER, Heat and Electricity Storage). Electron microscopy was carried out at the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zurich and at the Empa Electron Microscopy Center.Notes and references
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed
.
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed
- C. K. Chan, R. N. Patel, M. J. O'Connell, B. A. Korgel and Y. Cui, ACS Nano, 2010, 4, 1443–1450 CrossRef CAS PubMed
- M. F. Oszajca, M. I. Bodnarchuk and M. V. Kovalenko, Chem. Mater., 2014, 26, 5422–5432 CrossRef CAS
- K. Kravchyk, L. Protesescu, M. I. Bodnarchuk, F. Krumeich, M. Yarema, M. Walter, C. Guntlin and M. V. Kovalenko, J. Am. Chem. Soc., 2013, 135, 4199–4202 CrossRef CAS PubMed
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H.-W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed
- A. Jahel, C. M. Ghimbeu, L. Monconduit and C. Vix-Guterl, Adv. Energy Mater., 2014, 4, 1400025 CrossRef
- J. Liu, Y. Wen, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 6387–6392 CrossRef CAS PubMed
- M. He, M. Walter, K. V. Kravchyk, R. Erni, R. Widmer and M. V. Kovalenko, Nanoscale, 2015, 7, 455–459 RSC
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255–1262 CrossRef CAS PubMed
- C. C. Li, Q. H. Li, L. B. Chen and T. H. Wang, J. Mater. Chem., 2011, 21, 11867–11872 RSC
- C. Guo, L. Wang, Y. Zhu, D. Wang, Q. Yang and Y. Qian, Nanoscale, 2015, 10123–10129 RSC
- G. Zeng, N. Shi, M. Hess, X. Chen, W. Cheng, T. Fan and M. Niederberger, ACS Nano, 2015, 9, 4227–4235 CrossRef CAS PubMed
- J. Zhang, K. Wang, Q. Xu, Y. Zhou, F. Cheng and S. Guo, ACS Nano, 2015, 9, 3369–3376 CrossRef CAS PubMed
- X.-Y. Yu, H. Hu, Y. Wang, H. Chen and X. W. Lou, Angew. Chem., Int. Ed., 2015, 7395–7398 CrossRef CAS PubMed
- J. Zhou, J. Qin, X. Zhang, C. Shi, E. Liu, J. Li, N. Zhao and C. He, ACS Nano, 2015, 9, 3837–3848 CrossRef CAS PubMed
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826–4830 CrossRef CAS PubMed
- J. S. Cho, Y. J. Hong and Y. C. Kang, ACS Nano, 2015, 9, 4026–4035 CrossRef CAS PubMed
- L. Wang, J. Liang, Y. Zhu, T. Mei, X. Zhang, Q. Yang and Y. Qian, Nanoscale, 2013, 5, 3627–3631 RSC
- J. B. Cook, E. Detsi, Y. Liu, Y.-L. Liang, H.-S. Kim, X. Petrissans, B. Dunn and S. H. Tolbert, ACS Appl. Mater. Interfaces, 2017, 9, 293–303 CAS
- Y. Jin, Y. Tan, X. Hu, B. Zhu, Q. Zheng, Z. Zhang, G. Zhu, Q. Yu, Z. Jin and J. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 15388–15393 CAS
- H. Zhang, X. Huang, O. Noonan, L. Zhou and C. Yu, Adv. Funct. Mater., 2017, 27, 1606023 CrossRef
- H. Ying and W.-Q. Han, Adv. Sci., 2017, 4, 1700298 CrossRef PubMed
- A. D. W. Todd, R. E. Mar and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A597–A604 CrossRef CAS
- S.-I. Lee, S. Yoon, C.-M. Park, J.-M. Lee, H. Kim, D. Im, S.-G. Doo and H.-J. Sohn, Electrochim. Acta, 2008, 54, 364–369 CrossRef CAS
- P. P. Ferguson, A. D. W. Todd and J. R. Dahn, Electrochem. Commun., 2008, 10, 25–31 CrossRef CAS
- P. P. Ferguson, M. L. Martine, A. E. George and J. R. Dahn, J. Power Sources, 2009, 194, 794–800 CrossRef CAS
- Z. Chen, J. Qian, X. Ai, Y. Cao and H. Yang, J. Power Sources, 2009, 189, 730–732 CrossRef CAS
- X.-Y. Fan, F.-S. Ke, G.-Z. Wei, L. Huang and S.-G. Sun, J. Alloys Compd., 2009, 476, 70–73 CrossRef CAS
- L. Huang, J.-S. Cai, Y. He, F.-S. Ke and S.-G. Sun, Electrochem. Commun., 2009, 11, 950–953 CrossRef CAS
- F. Nacimiento, R. Alcántara and J. L. Tirado, J. Electrochem. Soc., 2010, 157, A666–A671 CrossRef CAS
- J. He, H. Zhao, J. Wang, J. Wang and J. Chen, J. Alloys Compd., 2010, 508, 629–635 CrossRef CAS
- X.-L. Wang, W.-Q. Han, J. Chen and J. Graetz, ACS Appl. Mater. Interfaces, 2010, 2, 1548–1551 CAS
- F. Nacimiento, P. Lavela, J. Tirado and J. Jiménez-Mateos, J. Solid State Electrochem., 2012, 16, 953–962 CrossRef CAS
- G. Ferrara, L. Damen, C. Arbizzani, R. Inguanta, S. Piazza, C. Sunseri and M. Mastragostino, J. Power Sources, 2011, 196, 1469–1473 CrossRef CAS
- Z. Du and S. Zhang, J. Phys. Chem. C, 2011, 115, 23603–23609 CAS
- L.-J. Xue, Y.-F. Xu, L. Huang, F.-S. Ke, Y. He, Y.-X. Wang, G.-Z. Wei, J.-T. Li and S.-G. Sun, Electrochim. Acta, 2011, 56, 5979–5987 CrossRef CAS
- M.-Y. Li, C.-L. Liu, M.-R. Shi and W.-S. Dong, Electrochim. Acta, 2011, 56, 3023–3028 CrossRef CAS
- D.-H. Nam, R.-H. Kim, C.-L. Lee and H.-S. Kwon, J. Electrochem.
Soc., 2012, 159, A1822–A1826 CrossRef CAS
- G. Ferrara, C. Arbizzani, L. Damen, M. Guidotti, M. Lazzari, F. G. Vergottini, R. Inguanta, S. Piazza, C. Sunseri and M. Mastragostino, J. Power Sources, 2012, 211, 103–107 CrossRef CAS
- R. M. Gnanamuthu, Y. N. Jo and C. W. Lee, J. Alloys Compd., 2013, 564, 95–99 CrossRef CAS
- J. R. Gonzalez, F. Nacimiento, R. Alcantara, G. F. Ortiz and J. L. Tirado, CrystEngComm, 2013, 15, 9196–9202 RSC
- X. Liu, J. Xie, H. Zhao, P. Lv, K. Wang, Z. Feng and K. Świerczek, Solid State Ionics, 2015, 269, 86–92 CrossRef CAS
- C. Wu, J. Maier and Y. Yu, Adv. Funct. Mater., 2015, 25, 3488–3496 CrossRef CAS
- B.-O. Jang, S.-H. Park and W.-J. Lee, J. Alloys Compd., 2013, 574, 325–330 CrossRef CAS
- X. Li, X. He, Y. Xu, L. Huang, J. Li, S. Sun and J. Zhao, J. Mater. Chem. A, 2015, 3, 3794–3800 CAS
- N. Mahmood, C. Zhang, F. Liu, J. Zhu and Y. Hou, ACS Nano, 2013, 7, 10307–10318 CrossRef CAS PubMed
- N. Mahmood, J. Zhu, S. Rehman, Q. Li and Y. Hou, Nano Lett., 2015, 15, 755–765 CrossRef CAS PubMed
- G. O. Park, J. Yoon, J. K. Shon, Y. S. Choi, J. G. Won, S. B. Park, K. H. Kim, H. Kim, W.-S. Yoon and J. M. Kim, Adv. Funct. Mater., 2016, 26, 2800–2808 CrossRef CAS
- X. Shi, H. Song, A. Li, X. Chen, J. Zhou and Z. Ma, J. Mater. Chem. A, 2017, 5, 5873–5879 CAS
- J. Zhu, D. Wang, T. Liu and C. Guo, Electrochim. Acta, 2014, 125, 347–353 CrossRef CAS
- N. Tamura, Y. Kato, A. Mikami, M. Kamino, S. Matsuta and S. Fujitani, J. Electrochem. Soc., 2006, 153, A1626–A1632 CrossRef CAS
- C. M. Ionica-Bousquet, P. E. Lippens, L. Aldon, J. Olivier-Fourcade and J. C. Jumas, Chem. Mater., 2006, 18, 6442–6447 CrossRef CAS
- Y. Xu, M. Zhou and Y. Lei, Adv. Energy Mater., 2016, 6, 1502514 CrossRef
- W. Li, X. Sun and Y. Yu, Small Methods, 2017, 1, 1600037 CrossRef
- B. Zhang, X.-S. Li, C.-L. Liu, Z.-H. Liu and W.-S. Dong, RSC Adv., 2015, 5, 53586–53591 RSC
- M. Walter, R. Erni and M. V. Kovalenko, Sci. Rep., 2015, 5, 8418 CrossRef CAS PubMed
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2011, 28, 965–976 CrossRef PubMed
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CAS
- A. M. Chockla, K. C. Klavetter, C. B. Mullins and B. A. Korgel, Chem. Mater., 2012, 24, 3738–3745 CrossRef CAS
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed
- Z. Yang, A. A. Gewirth and L. Trahey, ACS Appl. Mater. Interfaces, 2015, 7, 6557–6566 CAS
- I. A. Courtney and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 2045–2052 CrossRef CAS
- X. Huang, S. Cui, J. Chang, P. B. Hallac, C. R. Fell, Y. Luo, B. Metz, J. Jiang, P. T. Hurley and J. Chen, Angew. Chem., Int. Ed., 2015, 54, 1490–1493 CrossRef CAS PubMed
- N. Zhang, Q. Zhao, X. Han, J. Yang and J. Chen, Nanoscale, 2014, 6, 2827–2832 RSC
- Z. Zhu, S. Wang, J. Du, Q. Jin, T. Zhang, F. Cheng and J. Chen, Nano Lett., 2014, 14, 153–157 CrossRef CAS PubMed
- J. Liu, Y. Wen, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 6387–6392 CrossRef CAS PubMed
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470–474 CrossRef CAS PubMed
- J. Hwang, S. H. Woo, J. Shim, C. Jo, K. T. Lee and J. Lee, ACS Nano, 2013, 7, 1036–1044 CrossRef CAS PubMed
- S. R. Sivakkumar, J. Y. Nerkar and A. G. Pandolfo, Electrochim. Acta, 2010, 55, 3330–3335 CrossRef CAS
- F. Ding, W. Xu, D. Choi, W. Wang, X. Li, M. H. Engelhard, X. Chen, Z. Yang and J.-G. Zhang, J. Mater. Chem., 2012, 22, 12745–12751 RSC
-
P. Gevorkian, Large-Scale Solar Power Systems: Construction and Economics, Cambridge University Press, 2012 Search PubMed
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed
-
D. Foster, J. Wolfenstine, J. Read and J. L. Allen, Performance of Sony's Alloy Based Li-Ion Battery, DTIC Document, 2008 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr07309g |
| ‡ Present address: Department of Chemistry and Biochemistry, Montana State University, 59717 Montana, USA. |
| This journal is © The Royal Society of Chemistry 2018 |