
Self-assembled polyoxometalate–dendrimer structures for selective photocatalysis†

Abstract
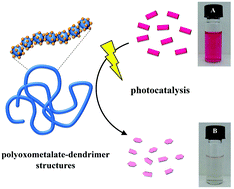
We present a novel, self-assembled nanostructure with selective photocatalytic activity formed from anionic polyoxometalate clusters and cationic dendrimers by electrostatic self-assembly. The association of the components in aqueous solution is driven by ionic interaction and steric factors yielding stable aggregates of a defined size with a coil-like structure. The assemblies show high potential for the application in solar-energy conversion systems due to their enhanced and substrate specific photocatalytic activity.
 Please wait while we load your content...
Please wait while we load your content...

