
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduced sintering of mass-selected Au clusters on SiO2 by alloying with Ti: an aberration-corrected STEM and computational study†
Yubiao
Niu
a,
Philomena
Schlexer
b,
Bela
Sebok
c,
Ib
Chorkendorff
 c,
Gianfranco
Pacchioni
b and
Richard E.
Palmer
*d
c,
Gianfranco
Pacchioni
b and
Richard E.
Palmer
*d
aNanoscale Physics Research Laboratory, School of Physics and Astronomy, University of Birmingham, B15 2TT, Birmingham, UK
bDipartimento di Scienza dei Materiali, Università di Milano-Bicocca, 20125 Milano, Italy
cDepartment of Physics, SurfCat, Technical University of Denmark, DK-2800, Kgs. Lyngby, Denmark
dCollege of Engineering, Swansea University, Bay Campus, Swansea, SA1 8EN, UK. E-mail: r.e.palmer@swansea.ac.uk
First published on 12th January 2018
Abstract
Au nanoparticles represent the most remarkable example of a size effect in heterogeneous catalysis. However, a major issue hindering the use of Au nanoparticles in technological applications is their rapid sintering. We explore the potential of stabilizing Au nanoclusters on SiO2 by alloying them with a reactive metal, Ti. Mass-selected Au/Ti clusters (400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 amu) and Au2057 clusters (405229 amu) were produced with a magnetron sputtering, gas condensation cluster beam source in conjunction with a lateral time-of-flight mass filter, deposited onto a silica support and characterised by XPS and LEIS. The sintering dynamics of mass-selected Au and Au/Ti alloy nanoclusters were investigated in real space and real time with atomic resolution aberration-corrected HAADF-STEM imaging, supported by model DFT calculations. A strong anchoring effect was revealed in the case of the Au/Ti clusters, because of a much increased local interaction with the support (by a factor 5 in the simulations), which strongly inhibits sintering, especially when the clusters are more than ∼0.60 nm apart. Heating the clusters at 100 °C for 1 h in a mixture of O2 and CO, to simulate CO oxidation conditions, led to some segregation in the Au/Ti clusters, but in line with the model computational investigation, Au atoms were still present on the surface. Thus size-selected, deposited nanoalloy Au/Ti clusters appear to be promising candidates for sustainable gold-based nanocatalysis.
000 amu) and Au2057 clusters (405229 amu) were produced with a magnetron sputtering, gas condensation cluster beam source in conjunction with a lateral time-of-flight mass filter, deposited onto a silica support and characterised by XPS and LEIS. The sintering dynamics of mass-selected Au and Au/Ti alloy nanoclusters were investigated in real space and real time with atomic resolution aberration-corrected HAADF-STEM imaging, supported by model DFT calculations. A strong anchoring effect was revealed in the case of the Au/Ti clusters, because of a much increased local interaction with the support (by a factor 5 in the simulations), which strongly inhibits sintering, especially when the clusters are more than ∼0.60 nm apart. Heating the clusters at 100 °C for 1 h in a mixture of O2 and CO, to simulate CO oxidation conditions, led to some segregation in the Au/Ti clusters, but in line with the model computational investigation, Au atoms were still present on the surface. Thus size-selected, deposited nanoalloy Au/Ti clusters appear to be promising candidates for sustainable gold-based nanocatalysis.
1. Introduction
Gold (Au) is considered to be the noblest of all metals, long regarded as chemically rather inert1 and thus (more or less) catalytically inactive. Nevertheless a report from as long ago as 1906 states that a hot Au mesh can facilitate the combination of O2 and H2,2 while a paper from 1925 indicates that Au is capable of catalysing the oxidation of CO to CO2.3 But since Au is fairly expensive and the reported reaction rates were lower compared with other metals, Au was not considered as a viable catalyst until Sennewald et al.4 found that a Pd–Au catalyst was active for the oxidative acetoxylation of ethylene to vinyl acetate in 1965 and then Bond and Sermon5 demonstrated the activity of supported Au particles with diameters of 2–100 nm for the hydrogenation of 1-pentene at 100 °C in 1973. Later, in the 1980s, Haruta et al.6,7 discovered that Au nanoparticles, with diameters of 3–5 nm hold up on reducible supports, were exceptionally active for CO oxidation even at −70 °C, and Hutchings8 demonstrated the catalytic activity of Au3+ in the hydrochlorination of acetylene to vinyl chloride. Since then, Au has attracted growing attention as a catalyst and become the preeminent example of a size effect in heterogeneous catalysis.9–11As CO oxidation is one of the simplest reactions which Au can catalyse, and CO is frequently used as a probe molecule in surface science, CO oxidation on TiO2 supported Au nanoparticles has become a prototypical model system in Au catalysis,12–15 and numerous studies have been reported regarding the reaction mechanisms,16–18 active sites,11,19–21 and active species.22,23 Looking at the experimentally measured catalytic activities of Au nanoparticles on different supports in the CO oxidation reaction, two main conclusions can be drawn. The Au nanoparticles are not active above a size of approx. 5 nm and on some oxide supports, such as SiO2, they are not active at all, regardless of size.11
Although supported Au nanoparticles have genuine potential in technological applications, a major issue hindering their implementation is rapid sintering. It has been shown that Au atoms are mobile and can migrate to form 3D islands on a TiO2 surface even at 150–160 K.24,25 Also on silica, Au particles tend to sinter quickly, unless defects are present on the silica surface to which the Au particles bind more strongly.26 However, the defects have to be present on the surface before the cluster deposition and are unlikely to be formed in situ.27 According to the work of Yang et al.,28 Au clusters on TiO2 sinter by Ostwald ripening between 300 K and 410 K and the sintering is accelerated by the presence of a mixture of CO and O2. The sintering modes of mass-selected Au nanoclusters deposited on amorphous carbon were studied by Hu et al. They found that the sintering process is size-dependent; Au561±13 and Au923±20 clusters exhibit Ostwald ripening, whereas Au2057±45 ripens through cluster diffusion and coalescence (Smoluchowski ripening).29
The stabilisation of supported nanoparticles against sintering has attracted significant research effort.30,31 Two pathways are usually described: alloying the particles, or encapsulating them within an oxide or organic shell. The latter may hinder the use of nanoparticles in catalytic applications because of surface blocking, but alloying may provide a route to nanoparticles which are both stable and active. In particular, the investigations of mass-selected alloy nanoparticles synthesised in the gas phase have already shown the unique properties of such materials. Examples include increased activity in the oxygen reduction reaction (ORR) for case of PtxY and PtxGd nanoparticles as a result of strain32,33 and the dynamic behavior of CuZn nanoparticles under methanol synthesis reaction conditions.34 With regards to sintering, it has been shown that alloying Au with Ir, Cu or Ag increases the stability of chemically synthesized nanoparticles on oxide supports.35–41 However, studies of the effects of alloying on the stability of mass-selected nanoparticles against sintering have been lacking to date.
Here we show that the sintering rate of mass-selected Au/Ti alloy nanoclusters, synthesised in a magnetron sputtering, gas condensation cluster beam source42,43 is much lower than that of mass-selected pure Au nanoclusters on silica. The composition of the clusters was characterised by X-ray Photoelectron Spectroscopy (XPS) and Low Energy Ion Scattering (LEIS) while aberration-corrected Scanning Transmission Electron Microscopy (STEM) was used to investigate the sintering process via direct real space imaging with atomic resolution. Compementary ab initio calculations confirm stronger binding between alloyed Au/Ti clusters and the SiO2 surface compared with pure Au clusters.
2. Experimental and computational details
2.1 Experimental details
Au2057 (405229 amu) and Au/Ti (400000 amu) nanoclusters were produced with a DC magnetron sputtering, gas condensation cluster beam source43 from Au (PI-KEM, 99.99%) and Au/Ti (PI-KEM, 50/50 at%, 99.99%) targets, respectively. The clusters with a specific mass were selected by a lateral time-of-flight mass filter42 before deposition onto the support with a kinetic energy of approx. 0.5 eV per atom. The mass-selected clusters were deposited onto thin silica films suspended on copper TEM grids (EMS, USA) and onto solid Si substrates terminated with a 50 nm thermally grown SiO2 layer. These substrates were equipped with heater elements and a Pt resistance temperature detector (RTD) evaporated on their back side. The loading of all the samples was approx. 10% (projected surface area coverage), i.e., approx. 10% of the surface covered by clusters. Before deposition the Si/SiO2 substrates were sputtered by an Ar+ beam from the cluster source, with a beam current of 1 nA for 500 s (at 1000 eV Ar+) in order to clean the surface and create defects to act as anchor points for the clusters. After deposition on the TEM supports, the clusters were imaged with a 200 kV aberration-corrected JEOL 2100F Scanning Transmission Electron Microscopy (STEM) in high-angle annular dark field (HAADF) mode after transfer under atmospheric conditions. HAADF images were acquired with inner and outer detector angles of 62 and 164 mrad (camera length 10 cm), and probe convergence angle of 19 mrad. The Si/SiO2 samples were characterized with XPS (non-monochromatized Mg Kα line from a SPECS XR50 X-ray gun, using an OMICRON NanoSAM 7 channel energy analyzer) and LEIS (1 KeV He+ from an Omicron ISE100 ion gun using the same energy analyzer), again after transfer under atmospheric conditions. The silicon supports were heated in 1 bar of O2:CO = 4:1 to approx. 370 K (100 °C) in order to simulate the effect of CO oxidation conditions. We estimate the error in temperature here to be around ±15 K. After heating the supports were characterized again with XPS and LEIS to investigate the surface composition of the clusters and the effect of the heating.
2.2 Computational details
We performed spin polarized density functional theory (DFT) calculations with the Vienna Ab Initio Simulation Package (VASP 5.2)44–47 using the Perdew, Burke and Ernzerhof (PBE) exchange–correlation functional.48,49 Electron-ion interactions were described via the projector augmented wave (PAW) method.50,51 O(2s, 2p), Si(3s, 3p), Au(5d, 6s) and Ti(3d, 4s, 4p) states were treated explicitly. For electronic relaxations, we used the blocked Davidson iteration scheme.52,53 Full geometric structure optimizations were performed, allowing all ions to relax until forces were smaller than |0.01| eV Å−1. The lattice parameters were kept fixed during structure optimizations.To represent the silica surface, we used a periodic fully hydroxylated α-quartz (001) surface, as described in our previous study.54 A (3 × 3) surface super cell was used with lattice parameters of a = b = 15.11 Å and γ = 120° and with a slab thickness of 9 layers of [SiO4] tetrahedra. The slabs were separated by more than 20 Å of vacuum to ensure enough space for the metal clusters. Wave functions were expanded in a plane wave basis up to a kinetic energy of 400 eV. A Γ-centred K-point grid in the Monkhorst–Pack scheme55 was used, which was set to the Γ-point.
As dispersion forces can be important for the cluster-support interaction, we applied the semi-empirical dispersion correction proposed by Grimme,56 known as the DFT-D2 approach. As the DFT-D2 approach is assumed to overestimate the dispersion in oxides, we changed the C6 and R0 parameters as suggested by Tosoni and Sauer.57 The resulting approach is called DFT-D2′. Atomic charges q were determined via the Bader decomposition scheme.58–60 The adsorption energies of the clusters, EADS, are defined in eqn (1), where E(AuxTiy) are the metal clusters in the gas-phase and S is the support.
| EADS(AuxTiy/S) = E(AuxTiy/S) − E(AuxTiy) − E(S) | (1) |
3. Results and discussion
3.1 XPS and LEIS of the nanoclusters
XPS was used to provide information on the composition of the deposited clusters. Typical XPS spectra are shown in Fig. 1 for both the Au2057 and Au/Ti nanoclusters deposited on Si/SiO2 supports, as received after transport under atmospheric conditions, and also after heating in a mixture of O2 and CO in order to simulate the effect of CO oxidation conditions. In all samples the Si in SiO2 binding state, originating from the substrate, can be detected. Traces of carbonaceous contaminants from atmospheric exposure are also visible in the spectra. In case of the Au2057 nanocluster sample, no significant changes are visible in the XPS spectra taken before and after heating in O2/CO (Fig. 1A). In the case of the Au/Ti nanoclusters, the relative intensity of the Au signal compared with the Si signal (normalized peak size) in the spectrum decreases after heating. This is consistent with a segregation process in which more Ti is drawn to the cluster surface, which is underlined by the change of the Au:Ti ratio determined from the spectra. The as received clusters show a surface Au:Ti ratio of 60:40, while after heating the ratio is approx. 40:60. Some Ti is present on the surface in the form of TiO2 regardless of the heating (see detailed spectra in ESI, Fig. S1†).
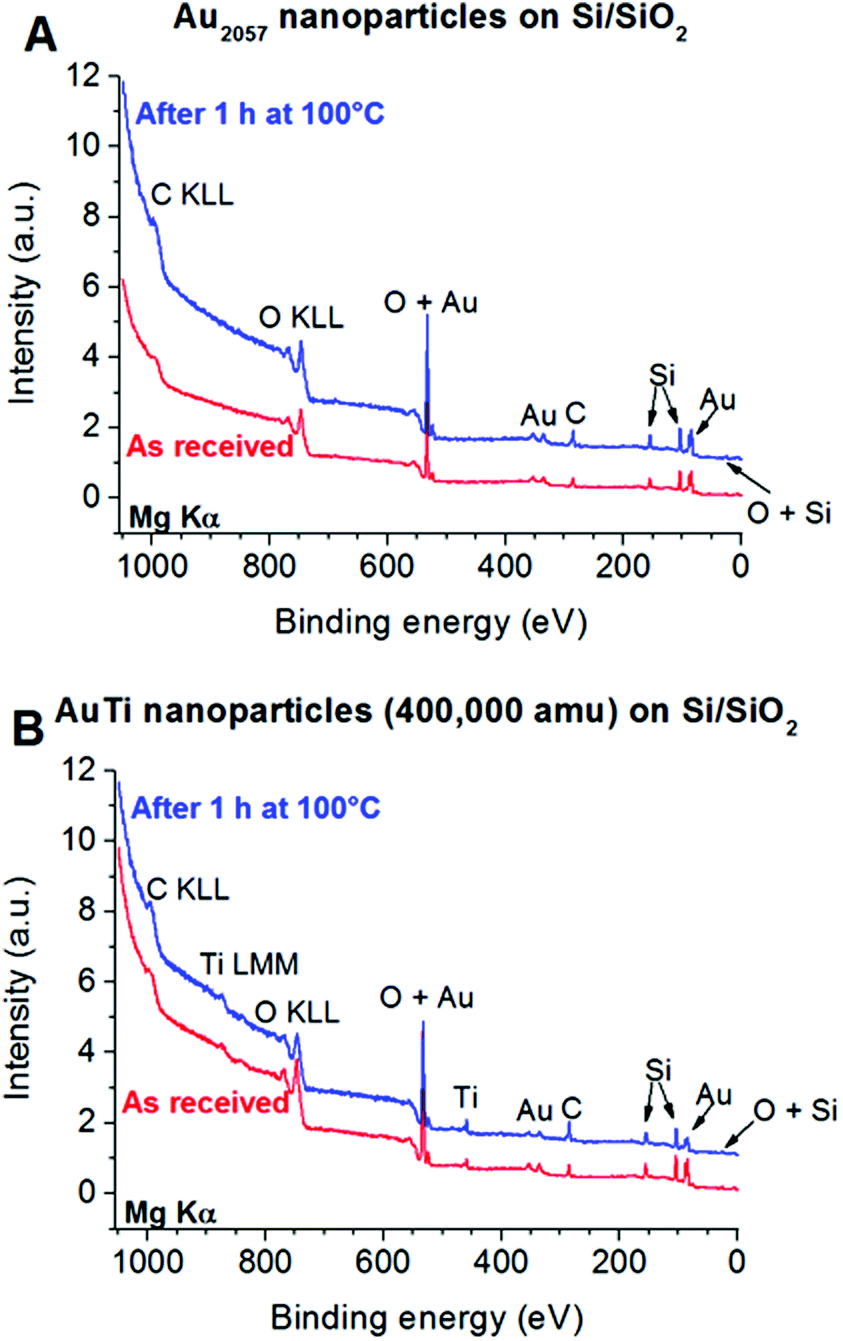 | ||
| Fig. 1 XPS spectra of Au2057 (A) and Au/Ti (B) nanoclusters deposited on Si/SiO2 slabs before and after 100 °C for 1 h in 1 bar of O2:CO = 4:1 mixture. Spectra were calibrated for Au 4f7/2 line to appear at binding energy of 84 eV and were normalized with the area of the Si 2s peak. The plots were offset in y direction for better visibility. | ||
In order to further investigate the surface of the samples, LEIS spectra were recorded (Fig. 2) characterizing the outermost layer of the sample surface. In both Au and Au/Ti cluster samples, O, Si and Au are detectable on the surface before heating, as expected for Au-containing clusters deposited on a SiO2 surface. In case of the sample with Au2057 clusters (Fig. 2A), small additional contaminant(s) can be detected having a mass of approx. 39 amu (Cl, K or Na), which could originate from the handling of the sample. No such contaminant could be detected in case of the sample with Au/Ti clusters, since Ti is clearly visible in the same region (Fig. 2B). After heating the samples, the Si and O peaks disappear from the spectra. As XPS confirms the presence of SiO2, this disappearance is most probably the effect of carbonaceous contaminants on the surface completely covering the support. Nevertheless, in both samples Au is still clearly visible at the outermost surface.
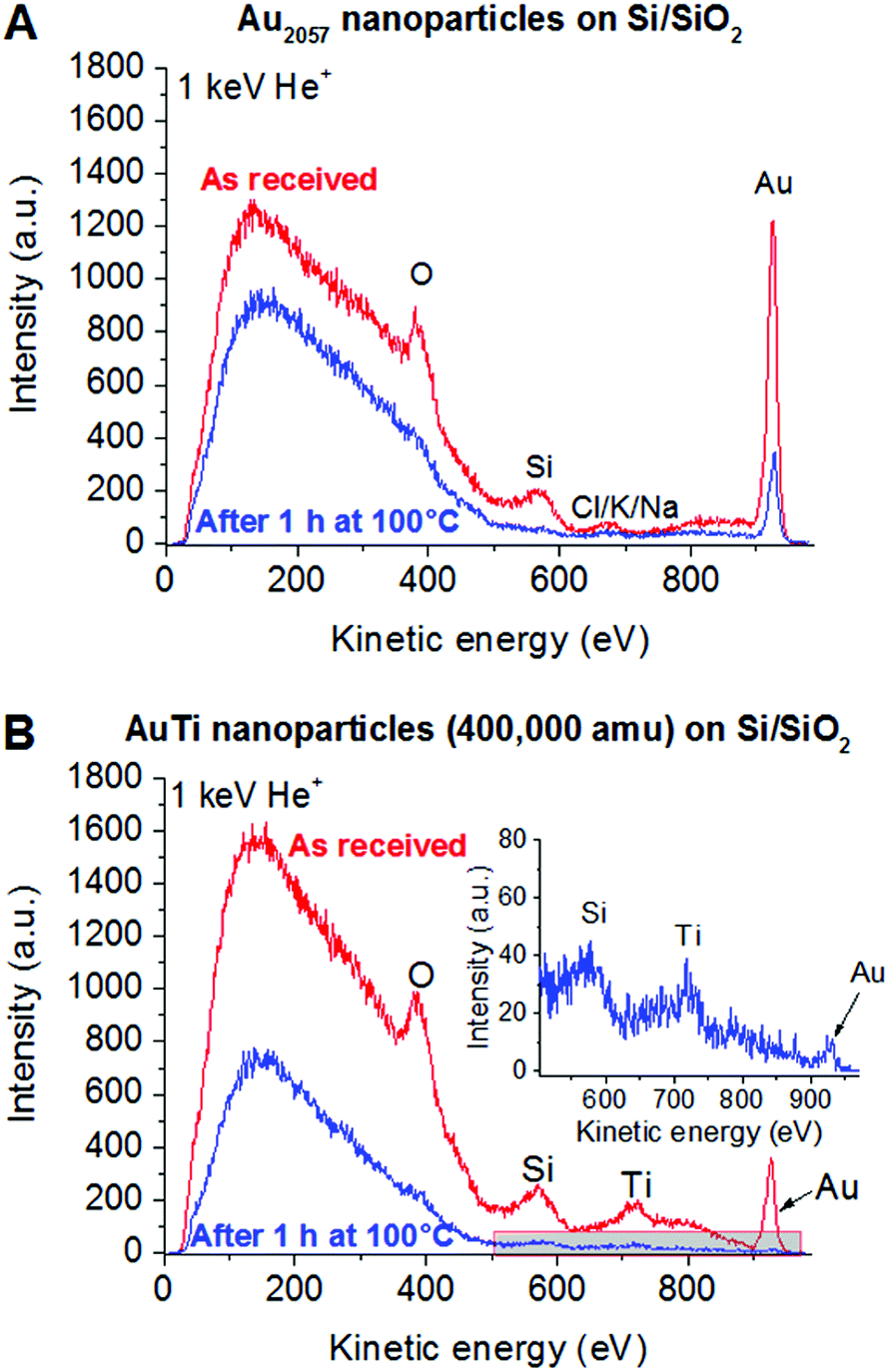 | ||
| Fig. 2 LEIS spectra of Au2057 (A) and Au/Ti (B) nanoclusters deposited on Si/SiO2 slabs before and after (inset) 100 °C for 1 h in 1 bar of O2:CO = 4:1 mixture taken with 1 keV He+ ions. | ||
Based on the XPS and LEIS measurements, a segregation process upon heating in O2/CO is changing the surface composition of the Au/Ti nanoclusters, but in line with the computational investigation (see below) Au atoms are still present on the surface, thus opening up the possibility of Au catalysis using the Au/Ti clusters under realistic reaction conditions.
3.2 STEM study of the sintering process
The mean diameters of Au2057 and Au/Ti (400k amu) clusters deposited onto thin silica film were measured, based on projected surface area, and found to be 4.07 nm and 3.94 nm, respectively. The shape of Au2057 clusters can be obtained from the relationship between the cluster diameter (D) and the number of Au atoms (N).61 The shape of Au cluster would be pseudospherical, if their D ∼ N1/3 relations correspond with the spherical geometric model, Ds = κN1/3. Here, κ = 0.328 nm, is calculated from the experimental data.62 This value is corresponding well with twice of the Wigner–Seitz radius, rws = 0.165 nm.63,64 The diameters of the Au2057 clusters fit a quasi-spherical geometric model well, indicating that they do not relax substantially to a hemispherical shape on the surface.To evaluate whether the addition of Ti enhances the stability of the alloy clusters compared with the pure Au clusters, and to shed light on the sintering mechanism(s), the process of sintering induced by electron beam irradiation was investigated with the aberration-corrected STEM. Fig. 3 shows sequential images of Au and Au/Ti cluster dimers exposed to the electron beam.
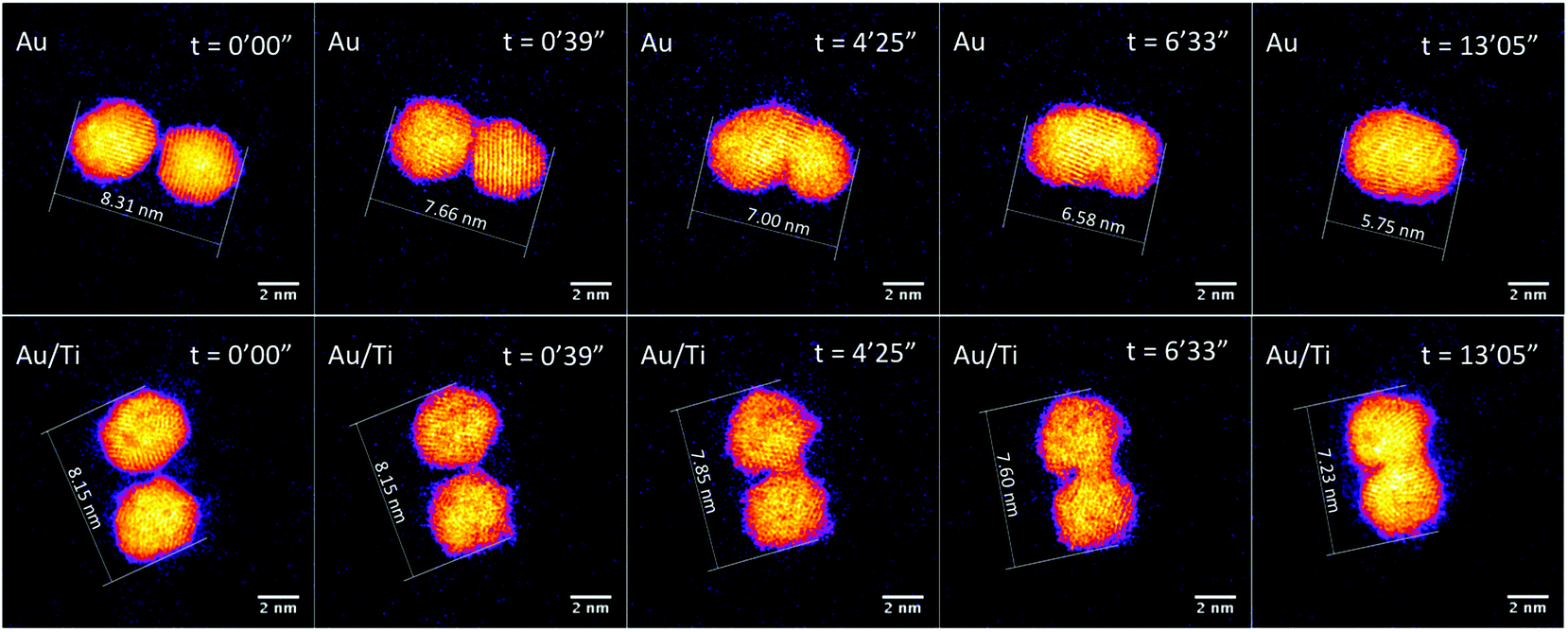 | ||
| Fig. 3 STEM images of dimers of Au2057 and of Au/Ti clusters continuously exposed to electron beam irradiation, with an acquisition time of 1.3 seconds per frame and a dose of 6.3 × 103 e− per Ångstrom2 per frame. At the beginning of imaging, the gap sizes between the Au2057 and Au/Ti cluster pairs are ∼0.30 nm and ∼0.25 nm, respectively. | ||
The observed sintering process of the Au clusters can be divided into two phases according to the STEM images and the change of the measured major axis of the Au dimer. In the first phase, the two Au clusters are seen to move toward each other once they are exposed to the electron beam driven by surface plasmon coupling,65,66 which can be confirmed by the major axis of the Au dimer shrinking from 8.31 nm to 7.66 nm after 39 s, as plotted in Fig. 4 (the cluster migration can also be seen in the ESI in Fig. S2†). The cluster migration leads the two initially separated clusters to make contact with each other. Afterwards, in the second phase, a process of coalescence, presumably driven by peripheral atom diffusion, decreases the surface area of the dimer. Two different shrink rates of the major axis of the dimer can be found in these two phases of sintering, which are ∼1.00 nm min−1 and ∼0.17 nm min−1 for the first and second phases, respectively (Fig. 4). Thus the sintering behavior of the Au dimer starts at a relatively high rate with the two clusters quickly migrating towards each other. After about 50 s, once the clusters collide, the second and slower phase of sintering begins and the rate of peripheral Au atom diffusion regulates the rate of sintering. In the case of the Au2057 clusters, 8 pairs of Au cluster dimers were imaged with the STEM, and 6 pairs of them showed similar sintering behavior as that discussed above. The clusters in the other 2 pairs of dimers with a gap size of ∼0.8 nm in between were simply found to move away from each other during imaging.
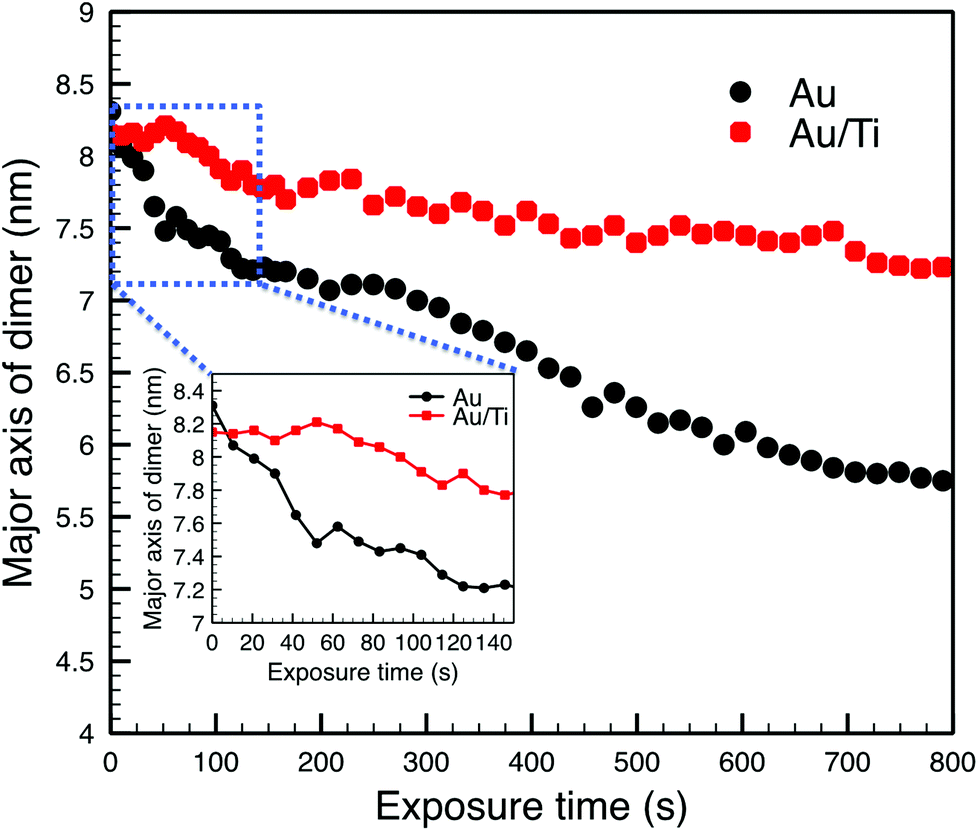 | ||
| Fig. 4 The length of the major axis of the cluster dimers as a function of the time of exposure to the electron beam. The inset shows an enlarged view of the graph in blue square. | ||
Compared with the Au clusters, the Au/Ti clusters show different sintering behavior, although once more it can be divided into two phases. In the first phase, the Au/Ti clusters remain at their original positions instead of quickly migrating towards each other like the Au clusters. Thus the Au/Ti cluster dimer retains the same length of major axis (8.15 nm) after being exposed to the electron beam for 39 s. In this first phase, only peripheral atom diffusion is found. However, once diffusing atoms found in the gap between the two clusters make a first physical connection between the clusters, the second phase of sintering is initiated. In this phase, coalescence takes place around the bridge formed in between, but the coalescence rate here (0.08 nm min−1) is much lower than that of the Au cluster dimer, Fig. 4 (also be confirmed in Fig. S3 and S5 in the ESI†).
The different sintering behavior of Au/Ti clusters compared with Au clusters on silica can be attributed to the strong interaction between Ti and the lattice oxygen of the silica support, which tends to anchor the clusters in the dimer against sintering. However, the anchoring effect is insufficient to prevent coalescence once two clusters come into contact, so it can slow down the coalescence process but cannot prevent it completely. If the distance between two neighbouring clusters is large enough (see below) that the diffusing atoms cannot “build a bridge” between them, then sintering is exceptionally slow.
Fig. 5 (see also Fig. S4 in the ESI†) shows a Au/Ti cluster dimer with a slightly larger gap than that in Fig. 3. It can be seen that the two clusters do not sinter after electron exposure for as long as 13 min, and basically the major axis of this dimer retains the same value. Peripheral atom diffusion is again found during electron exposure. At 1 min 28 s, the atoms highlighted by arrows can be found located between the two clusters, and then disappear 3 s later by moving away from the dimer area or binding to one of the two clusters. It is further observed that the cluster on the right exhibits a transient asperity due to the accumulation of diffusing atoms at 3 min 33 s. However, the distance between the clusters is too large for a contact bridge to be formed, and the asperity has decayed at 3 min 57 s. This confirms that sintering does not happen between Au/Ti clusters if the cluster distance is too large for the diffusing atoms to build a bridge in between. In future, experimental studies of hundreds of such dimer pairs may lead to the precise measurement of a critical distance for sintering by bridge formation, dependent on temperature and other key parameters (including in the case of the electron beam experiments, the current). In the case of Au/Ti clusters, 14 pairs of Au/Ti clusters dimers were imaged with STEM, and all of them showed similar behavior against sintering.
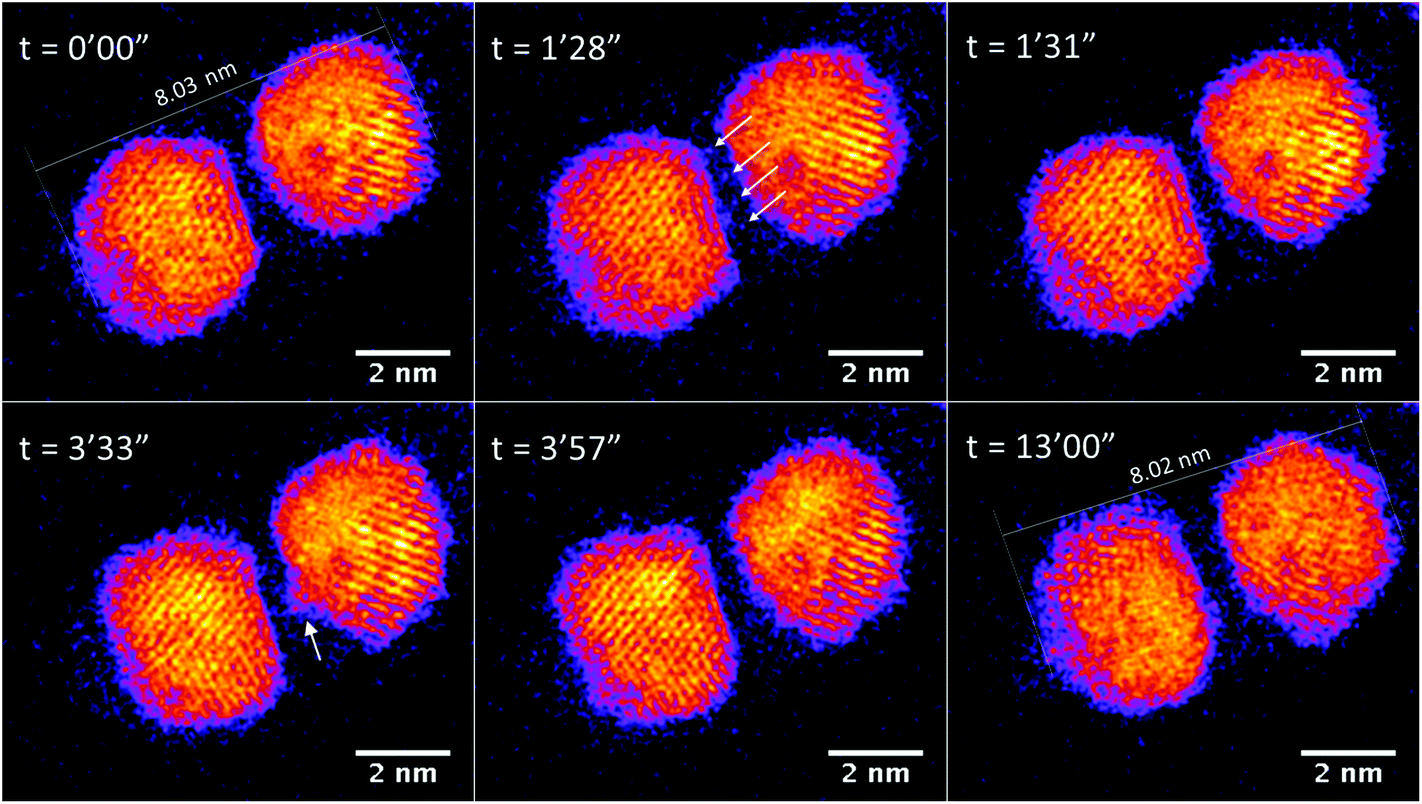 | ||
| Fig. 5 STEM images of Au/Ti cluster dimers, with a larger gap (∼0.65 nm) between than Fig. 3, continuously exposed to electron beam irradiation with acquisition time of 1.3 seconds per frame and a dose of 9.8 × 103 e− per Angstrom2 per frame. | ||
3.3 DFT calculations of surface anchoring
The size of the observed particles (around 2000 atoms) is such that we cannot directly model them with DFT calculations. It is likely that these particles will adopt shapes derived from a Wulff construction, such as a truncated octahedron dominated by (100) and (111) surfaces. To investigate further the role of Ti atoms against sintering, we considered, amongst others, the adsorption of Au20 and Au10Ti10 tetrahedral clusters which exhibit (111) faces only. While these clusters are smaller than in the experiment, the nature of the bonding with the surface is rather local, and the essence of the interaction between cluster and support is sufficiently well represented by the chosen model. Before adsorbing the clusters on the surface, we optimized the geometries of the free-standing clusters. With photoelectron spectroscopy and relativistic density functional theory simulations, Li et al.67 proposed that Au20 prefers a tetrahedral shape. Wang and Palmer68 found that this tetrahedral structure is observed, amongst other isomers, when size-selected Au20 clusters soft-landed were observed by aberration-corrected STEM on amorphous carbon film. Other experimental69 and computational70 studies also identify the tetrahedral geometry. In our simulations we also find that the tetrahedral structure is the lowest energy geometry for free Au20 clusters. For the bimetallic Au10Ti10 cluster, we considered this and other structures, while also testing different distributions of the two elements within the clusters. We considered several cluster isomers, but the potential energy surface (PES) possesses a very large number of local minima, which cannot be extensively explored without a global search algorithm. In this respect, our results are representative of some potential structures, but we do not pretend to have identified the global minimum on the PES. We find that the best cluster shape for the Au10Ti10 cluster is not in fact a pyramid, but a compact shape with Ti atoms sitting largely inside the cluster and a majority of Au atoms in the outer layer.After the optimization of the free-standing clusters, the most stable cluster isomers were deposited on the fully hydroxylated α-quartz (001) surface. This choice is dictated by the assumption that under experimental conditions the silica surface is not hydroxyl-free. Fully dehydroxylated silica surfaces can be obtained only after thermal treatment above 600 °C,71 and SiO2 surfaces get partially hydroxylated even under high vacuum conditions almost instantly.72 Since our samples are exposed to air, it is likely that a given concentration of OH groups will be present. The density of the OH groups in our model is probably higher that in the real samples, but no quantitative assessment is possible. In any case, in a previous study on tiny AuTi bimetallic clusters we considered both hydroxylated and de-hydroxylated silica surfaces, and we found that the AuTi bimetallic clusters interact strongly also with the de-hydroxylated surface. In this case oxygen atoms are extracted from the surface to bind strongly with the Ti component of the cluster.73 The structure of the hydroxylated silica surface has been described in a previous study.27 The resulting structures are shown in Fig. 6, and relevant adsorption parameters are summarized in Table 1.
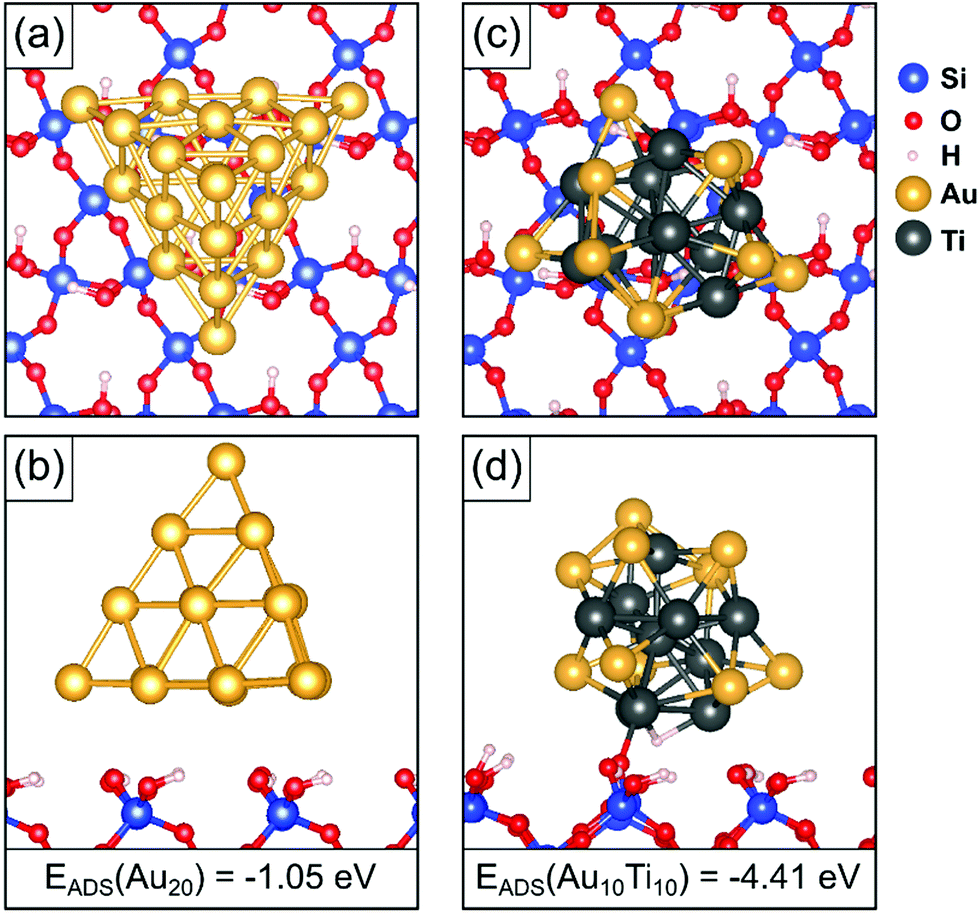 | ||
| Fig. 6 (a) Top and (b) side views of Au20 supported on the fully hydroxylated α-quartz (001) surface from DFT simulations. (c) Top view and (d) side view of Au10Ti10 supported on the same surface. | ||
| E ADS | q AVG(Au) | q AVG(Ti) | q TOT(cluster) | |
|---|---|---|---|---|
| Au20 free-standing | — | 0.00 | — | 0.00 |
| Au20 on silica | −1.05 | 0.00 | — | 0.00 |
| Au10Ti10 free-standing | — | −0.77 | +0.77 | 0.00 |
| Au10Ti10 on silica | −4.41 | −0.79 | +0.93 | +1.39 |
We find that the Au20 cluster exhibits a relatively low adsorption energy on the silica surface, only −1.05 eV, Table 1. The atoms in the supported Au clusters show an average Bader charge close to zero, indicating the absence of chemical interaction with the surface. The relatively weak binding of the Au cluster on the surface is thus largely due to dispersion forces.
The Au10Ti10 bimetallic cluster, on the other hand, exhibits a much larger adsorption energy of −4.41 eV. Only around 20% of the adsorption energy is due to dispersion forces. The large adsorption energy is due to a spontaneous local reaction of the cluster with the surface, via hydrogen reverse spillover. This can be seen in the side view in Fig. 6(d). The surface ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O–H group is split, enabling the H atom to bind to the cluster at a Ti hollow site, while the residual Si–O˙ group binds to an adjacent Ti atom of the cluster, with a resulting Ti–O bond length of 1.96 Å. This local reaction anchors the cluster to the support via the formation of a Si–O–Au10Ti10 complex. We expect this strong local binding interaction to anchor the AuTi bimetallic clusters to the hydroxylated silica surface, and to operate independently of the cluster size, leading to reduced sintering, as observed in the experiments.
Si–O–H group is split, enabling the H atom to bind to the cluster at a Ti hollow site, while the residual Si–O˙ group binds to an adjacent Ti atom of the cluster, with a resulting Ti–O bond length of 1.96 Å. This local reaction anchors the cluster to the support via the formation of a Si–O–Au10Ti10 complex. We expect this strong local binding interaction to anchor the AuTi bimetallic clusters to the hydroxylated silica surface, and to operate independently of the cluster size, leading to reduced sintering, as observed in the experiments.
Of course, in the experimental situation the clusters are exposed to ambient conditions between deposition and the sintering experiments. Thus we may expect some oxidation of the Ti atoms at the periphery of the Au/Ti clusters, after the removal of the sample out of the deposition chamber, as the XPS confirms that Ti is present on the surface in the form of TiO2. However, during and directly after the deposition of the clusters in UHV, the metallic clusters can react with the silica surface, as shown in the calculations.
4. Conclusions
Size-selected Au/Ti nanoalloy clusters (400, 000 amu) and pure Au2057 (405, 229 amu) clusters were produced by gas-phase synthesis with a magnetron sputtering, gas condensation cluster beam source and deposited onto silica supports. Chemical characterisation of the deposited clusters was provided by XPS and LEIS measurements. Upon heating the clusters in an O2/CO environment to simulate catalytic reaction conditions, the surface composition of the Au/Ti clusters was changed by a segregation process, but both Au and Ti were still visible on the surface. Thus the supported nanoalloy Au/Ti clusters are considered as viable candidates for Au-based catalysis.The sintering behavior of the clusters under electron beam annealing was explored by aberration-corrected STEM imaging in real space and real time. Two neighbouring Au2057 clusters in a dimer were found to quickly migrate and, in a second slower phase, coalesce with each other. In contrast, Au/Ti dimers showed a strong anchoring effect against sintering due to the presence of the reactive Ti atoms, most notably when the gap between them exceeded 0.60 nm. Sintering can still happen if two Au/Ti clusters are extremely close to each other, but this is due to atom diffusion between the two clusters instead of cluster migration. Sintering is expected to be exceedingly slow if the distance between the Au/Ti clusters is large enough and the diffusing atoms cannot “build a bridge” in between.
DFT calculations show that, in model bimetallic clusters (20 atoms), the Au atoms prefer a position at the surface of the nanoclusters, in good agreement with the outcome of the LEIS experiments. The calculations furthermore show that Au clusters can be bonded much more strongly (by a factor of 5) to a silica support by alloying them with Ti. This effect is due to the increased reactivity of the Au/Ti bimetallic clusters when they present a surface containing a reactive metal (Ti). Future computations may address the role of the oxidation of the bimetallic Au/Ti clusters but the qualitative enhancement of surface anchoring against sintering is expected to be preserved. The presence of surface Au atoms in size-selected Au/Ti nanoalloy clusters and the anchoring effect due to Ti incorporation may open up new possibilities in Au-based nanocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
For funding of this work, we gratefully acknowledge the European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 607417 (Catsense), as well as an EPSRC Fellowship Grant and the Villum Foundation V-SUSTAIN grant 9455 to the Villum Center for the Science of Sustainable Fuels and Chemicals.References
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS
.
- W. A. Bone and R. V. Wheeler, Proc. R. Soc. A, 1906, 77, 146–147 CrossRef
- W. A. Bone and G. W. Andrew, Proc. R. Soc. A, 1925, 109, 459–476 CrossRef CAS
-
K. Sennewald, W. Vogt and H. Glaser, PatentDE1244766B, 1967 Search PubMed
- G. C. Bond and P. A. Sermon, Gold Bull., 1973, 6, 102–105 CrossRef CAS
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS
- G. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS
- M. Haruta, Angew. Chem., Int. Ed., 2014, 53, 52–56 CrossRef CAS PubMed
- A. Taketoshi and M. Haruta, Chem. Lett., 2014, 43, 380–387 CrossRef CAS
- B. Hvolbæk, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Nørskov, Nano Today, 2007, 2, 14–18 CrossRef
- Y. G. Wang, D. C. Cantu, M. S. Lee, J. Li, V. A. Glezakou and R. Rousseau, J. Am. Chem. Soc., 2016, 138, 10467–10476 CrossRef CAS PubMed
- J. Saavedra, C. Powell, B. Panthi, C. J. Pursell and B. D. Chandler, J. Catal., 2013, 307, 37–47 CrossRef CAS
- K. Christmann, S. Schwede, S. Schubert and W. Kudernatsch, ChemPhysChem, 2010, 11, 1344–1363 CrossRef CAS PubMed
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed
- D. Widmann and R. J. Behm, Acc. Chem. Res., 2014, 47, 740–749 CrossRef CAS PubMed
- T. Fujitani and I. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 10144–10147 CrossRef CAS PubMed
- I. N. Remediakis, N. Lopez and J. K. Nørskov, Angew. Chem., Int. Ed., 2005, 117, 1858–1860 CrossRef
- Z. Duan and G. Henkelman, ACS Catal., 2015, 5, 1589–1595 CrossRef CAS
- I. X. Green, W. J. Tang, M. Neurock and J. T. Yates, Acc. Chem. Res., 2014, 47, 805–815 CrossRef CAS PubMed
- I. X. Green, W. J. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed
- P. Kast, G. Kučerová and R. J. Behm, Catal. Today, 2015, 244, 146–160 CrossRef CAS
- D. Widmann and R. J. Behm, Angew. Chem., Int. Ed., 2011, 50, 10241–10245 CrossRef CAS PubMed
- L. Zhang, R. Persaud and T. E. Madey, Phys. Rev. B: Condens. Matter, 1997, 56, 10549–10557 CrossRef CAS
- S. C. Parker, A. W. Grant, V. A. Bondzie and C. T. Campbell, Surf. Sci., 1999, 441, 10–20 CrossRef CAS
- G. M. Veith, A. R. Lupini, S. Rashkeev, S. J. Pennycook, D. R. Mullins, V. Schwartz, C. A. Bridges and N. J. Dudney, J. Catal., 2009, 262, 92–101 CrossRef CAS
- P. Schlexer and G. Pacchioni, Top. Catal., 2016, 60, 459–470 CrossRef
- F. Yang, M. S. Chen and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 254–260 CAS
- K. J. Hu, S. R. Plant, P. R. Ellis, C. M. Brown, P. T. Bishop and R. E. Palmer, J. Am. Chem. Soc., 2015, 137, 15161–15168 CrossRef CAS PubMed
- A. Cao, R. Lu and G. Veser, Phys. Chem. Chem. Phys., 2010, 12, 13499–13510 RSC
-
Z. Ma and S. Dai, Heterogeneous Gold Catalysts and Catalysis, Royal Soc Chemistry, Cambridge, 2014 Search PubMed
- P. Hernandez-Fernandez, F. Masini, D. N. McCarthy, C. E. Strebel, D. Friebel, D. Deiana, P. Malacrida, A. Nierhoff, A. Bodin, A. M. Wise, J. H. Nielsen, T. W. Hansen, A. Nilsson, I. E. L. Stephens and I. Chorkendorff, Nat. Chem., 2014, 6, 732–738 CrossRef CAS PubMed
- A. Velazquez-Palenzuela, F. Masini, A. F. Pedersen, M. Escudero-Escribano, D. Deiana, P. Malacrida, T. W. Hansen, D. Friebel, A. Nilsson, I. E. L. Stephens and I. Chorkendorff, J. Catal., 2015, 328, 297–307 CrossRef CAS
- C. Holse, C. F. Elkjaer, A. Nierhoff, J. Sehested, I. Chorkendorff, S. Helveg and J. H. Nielsen, J. Phys. Chem. C, 2015, 119, 2804–2812 CAS
- A. Gomez-Cortes, G. Diaz, R. Zanella, H. Ramirez, P. Santiago and J. M. Saniger, J. Phys. Chem. C, 2009, 113, 9710–9720 CAS
- Y. J. Guan and E. J. M. Hensen, J. Catal., 2013, 305, 135–145 CrossRef CAS
- C. W. Han, P. Majumdar, E. E. Marinero, A. Aguilar-Tapia, R. Zanella, J. Greeley and V. Ortalan, Nano Lett., 2015, 15, 8141–8147 CrossRef CAS PubMed
- X. Y. Liu, A. Q. Wang, X. D. Wang, C. Y. Mou and T. Zhang, Chem. Commun., 2008, 3187–3189 RSC
- X. Y. Liu, A. Q. Wang, X. F. Yang, T. Zhang, C. Y. Mou, D. S. Su and J. Li, Chem. Mater., 2009, 21, 410–418 CrossRef CAS
- G. C. Ma, A. Binder, M. F. Chi, C. Liu, R. C. Jin, D. E. Jiang, J. Fan and S. Dai, Chem. Commun., 2012, 48, 11413–11415 RSC
- A. Sandoval, C. Louis and R. Zanella, Appl. Catal., B, 2013, 140, 363–377 CrossRef
- B. von Issendorff and R. E. Palmer, Rev. Sci. Instrum., 1999, 70, 4497 CrossRef CAS
- S. Pratontep, S. J. Carroll, C. Xirouchaki, M. Streun and R. E. Palmer, Rev. Sci. Instrum., 2005, 76, 045103 CrossRef
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558–561 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251–14269 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS
- P. E. Blochl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59, 1758–1775 CrossRef CAS
-
G. H. F. Diercksen and S. Wilson, Methods in Computational Molecular Physics, Springer Netherlands, 1983 Search PubMed
-
C. Moler and I. Shavitt, Report on the Workshop: Numerical Algorithms in Chemistry: Algebraic Methods, Lawrence Berkeley Laboratory, University of California, 1978 Search PubMed
- P. Schlexer and G. Pacchioni, J. Phys.: Condens. Matter, 2016, 28, 364005 CrossRef PubMed
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter, 1976, 13, 5188–5192 CrossRef
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- S. Tosoni and J. Sauer, Phys. Chem. Chem. Phys., 2010, 12, 14330–14340 RSC
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed
- N. Young, Z. Li, Y. Chen, S. Palomba, M. Di Vece and R. Palmer, Phys. Rev. Lett., 2008, 101, 246103 CrossRef CAS PubMed
- K. P. Arkill, J. M. Mantell, S. R. Plant, P. Verkade and R. E. Palmer, Sci. Rep., 2015, 5, 9234 CrossRef CAS PubMed
- Y. Han, D. S. He, Y. Liu, S. Xie, T. Tsukuda and Z. Y. Li, Small, 2012, 8, 2361–2364 CrossRef CAS PubMed
-
B. Smirnov, Clusters and small particles: in gases and plasmas, Springer, New York, 2000 Search PubMed
- P. E. Batson, Phys. Rev. Lett., 1982, 49, 936–940 CrossRef CAS
- P. E. Batson, A. Reyes-Coronado, R. G. Barrera, A. Rivacoba, P. M. Echenique and J. Aizpurua, Nano Lett., 2011, 11, 3388–3393 CrossRef CAS PubMed
- J. Li, X. Li, H. J. Zhai and L. S. Wang, Science, 2003, 299, 864–867 CrossRef CAS PubMed
- Z. W. Wang and R. E. Palmer, Nanoscale, 2012, 4, 4947–4949 RSC
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed
- L. M. Molina and B. Hammer, J. Catal., 2005, 233, 399–404 CrossRef CAS
- C. Armistead, A. Tyler, F. Hambleton, S. Mitchell and J. A. Hockey, J. Phys. Chem., 1969, 73, 3947–3953 CrossRef CAS
- A. S. D'Souza and C. G. Pantano, J. Am. Ceram. Soc., 1999, 82, 1289–1293 CrossRef
- P. Schlexer and G. Pacchioni, J. Phys. Chem. C, 2017, 14717–14724 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr06323g |
| This journal is © The Royal Society of Chemistry 2018 |