
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic self-assembly of DNA minor groove-binding ligand DB921 into nanotubes triggered by an alkali halide†
R.
Mizuta
ab,
J. M.
Devos
a,
J.
Webster
 ab,
W. L.
Ling
c,
T.
Narayanan
b,
A.
Round
df,
D.
Munnur
abe,
E.
Mossou
af,
A. A.
Farahat
gh,
D. W.
Boykin
g,
W. D.
Wilson
g,
S.
Neidle
e,
R.
Schweins
a,
P.
Rannou
i,
M.
Haertlein
a,
V. T.
Forsyth
af and
E. P.
Mitchell
*bf
ab,
W. L.
Ling
c,
T.
Narayanan
b,
A.
Round
df,
D.
Munnur
abe,
E.
Mossou
af,
A. A.
Farahat
gh,
D. W.
Boykin
g,
W. D.
Wilson
g,
S.
Neidle
e,
R.
Schweins
a,
P.
Rannou
i,
M.
Haertlein
a,
V. T.
Forsyth
af and
E. P.
Mitchell
*bf
aInstitut Laue-Langevin, 71 Avenue des Martyrs, 38000 Grenoble, France
bEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France. E-mail: mitchell@esrf.eu
cUniv. Grenoble Alpes, CEA, CNRS, IBS, F-38000 Grenoble, France
dEuropean Molecular Biology Laboratory, 71 Avenue des Martyrs, 38000 Grenoble, France
eSchool of Pharmacy, University College London, Brunswick Square, London, WC1N 1AX, UK
fFaculty of Natural Sciences, Keele University, Staffordshire, ST5 5BG, UK
gDepartment of Chemistry, Georgia State University, Atlanta, GA 30303, USA
hDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt
iUniv. Grenoble Alpes, CNRS, CEA, INAC-SyMMES, 38000 Grenoble, France
First published on 8th March 2018
Abstract
We describe a novel self-assembling supramolecular nanotube system formed by a heterocyclic cationic molecule which was originally designed for its potential as an antiparasitic and DNA sequence recognition agent. Our structural characterisation work indicates that the nanotubes form via a hierarchical assembly mechanism that can be triggered and tuned by well-defined concentrations of simple alkali halide salts in water. The nanotubes assembled in NaCl have inner and outer diameters of ca. 22 nm and 26 nm respectively, with lengths that reach into several microns. Our results suggest the tubes consist of DB921 molecules stacked along the direction of the nanotube long axis. The tubes are stabilised by face-to-face π–π stacking and ionic interactions between the charged amidinium groups of the ligand and the negative halide ions. The assembly process of the nanotubes was followed using small-angle X-ray and neutron scattering, transmission electron microscopy and ultraviolet/visible spectroscopy. Our data demonstrate that assembly occurs through the formation of intermediate ribbon-like structures that in turn form helices that tighten and compact to form the final stable filament. This assembly process was tested using different alkali–metal salts, showing a strong preference for chloride or bromide anions and with little dependency on the type of cation. Our data further demonstrates the existence of a critical anion concentration above which the rate of self-assembly is greatly enhanced.
1. Introduction
Self-assembling supramolecular systems enable the spontaneous formation of complex nanoscale structures. These systems present a range of accessible morphologies, often with well-defined dimensional parameters and physical properties that allow for numerous potential applications, making such materials highly desirable across various disciplines of science and technology. The ability to initiate and control their assembly further offers considerable potential for application. In particular, the nanotube architecture has become ubiquitous throughout many classes of self-assembling systems, leading to their use in fields such as nano-templating and bottom-up nanofabrication,1 biological delivery devices,2 nanoscale wires and optical devices.3Previously reported systems have shown that such self-assembly typically arises from a subtle interplay between various non-covalent interactions including ionic interactions, hydrogen bonding, π–π stacking of aromatic groups, solvent interactions and steric hindrances.4–6 Therefore, a thorough understanding of these governing interactions is paramount in achieving the necessary control over the dynamics of the self-assembly and resulting morphology for potential applications. Owing to the extensive number of parameters that influence the assembly, it remains a difficult task to achieve such control. However, recent examples demonstrate promising developments in the field towards materials with tailored functionalities.7–10
Small organic molecules that recognise and bind specifically to the minor groove of DNA, often based on planar heterocyclic ring structures, have been studied extensively.11–15 Interest in these molecules arises from their strong biotechnological potential and as candidate anti-parasitic and DNA sequence recognition agents via specific curvature of the molecules fitting the grooves in DNA helices. However, to date none of these small molecules have been shown to possess self-assembling properties. The biscationic form of 2-(4′-amidinobiphenyl-4-yl)-1H-benzimidazole-5-amidine, one member of this family of compounds and named hereinafter as DB921, consists of three connected rings, terminated at each end of the planar molecule by charged amidinium groups, shown in Fig. 1.16,17
 | ||
| Fig. 1 Chemical structure of the heterocyclic cationic DB921. | ||
Here we report a novel hierarchical self-assembling system of nanotubes, formed by DB921 in the presence of suitable alkali-halide salts. We present a multi-disciplinary study conducted using complementary techniques of solution-state small-angle X-ray and neutron scattering (SAXS and SANS respectively), negative stain and cryo-transmission electron microscopy (cryo-TEM), UV-vis spectroscopy and X-ray fibre diffraction, and propose an assembly mechanism and a possible atomic-level model for the nanotubes.
2. Experimental
2.1 Nanotube preparation
DB921 was synthesised as previously published with acetate counter ions.16 The resulting powder was stored at 4 °C away from light until use. A stock solution of DB921, with double the desired concentration (i.e. 12 mM) for the final nanotube solution, was prepared with 8% DMSO (v/v) in order to improve solubility, and deionised water. DB921 powder was firstly dissolved in DMSO and mixed with a vortex mixer at approximately 1000 rpm for 30 seconds. Deionised water was subsequently added to make a solution containing 8% (v/v) DMSO and 12 mM DB921 and mixed again with a vortex mixer at approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for a minimum of 5 minutes. The resulting solution was centrifuged at 10000 rpm for 10 minutes and the supernatant extracted. Samples were then centrifuged to remove any excess insoluble material from the saturated DB921 solution. Aqueous NaCl solutions were also made at double the required final concentration. Equal volumes of the DB921 and NaCl solutions were mixed for a two-fold dilution to initiate nanotube assembly. All nanotube preparation was carried out at room temperature. Formation of nanotubes is associated with a phase separation with a darker yellow phase containing the nanotubes (see ESI†).
000 rpm for a minimum of 5 minutes. The resulting solution was centrifuged at 10000 rpm for 10 minutes and the supernatant extracted. Samples were then centrifuged to remove any excess insoluble material from the saturated DB921 solution. Aqueous NaCl solutions were also made at double the required final concentration. Equal volumes of the DB921 and NaCl solutions were mixed for a two-fold dilution to initiate nanotube assembly. All nanotube preparation was carried out at room temperature. Formation of nanotubes is associated with a phase separation with a darker yellow phase containing the nanotubes (see ESI†).
The same protocol was used for the various salts tested in this study. The final samples were saturated solutions of DB921 with 4% (v/v) DMSO and the desired final concentration of the various salts tested in the experiments described in this paper. Regarding the sample prepared in D2O for the SANS experiment, the same protocol was followed apart from using deuterated DMSO and D2O.
For the tests on assembly at different pH, both DB921 stock solution and NaCl solution were pH-corrected; different buffers were used for different pH ranges. For pH 4–6: acetate buffer. For pH 7.8: HEPES buffer. For pH 9: glycine buffer.
2.2 Transmission electron microscopy
For negative stain TEM, ∼4 μl of the sample was applied to a mica sheet covered with a film of evaporated carbon. The carbon film was then floated off the mica in 2% sodium silico-tungstate (SST) and retrieved onto a 400-mesh copper electron microscopy grid. Imaging was performed on a Philips CM12 transmission electron microscope at 120 kV, an FEI T12 at 120 kV or an FEI F20 at 200 kV. Images were recorded on an Orius 832 CCD camera (CM12 and T12) or an Eagle digital camera (F20).For cryo-TEM, ∼4 μl of the sample was applied to a glow-discharged Quantifoil grid and plunge frozen in liquid ethane using a Vitrobot. Frozen grids were observed on an FEI F20 microscope at 200 kV or on an FEI Polara microscope at 300 kV. Images on the Polara were recorded on an Ultrascan 4000 CCD camera. Nanotube dimensions were based on an average of 100 nanotube measurements from the cryo-TEM images.
2.3 Solution SAXS
Solution SAXS was conducted at the automated BM29 beamline for Bio-SAXS at the European Synchrotron Radiation Facility (ESRF, Grenoble).18,19 35 μl of sample solution, maintained at 20 °C, was pumped through a 1.5 mm diameter quartz capillary and continuously flowed during the data collection in order to minimise radiation damage. A sample to detector distance of 2.87 m with an X-ray wavelength of 0.9919 Å was employed with a sample exposure time of 1 second. Buffer measurements were conducted before and after each sample measurement for subsequent background subtraction using 35 μl volumes for each. Raw two-dimensional scattering data was radially averaged about the beam centre in order to obtain one-dimensional profiles of scattered intensity as a function of the magnitude of scattering vector (Q). Time-resolved SAXS spectra were also measured on BM29. Nanotube initiation, sample loading and beamline interlock took less than 5 minutes.Higher resolution SAXS measurements over an extended Q range were performed on selected samples at the ESRF ID02 beamline.20 Two sample-to-detector distances of 1.2 m and 10 m were used to cover the Q range from 0.007 nm−1 to 7 nm−1. The data reduction procedure was similar to BM29 measurements and intensities were normalised to an absolute scale. Fitting of the one-dimensional data was performed using the software SASView version 2.2.1. with the core cylinder model.21 Parameters derived from the fitting were inner and outer cylinder radii and the polydispersity of the outer radius defined as the standard deviation divided by the mean value. Here, the polydispersity was better described by a log-normal size distribution of outer radius and the wall thickness defined by the chemical structure was assumed to be uniform.
2.4 Solution SANS
SANS data were collected on instrument D11 at the ILL.22 The wavelength used was 6 Å with a spread of 9%. Two instrument configurations were used, with sample-detector distances of 1.2 m and 8 m and corresponding collimation lengths of 4 m and 8 m respectively, resulting in a Q range from 0.071 nm−1 to 5 nm−1. A beam-size of 7 × 10 mm was used. Data were put on absolute scale using the secondary calibration standard H2O (of 1 mm path length). Standard reduction procedures were followed using the ILL software LAMP.23 Data fitting was performed using SASVIEW.2.5 Fibre diffraction
Fibres of nanotubes were synthesised from pellets, made from a solution of fully assembled nanotubes, spun down at 100000 rpm in a Beckman Coulter Air-Driven Ultracentrifuge. 2 μl droplets of sedimented nanotube pellet were dispensed in between two glass rods and slowly pulled apart. A stretched droplet was allowed to dry over a minimum period of 24 hours. The fibre diffraction was conducted on ID23-2, the Gemini Macromolecular Crystallography Beamline (ESRF, Grenoble).24 Data were collected to 3 Å resolution using X-rays of wavelength of 0.873 Å with an exposure time of 1.0 second, using a PILATUS2 3 M detector at a sample to detector distance of 377.8 mm.
2.6 UV-Vis spectroscopy
UV-vis spectra were obtained using a Shimadzu UV – 2401PC Spectrophotometer. 80 μl of sample were dispensed in Hellma Quartz SUPRASIL cuvettes, model 105.202-QS with a path length of 10 mm. All spectra were obtained having taken a background baseline.2.7 Modelling
Molecular modelling of an individual DB921 molecule was performed using the Accelrys Materials Studio package (www.accelrys.com). The length of a single DB921 in water was extracted from a geometry-optimised structure, calculated using the semi-empirical molecular-orbital theory module VAMP. The semi-empirical approach was achieved through an NDDO approximation, namely AM1 (Austin Model 1). A COSMO solvation scheme was employed with the medium dielectric set to that of water, 78.54 (no units).3. Results and discussion
3.1 Nanotube morphology
The structural characterisation studies show that the mature nanotubes formed with NaCl have an outer diameter (DO) and inner diameter (DI) of about 26 nm and 22 nm respectively, as detailed in Table 1. Fig. 2(a) shows the azimuthally averaged high-resolution SAXS curves measured (using the ID02 beamline at ESRF) from two nanotube solutions which display characteristic oscillations of cylindrical structures. Up to ten intensity maxima are observed, indicating a narrow diameter distribution of the tubes. In addition, the minimum in the high Q region (∼3.5 nm−1) defines the thickness of the wall. The SAXS curves were fitted with a core–shell cylinder model with the contrast of the core being the same as the solvent (i.e. a hollow cylinder). This modelling showed that the polydispersity, defined as the ratio of standard deviation and mean diameter using a log-normal size distribution, is about 6%. The wall thickness was assumed to be uniform since it is defined by the molecular architecture. As can be seen in Fig. 2(a), the model accurately describes the data over the broad Q range but at lower Q range, the interactions or entanglements between tubes cannot be excluded. The best-fit length was about 2000 nm which is outside the Q range of measurement.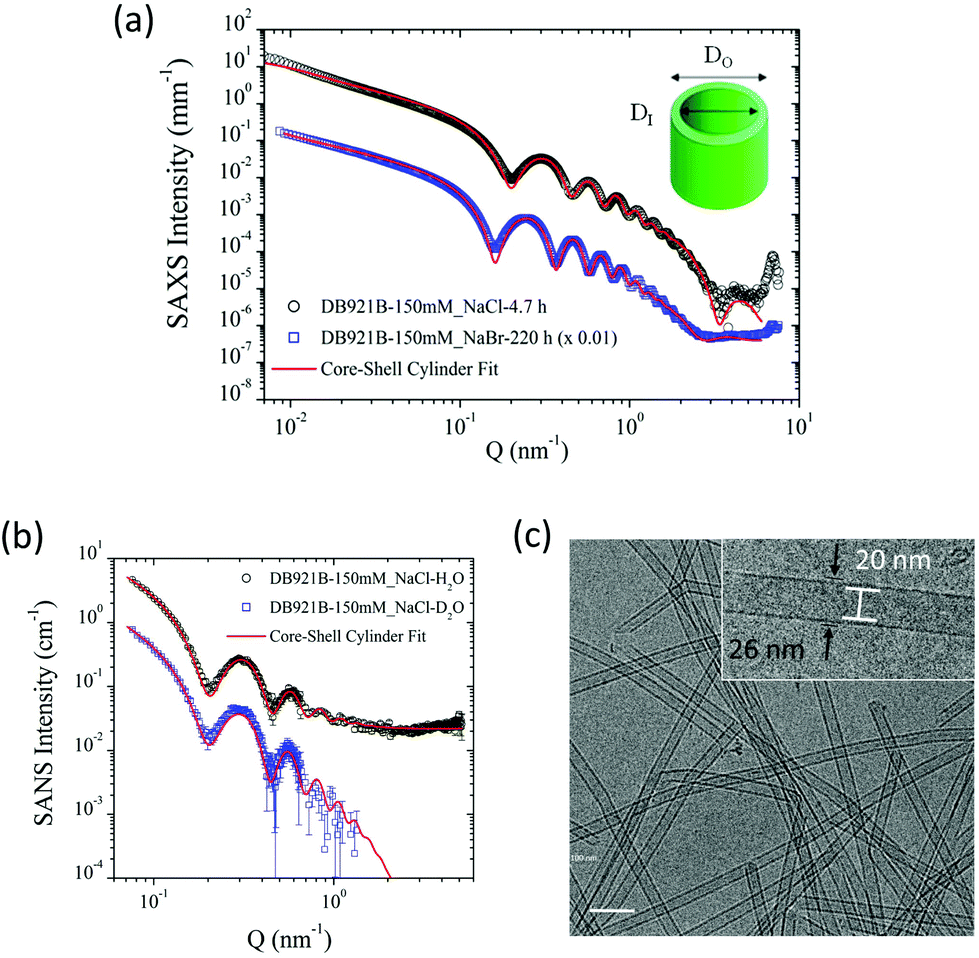 | ||
| Fig. 2 (a) SAXS (beamline ID02) intensity of fully assembled nanotubes from 150 mM NaCl and NaBr salt, fitted with a core–shell cylinder model. All SAXS intensities are shown as a function of the modulus of the scattering vector Q. Inset: DI and DO denote the inner and outer tube diameters respectively. (b) SANS (instrument D11) intensity of fully assembled nanotubes in 150 mM NaCl in water and deuterated water, fitted with a core–shell cylinder model. (c) Cryo-TEM image of fully assembled nanotubes from 150 mM NaCl. Inset: Average inner and outer diameters are found to be 20.1 nm and 25.7 nm, respectively. | ||
| Characterisation technique | D I [nm] | D O [nm] | Wall Thickness [nm] |
|---|---|---|---|
| SAXS NaCl | 22.4 | 26.1 | 1.85 |
| SANS NaCl (H2O solution) | 22.4 | 26.2 | 1.9 |
| SANS NaCl (D2O solution) | 23.0 | 26.8 | 1.9 |
| Cryo-TEM NaCl | 20 | 26 | 3 |
| SAXS NaBr | 27.8 | 32.0 | 2.1 |
SANS data on mature nanotubes formed in the presence of NaCl were recorded on D11 at ILL in both H2O and D2O solutions as shown in Fig. 2(b). Both sets of SANS data could be fitted as a core–shell cylinder model and were consistent with the SAXS data analysis shown in Fig. 2(a). The incoherent background in the high Q region makes an independent determination of the wall thickness uncertain and therefore it was fixed at the SAXS value (1.9 nm). As can be seen in Table 1, the outer diameter deduced from the SANS model is consistent with the SAXS value.
The results highlight the remarkable monodispersity in nanotube cross-section, which is also evident in the cryo-TEM image of the nanotubes shown in Fig. 2(c). High degrees of monodispersity in cross-sectional dimensions have previously been reported in other similar self-assembled tubular systems.25,26 In such systems, the wall-thicknesses often correspond to a monolayer formed of the constituent molecules. This is particularly true for bolaamphiphilic molecules that enable hydrophilic groups to arrange on the inner and outer tube surfaces, with hydrophobic groups encased within the wall.26,27 In a similar manner, DB921 molecules may adopt an orientation with the hydrophilic, terminal amidinium groups pointing towards the inner and outer walls of the tube, thus sequestering the hydrophobic aromatic backbone. Furthermore, halide counterions are expected to condense around the positively charged amidinium groups at each end to achieve charge neutralisation.7 Therefore, we propose that the bulk of the nanotube wall is composed of a monolayer of DB921, with additional layers of halide ions decorating the inner and outer surfaces, screening the electrostatic repulsion while potentially facilitating π-stacking by bridging across adjacent amidinium ions.
For the present system, the calculated length of a single DB921 molecule of 1.92 nm, combined with the diameter of two chloride ions (2 × 0.36 nm) associated at either end, would suggest a wall-thickness value of 2.64 nm. The wall thickness deduced from SAXS in solution of 1.85 nm, where the counterion profile may be more diffuse, is comparable to the size of the DB921 molecule. The TEM measurements suggest a larger thickness of 3 nm. The larger wall thickness measured from cryo-TEM might be due to the subjectivity in determining the wall boundary during the measurements and the defocusing necessary to obtain sufficient contrast to clearly discerning the nanotubes. The SAXS/SANS dimensions were therefore used for modelling. SAXS measurements (ESRF beamline BM29) were made on samples aged by 1 month at 4 °C, which confirmed long-term stability of the nanotubes (data not shown).
Nanotube formation was also tested using various cation/anion combinations from the alkali metal/halide element groups. The results show that nanotube formation is independent of the alkali metal used, where the closely aligned scattering features (BM29 beamline) shown in Fig. 3(a) indicate very similar nanotube cross sections. Negative stain TEM also shows the consistency of the nanotube morphologies over the alkali–metal series (data not shown). Formation was also investigated using various halide ions. The results shown in Fig. 3(b) demonstrate that both chloride and bromide ions allow the formation of nanotubes. However, tube formation does not occur with either fluoride or iodide, even following a month of incubation. The results show that chloride or bromide ions are an essential component of the nanotubes, their negative charges counterbalancing the positive amidinium charges. Given that the nanotube formation is selective for only two of the halide ions, an effect of radius is implied where only chloride and bromide have radii appropriate to chelate the amidinium groups and allow tube formation. It is not unreasonable to consider that the ions could bridge between adjacent amidinium groups and actively hold together the DB921 molecular network in addition to balancing the DB921 positive charges.
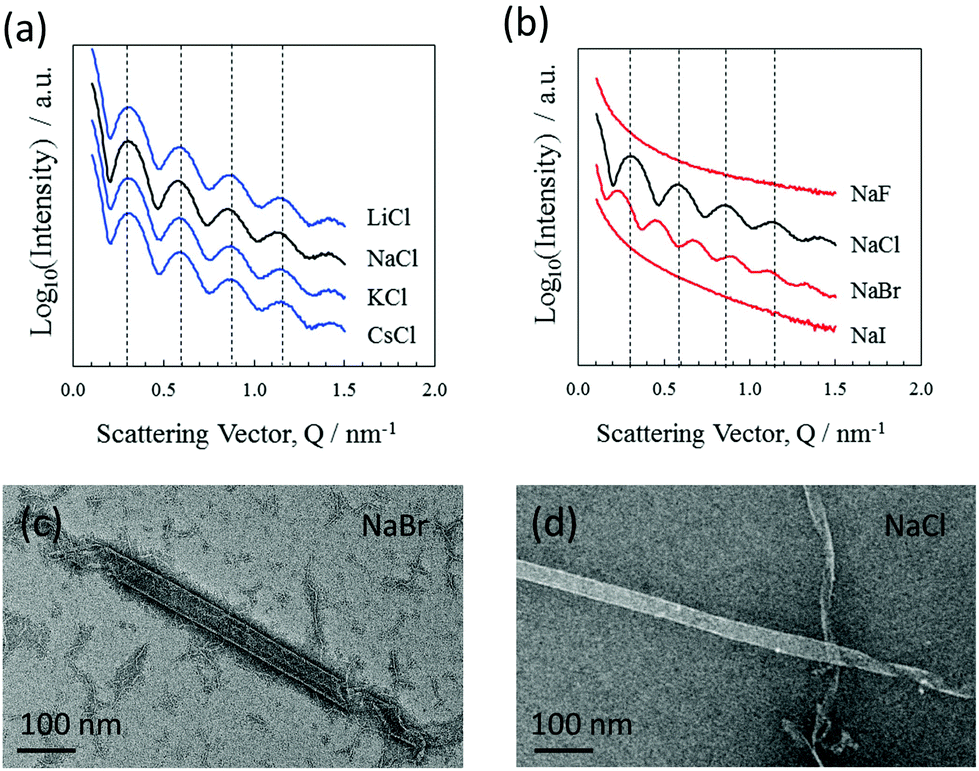 | ||
| Fig. 3 (a) SAXS intensity (BM29) of nanotubes as formed with varying alkali chlorides. SAXS intensities were measured at 67, 87, 73 and 79 minutes for Li, Na, K and Cs respectively. Dashed lines indicate close alignment of the peaks and troughs, caused by similar cross-sectional nanotube dimensions for all of the salts. (b) SAXS intensities (BM29) of nanotubes as formed with varying sodium halides. Characteristic oscillations were not observed for NaF and NaI. The peaks and troughs for NaBr do not align with those of NaCl as indicated by the dashed lines, which are aligned with the NaCl peaks. (c,d) Negative stain TEM images of nanotubes assembled in NaBr (c) and NaCl (d). | ||
SAXS curves, shown in Fig. 2(a) and as measured from nanotubes formed with bromide ions, yield a diameter of 32.0 nm when fitted with a similar model. This diameter is larger than that found for the chloride equivalent (the minimum shifts to smaller Q values), while the wall thickness remains similar. This finding further emphasises the involvement of anions likely forming a diffuse outer shell and influencing the bending energy to form a curved surface. Negative stain TEM images shown in Fig. 3(c) also show this increase in tube diameter. A similar effect of counterions modulating the structure and size has been reported in the case of peptide nanotubes7 and discussed in the context of controlling the chiral architecture of self-assembled amphiphilic systems.28
Nanotube assembly was further examined across a pH range from 4 to 9. Assembly was performed with high chloride concentration (150 mM) with SAXS spectra recorded 2.5 hours after initiation (ESI†). The results show no pH dependence in this pH range.
3.2 Assembly mechanism
A more detailed time-resolved study of the assembly of NaCl nanotubes was conducted using a combination of negative stain-TEM and solution-state SAXS (BM29 beamline).A time-resolved study was performed by negative stain-EM to image the assembly process. For convenience, nanotubes were assembled in a lower concentration of the DB921 of 4.5 mM, enabling a longer assembly time of several days. Times were measured from the initiation of nanotube formation by mixing the DB921 and sodium chloride solutions. Fig. 4(a)–(c) shows the sequential formation of intermediates before complete nanotube formation. Shortly after (40 minutes) the assembly is initiated, short fibrillar structures of width 2 to 5 nm and lengths of 50 to 100 nm are seen as in Fig. 4(a), left. Additionally, the sample is inter-dispersed with longer fibrils with widths varying between 20 to 30 nm and lengths of 200 to 300 nm (Fig. 4(a), right).
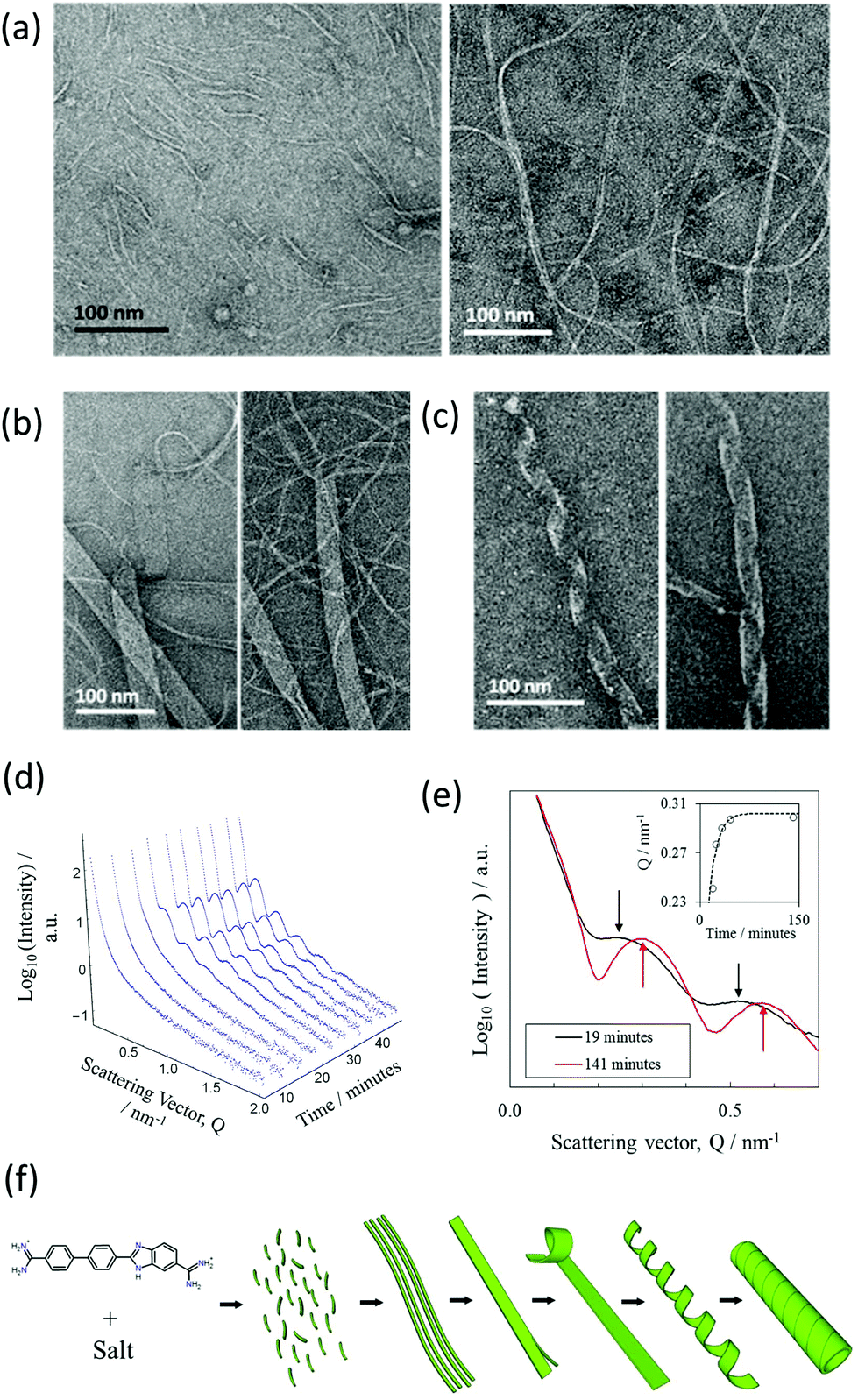 | ||
| Fig. 4 Evolution of the nanotubes with time. (a)–(c) Negative stain TEM images at time stamps 40 minutes, 24 hours and 48 hours after tube initiation. (d) Time-resolved SAXS (BM29) for the growth of the nanotubes with a saturated DB921 solution and NaCl concentration of 70 mM. (e) A comparison of maturing nanotubes in 70 mM NaCl in the earlier stages of growth (19 minutes) against a mature sample (141 minutes) showing the minima becoming sharper indicating the narrowing of the distribution of nanotube diameter with time. Inset: The apparent shift in first SAXS peak to higher scattering vector over time. The plateau behaviour reflects the completion of assembly. A bounded exponential line of best fit is plotted with the data to indicate the underlying trend. (f) Schematic showing the proposed mechanism of nanotube formation from ribbons to helices to mature nanotubes. | ||
The coexistence of these two morphologies suggests that initial aggregation of the DB921 molecules leads to the formation of short and thin fibrils, which in turn aggregate to form elongated fibrils. Twenty-four hours post-initiation we observe a system of ribbons with widths of approximately 50 nm and lengths of up to several microns (Fig. 4(b)). In particular, thin fibrils that are similar to those displayed in Fig. 4(a) are seen splitting and branching out from the ends of the aggregated fibrils, packed together in a side-by-side manner. Approaching 48 hours, we observe such ribbons helically twisted into mature nanotubes (Fig. 4(c)). This final step of helical winding has been previously identified in many nanotube systems.28–33
A similar time-resolved study was also carried out using SAXS (beamline BM29). The scattering by nanotubes assembled in saturated solution of DB921 (with fixed NaCl concentration at 70 mM and with 4% (v/v) DMSO) was measured at multiple time stamps after initiation varying from a few minutes up to one hour. The resulting SAXS curves are shown in Fig. 4(d). Several salient features are noted. Firstly, there exists an induction period immediately after assembly is initiated, during which no characteristic maxima in the SAXS spectra are observed. Secondly, following this induction time, initially weak maxima emerge that subsequently evolve into the scattering pattern associated with mature nanotubes. At first, the oscillations are weak in amplitude, presumably due to the coexistence of different structural moieties in the solutions. As more nanotubes are formed, their contribution to the total SAXS intensity becomes dominant, characterised by the well-defined maxima. The apparent shift in positions of the fringe maxima to higher Q with time, as shown in Fig. 4(e), is an indication of the maturing of nanotube population. At the same time, the population of other structural moieties such as the ribbons and helices as observed by TEM deplete with time as they are transformed into nanotubes. Such a hierarchical assembly mechanism corroborates the time-resolved TEM data.
The induction period is attributed to the time taken for the assembly to proceed up to the formation of nanotubes. The lack of peaks at early times during the assembly is due to the fact that the characteristic SAXS oscillations strictly pertain to tubular structures. Therefore, all early intermediates have broad scattering features as observed at early times in Fig. 4(d); the onset of oscillations is observed once the ribbons begin to coil.34 Despite not being fully formed nanotubes, their average structure as seen by X-ray scattering would resemble that of a nanotube. Furthermore, the SAXS reveals an apparent decrease in the cross-sectional size over time before reaching a “saturation” limit (Fig. 4(e)). This is consistent with an assembly by helical twisting and packing seen by TEM, corresponding to the tightening of the ribbons as the packing approaches completion. The proposed hierarchical assembly mechanism based on these studies is shown schematically in Fig. 4(f).
3.3 Triggering nanotube formation
A further investigation was made into the dependence of assembly upon the concentration of NaCl. For this study, concentrations of NaCl were varied between 0 mM and 150 mM with a saturated solution of DB921. SAXS data were recorded (beamline BM29) on the maturing nanotube system. In concentrations of 30 mM or less of salt, formation of nanotubes was not observed even after 1 month of incubation; at concentrations of 50 mM or greater, nanotubes were formed within one month. Importantly, it was noticed that a critical concentration of NaCl (∼60 mM) exists beyond which the assembly rate rapidly increases across a very narrow (few mM) concentration range: 60 mM NaCl gave no tubes even after four hours, whereas 63 mM resulted in visible features corresponding to nanotubes in the SAXS data after 40 minutes (Fig. 5(a) and (b)). This remarkable acceleration in formation rate could allow a controlled triggering of assembly. At even higher concentrations of salt, the first visible effects were observed in as little as a few minutes after initiation (data not shown). This increasing assembly rate for concentrations of 63 mM to 150 mM and above are shown in Fig. 5(c) in which the shifting of the first SAXS peak with time is shown for different concentrations. It was found that above 110 mM, there was no further increase in assembly rate, showing saturation of NaCl in the system. | ||
| Fig. 5 SAXS intensities (BM29) of time-resolved nanotube formation at (a) 60 mM NaCl and (b) 63 mM NaCl against fixed DB921 concentration. The red arrow indicates the presence of assembled nanotubes after 40 minutes. Experiments were repeated several times with the same results. (c) The shift in the first SAXS peak to higher scattering vector over time for different NaCl concentrations. The curves shifting to earlier times indicates that the nanotube assembly proceeds more rapidly when the NaCl concentration is increased from 63 mM, up to around a maximum concentration of 110 mM. (d) Induction time vs. NaCl concentration, where the induction time was found through the time-dependent integrated SAXS intensity, fitted with the Avrami equation for nucleation and growth. | ||
Finally, the dependence of assembly rate on NaCl concentration is directly quantified by plotting the induction time as a function of NaCl concentration. Here, the induction time was found by fitting the time dependence of the integrated SAXS intensity over the entire Q range (IInt) with the Avrami equation used to describe polymer crystallisation,35 given by
| IInt = a(1 − exp{−[(t − τI)/b]n}). | (1) |
The fitting was performed with parameters a corresponding to the final plateau value of IInt, τI the corresponding induction time, b the growth-rate factor and n is a coefficient fixed to 1 in this case. As shown in Fig. 5(d), it can be seen that at its fastest assembly rate, the nanotube formation completes in approximately 5 minutes.
3.4 π–π Stacking of aromatic rings into nanotubes
The results described above in section 3.1 show that the mature nanotubes are thin-walled cylindrical objects that are assumed to be filled with largely disordered solvent molecules. X-ray diffraction studies of shear-aligned nanotubes were carried out, yielding rather poorly-aligned samples diffracting to limited resolution. Fig. 6(a) shows an X-ray diffraction pattern recorded from partially aligned nanotubes of DB921. The pattern shows a very pronounced diffraction peak at around 0.345 nm resolution, corresponding to a periodicity approximately perpendicular to the nanotube axis. We associate this feature with π–π stacking, likely to be parallel displaced geometry of the aromatic rings27,30,36 along the nanotube axis.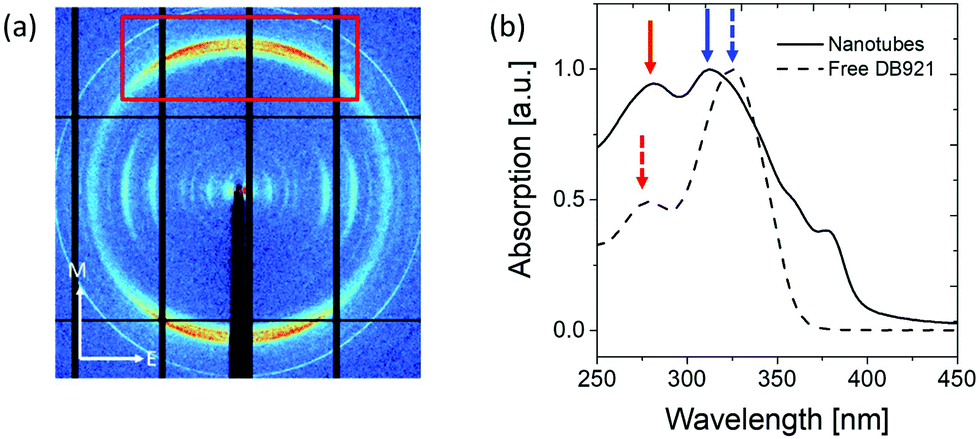 | ||
| Fig. 6 (a) X-ray diffraction pattern recorded from partially-aligned nanotubes. The long axes of the nanotubes are approximately aligned in the meridional (M) direction and perpendicular to the equatorial (E) direction. Strong diffraction at 0.345 nm resolution (red box) is observed. (b) UV-vis spectroscopy spectra on the unassembled DB921 molecules and assembled as mature nanotubes. Blue arrows indicate blue shift and red arrows indicate red shift in the absorption features. | ||
UV-vis spectra were obtained for the unassembled monomer and assembled nanotubes respectively as shown in Fig. 6(b). The characteristic absorptions of monomeric DB921 at 278 nm and 325 nm are found to be red-shifted to 283 nm and blue-shifted to 310 nm, respectively. Additional peaks are also noted at higher wavelengths of 359 nm and 378 nm. Similar spectral shifts are frequently observed in aggregated organic dye molecules which demonstrate face-to-face (H-type) or tail-to-tail (J-type) aggregation due to their high aromatic nature.37–40 A blue (hypsochromic) shift is attributed to H-type aggregation whilst a red (bathochromic) shift is characteristic of J-type aggregation. For the present system, a simple distinction between H- and J-type aggregation is likely to be insufficient given the complexity of the assembled DB921 molecules in the nanotube. However, the existence of both blue and red-shifted peaks suggests a combination of both H- and J-type configurations in the final nanotube, lending further evidence to the existence of π–π stacking interactions.
3.5 A possible structural model
The various structural characterisation techniques used in this study provide important insights to the nature and structure of these nanotubes. However, there are insufficient data to define a detailed molecular structure. The TEM results shown in Fig. 4 would suggest an overall spiral assembly. A possible consensus model for the chloride nanotube structure is illustrated in Fig. 7. The SAXS, SANS and TEM data define the overall shape and dimensions, as well as the assembly process, of the solvent-filled cylinder. It is suggested that the nanotube wall is primarily composed of a DB921 monolayer as deduced from SAXS, SANS and TEM values for the wall thickness and based on arguments of solvent interactions. Chloride ions condense into a diffuse profile on the inner and outer nanotube walls to compensate for the positive DB921 charge and providing further structural stability. It is acknowledged that, whilst Fig. 7 depicts a simplified radial arrangement of DB921 molecules within the wall, the true molecular orientation(s) may deviate from this via additional rotational degrees of freedom. | ||
| Fig. 7 Cartoon showing a possible structural model for the DB921 filaments. The top view shows side-by-side arrangement of DB921 mediated by intermolecular hydrogen bonds and diffuse halide counterions on the exterior and interior tube surfaces. The side view shows the π–π stacking (exagerated distance for purposes of clarity) tilted to the long axis of the tube, creating a spiral. The blue layer in the tube cross-section represents the bulk of the nanotube composed of aggregated DB921 molecules. The green layers represent diffuse halide counterions aggregated on the inner and outer nanotube walls for charge neutralisation. | ||
The diffraction and spectroscopy measurements suggest that the DB921 molecules stack in a regular way through π–π stacking of the aromatic rings. Together with the π–π stacking and the halide ion stabilising the assembly of DB921 in the nanotubes, each DB921 molecule has a hydrogen bond donor and acceptor symmetrically positioned on either side of the benzimidazole ring. Within the helix of side-to-side DB921 molecules this configuration could form N–H⋯N hydrogen bonds, further knitting together the assembly.41 Molecular curvature has been implicated previously in nanotube formation. A well-known example is the bent-core achiral molecules which form chiral liquid crystals.42,43 As noted earlier DB921 has an inherent curvature to the molecule that may be associated with its specific ability to form helical nanotubes described here from achiral building blocks.
3.6 Nanotube formation by similar minor-groove binding compounds
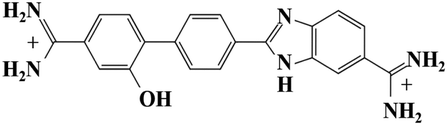
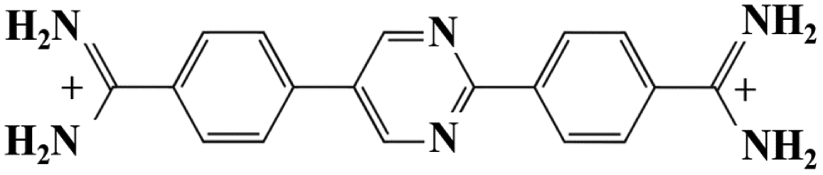
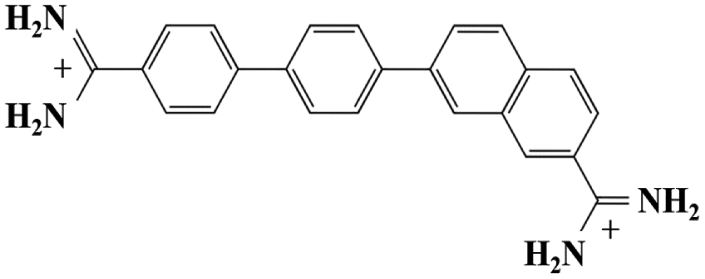
As mentioned in the introduction, DB921 is one of an extensive family of DNA minor-groove binding aromatic dications. In order to test the susceptibility of these compounds to form nanotubes, six similar compounds, as listed in Table 2, were tested using cryo-TEM under high (100–550 mM NaCl) salt conditions. Well ordered, uniform structures were not formed in any of the cases, showing that the ability to form nanotube structures appears to be related to the specific structure and configuration of the DB921 molecule (see ESI†).| DB985 |
|
| DB1963 |

|
| DB1242 |
|
| DB1055 |

|
| DB1883 |

|
| DB1177 |
|
Of particular interest in this series were compounds varying only by a single moiety on the aromatic ring structures. For example, the compound DB985 was examined. This molecule is identical to DB921 but with a hydroxyl group added at the ortho-position on the terminal phenyl ring. This suggests that the additional hydroxyl group sterically distorts the side-by-side alignment of the hydrogen-bonded molecules, preventing regular nanotube formation. Another example is DB1963 where one of the two amidinium groups is modified to be an amide, demonstrating the importance of the dication. A further example is DB1883 which is isostructural to the parent except for the absence of the imidazole nitrogen which is replaced with a carbon atom. This supports the hypothesis that the DB921 molecules are held together in a chain by N–H⋯N hydrogen bonds and that these are crucial in the formation of the regular nanotube structure.
4. Conclusions
Upon addition of NaCl, DB921 forms long, hollow nanotubes with an outer diameter and wall thickness of approximately 26 nm and 2 nm respectively, and lengths into the microns. A similar assembly is observed with the addition of NaBr, giving nanotubes with outer diameter of 32 nm. Nanotubes are not formed with other halogen group ions. The nature of the cation does not seem important to nanotube formation.Self-assembly is likely to be driven by a combination of electrostatic interactions, the formation of π–π stacking interactions and inter-molecular N–H⋯H hydrogen bonds, with additional halide ion stabilisation of the charged amidinium groups and controlling the curvature of helical ribbons.
The biophysical characterisation of these nanotubes has allowed a candidate structural model for the nanotubes to be presented. This is based on π–π stacked DB921 molecules, ordered in a helical fashion in the narrow walls of the filaments, with diffuse layers of halide ions screening the charged amidinium groups.
Time-resolved experiments demonstrate an elegant assembly process consisting of the formation of short fibril structures, which in turn associate to large-scale ribbons, in turn winding in a helical manner into the final hollow tubular structure.
It is noted that assembly is triggered by addition of chloride or bromide ions, with a change in size of the tubes according to the halide ion used. In recent years, the remarkable properties of self-assembled systems such as nanotubes have been manipulated for applications such as drug delivery and templating for bottom-up synthesis of nanoscale devices.44 The kinetic studies conducted have demonstrated that various parameters contribute to the assembly rates of the DB921 nanotubes, offering the promising possibility of controlled self-assembly. Adhesion of molecules to intermediate species prior to the completion of assembly may enable their encapsulation in the final tubules, allowing the function of a molecular- or drug-delivery system.
As previously mentioned, π–π interactions between aromatic rings appear to be highly prevalent in the final structure. Pi-conjugated molecules demonstrating extensive π–π stacking have been reported as promising organic semiconductors, in which the large degree of π-overlap enables effective charge transport.45,46 Our findings indicate that DB921 may be explored as a candidate for (opto)electronically active layers in devices for organic electronic applications.
Author contributions
The manuscript was written through the contribution of all authors. EPM/VTF conceived the study; all authors contributed to the overall study strategy and design. RM, JMD, JW, TN, AR, DM, EM, RS, EPM and VTF prepared nanotube samples and performed experimental work and X-ray and neutron analysis. WLL performed (cryo)-TEM work and analysis. DWB, WDW, AAF prepared the molecule. RM, SN, MH, TN, WLL, PR assisted in interpretation of structural results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the ESRF and ILL for the support of RM and JW during placements from Bath University (UK) to the EPN Campus (Grenoble, France). We acknowledge ESRF and ILL for access to X-ray and neutron scattering facilities. This work benefitted from SASView software, originally developed by the DANSE project under NSF award DM-0520547. This work used the platforms of the Grenoble Instruct-ERIC Centre (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The electron microscope facility is supported by the Rhône-Alpes Region, the Fondation Recherche Medicale (FRM), the fonds FEDER, the Centre National de la Recherche Scientifique (CNRS), the CEA, the University Grenoble Alpes, EMBL and the GIS-Infrastructures en Biologie Santé et Agronomie (IBISA). This research was supported, in part, by a US NIH grant (Grant No. GM111749) to WDW and DWB. This work was carried using the laboratory facilities of ILL's Life Sciences group – initially funded through EPSRC awards to VTF (grants EP/C015452/1, GR/R99393/01) for creation of the Deuteration Laboratory (D-Lab).References
- H. Li, J. D. Carter and T. H. E. LaBean, Mater. Today, 2009, 12, 24–32 CrossRef.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- G. A. Ozin, K. Hou, B. V. Lotsch, L. Cademartiri, D. P. Puzzo, F. Scotognella, A. Ghadimi and J. Thompson, Mater. Today, 2009, 12, 12–23 CrossRef CAS.
- J. V. Selinger, M. S. Spector and J. M. Schnur, J. Phys. Chem. B, 2001, 105, 7157–7169 CrossRef CAS.
- J. J. Cornelissen, A. E. Rowen, R. J. Nolte and N. A. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed.
- V. Castelletto, D. R. Nutt, I. W. Hamley, S. Bucak, C. Cenker and U. Olsson, Chem. Commun., 2010, 46, 6270–6272 RSC.
- F. Gobeaux, N. Fay, C. Tarabout, C. Mériadec, F. Meneau, M. Ligeti, D. A. Buisson, J. C. Cintrat, K. M. H. Nguyen, L. Perrin, C. Valéry, F. Artzner and M. Paternostre, J. Am. Chem. Soc., 2012, 134, 723–733 CrossRef CAS PubMed.
- Z. Shen, T. Wang and M. Liu, Chem. Commun., 2014, 50, 2096–2099 RSC.
- C. Valéry, F. Artzner and M. Paternostre, Soft Matter, 2011, 7, 9583–9594 RSC.
- W. Childers, A. K. Mehta, T. Q. Bui, Y. Liang and D. G. Lynn, Molecular Self Assembly: Advances and Applications, ed. A. D. Q. Li, Pan Stanford, 2012, ch. 1, pp. 1–36 Search PubMed.
- Y. Y. Liu, A. Kumar, S. Depauw, R. Nhili, M.-H. David-Cordonnier, M. P. Lee, M. A. Ismail, A. A. Farahat, M. Say, S. Chackal-Cateon, A. Batista-Parra, S. Neidle, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2011, 133, 10171–10183 CrossRef CAS PubMed.
- B. Nguyen, S. Neidle and W. D. Wilson, Acc. Chem. Res., 2009, 42, 11–21 CrossRef CAS PubMed.
- R. Nanjunda and W. D. Wilson, Curr. Protoc. Nucleic Acid Chem, 2012, ch. 8, 51:8.8:8.8.1–8.8.20 Search PubMed.
- Y. Miao, M. P. H. Lee, G. N. Parkinson, A. Batista-Parra, M. A. Ismail, S. Neidle, D. W. Boykin and W. D. Wilson, Biochemistry, 2005, 44, 14701–14708 CrossRef CAS PubMed.
- M. Munde, M. A. Ismail, R. Arafa, P. Peixoto, C. J. Collar, Y. Liu, L. Hu, M.-H. David-Cordonnier, A. Lansiaux, C. Bailly, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2007, 129, 13732–13743 CrossRef CAS PubMed.
- M. A. Ismail, A. Batista-Parra, Y. Mao, W. D. Wilson, T. Wenzler, R. Brun and D. W. Boykin, Bioorg. Med. Chem., 2005, 13, 6718–6726 CrossRef CAS PubMed.
- Y. Liu, Y. Chai, A. Kumar, R. R. Tidwell, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2012, 134, 5290–5299 CrossRef PubMed.
- P. Pernot, A. Round, R. Barrett, A. De Maria Antolinos, A. Gobbo, E. Gordon, J. Huet, J. Kieffer, M. Lentini, M. Mattenet, C. Morawe, C. Mueller-Dieckmann, S. Ohlsson, W. Schmid, J. Surr, P. Theveneau, L. Zerrad and S. McSweeney, J. Synchrotron Radiat., 2013, 20, 660–664 CrossRef CAS PubMed.
- M. E. Brennich, J. Kieffer, G. Bonamis, A. De Maria Antolinos, S. Hutin, P. Pernot and A. Round, Acta Crystallogr., Sect. D: Struct. Biol., 2016, D71, 67–75 Search PubMed.
- T. Narayanan, Structure from Diffraction Methods, ed. D. W. Bruce, D. O'Hare and R. I. Walton, John Wiley & Sons, Ltd, Chichester, 2014, ch. 5, pp. 259–324 Search PubMed.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-Rays, John Wiley & Sons, Inc., New York, 1955 Search PubMed.
- P. Linder and R. Schweins, Neutron News, 2010, 21, 15–18 CrossRef.
- LAMP, The Large Array Manipulation Program. http://www.ill.eu/data_treat/lamp/the-lamp-book/.
- D. Flot, T. Mairs, T. Giraud, M. Guijarro, M. Lesourd, V. Rey, D. van Brussel, C. Morawe, C. Borel, O. Hignette, J. Chavanne, D. Nurizzo, S. McSweeney and E. Mitchell, J. Synchrotron Radiat., 2010, 17, 107–118 CrossRef CAS PubMed.
- B. Jean, L. Oss-Ronen, P. Terech and Y. Talmon, Adv. Mater., 2005, 17, 728–731 CrossRef CAS.
- C. Valéry, M. Paternostre, B. Robert, T. Gulik-Krzywicki, T. Narayanan, J. C. Dedieu, G. Keller, M. L. Torres, R. Cherif-Cheikh, P. Calvo and F. Artzner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10258–10262 CrossRef PubMed.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed.
- A. Brizard, C. Aimé, T. Labrot, I. Huc, D. Berthier, F. Artzner, B. Desbat and R. Oda, J. Am. Chem. Soc., 2007, 129, 3754–3762 CrossRef CAS PubMed.
- N. Nakashima, S. Asakuma and T. Kunitake, J. Am. Chem. Soc., 1985, 107, 509–510 CrossRef CAS.
- Q. Hu, Y. Q. Wang, J. Jia, C. Wang, L. Feng, R. Dong, X. Sun and J. Hao, Soft Matter, 2012, 8, 11492–11498 RSC.
- G. J. Richards, J. P. Hill, J. Labuta, Y. Wakayama, M. Akada and K. Ariga, Phys. Chem. Chem. Phys., 2011, 13, 4868–4876 RSC.
- T. Shimizu, Bull. Chem. Soc. Jpn., 2008, 12, 1554–1566 CrossRef.
- D. Bong, T. Clark, J. Granja and M. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef PubMed.
- I. W. Hamley, Macromolecules, 2008, 41, 8948–8950 CrossRef.
- P. Panine, V. Urban, P. Boesecke and T. Narayanan, J. Appl. Crystallogr., 2003, 36, 991–994 CrossRef.
- P. Ma, Z. Bai, Y. Gao, Q. Wang, J. Kan, Y. Bian and J. Jiang, Soft Matter, 2011, 7, 3417–3422 RSC.
- A. Miyata, Y. Unuma and Y. Higashigaki, Bull. Chem. Soc. Jpn., 1991, 64, 2786–2791 CrossRef.
- U. Rösch, S. Yao, R. Wortmann and F. Würthner, Angew. Chem., Int. Ed., 2006, 45, 7026–7030 CrossRef PubMed.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef PubMed.
- N. C. Maiti, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1998, 102, 1528–1538 CrossRef CAS.
- W. Zieliński and A. Katrusiak, Cryst. Growth Des., 2013, 13, 696–700 Search PubMed.
- L. E. Hough, M. Spannuth, M. Nakata, D. A. Coleman, C. D. Jones, G. Dantlgraber, C. Tschierske, J. Watanabe, E. Körblova, D. M. Walba, J. E. Maclennan, M. A. Glaser and N. A. Clark, Science, 2009, 325, 456–460 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115(15), 7304–7373 CrossRef CAS PubMed.
- T. G. Barclay, K. Constantopoulos and J. Matisons, Chem. Rev., 2014, 114, 10217–10291 CrossRef CAS PubMed.
- A. N. Sokolov, T. Friščić and L. R. MacGillivray, J. Am. Chem. Soc., 2006, 128, 2806–2807 CrossRef CAS PubMed.
- K. Xiao, Y. Liu, T. Qi, W. Zhang, F. Wang, J. Gao, W. Qiu, Y. Ma, G. Cui, S. Chen, X. Zhan, G. Yu, J. Qin, W. Hu and D. Zhu, J. Am. Chem. Soc., 2005, 127, 13281–13286 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr03875e |
| This journal is © The Royal Society of Chemistry 2018 |