
Buried treasure: biosynthesis, structures and applications of cyclic peptides hidden in seed storage albumins

Abstract
Covering: 1999 up to the end of 2017
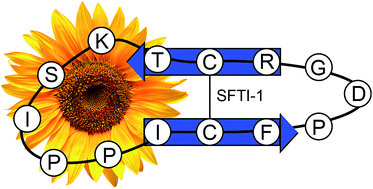
The small cyclic peptide SunFlower Trypsin Inhibitor-1 (SFTI-1) from sunflower seeds is the prototypic member of a novel family of natural products. The biosynthesis of these peptides is intriguing as their gene-encoded peptide backbone emerges from a precursor protein that also contains a seed storage albumin. The peptide sequence is cleaved out from the precursor and cyclised by the albumin-maturing enzymatic machinery. Three-dimensional solution NMR structures of a number of these peptides, and of the intact precursor protein preproalbumin with SFTI-1, have now been elucidated. Furthermore, the evolution of the family has been described and a detailed understanding of the biosynthetic steps, which are necessary to produce cyclic SFTI-1, is emerging. Macrocyclisation provides peptide stability and thus represents a key strategy in peptide drug development. Consequently the constrained structure of SFTI-1 has been explored as a template for protein engineering, for tuning selectivity towards clinically relevant proteases and for grafting in sequences with completely novel functions. Here we review the discovery of the SFTI-1 peptide family, their evolution, biosynthetic origin, and structural features, as well as highlight the potential applications of this unique class of natural products.
 Please wait while we load your content...
Please wait while we load your content...

