Recent progress in the isolation, bioactivity, biosynthesis, and total synthesis of natural spiroketals
Fu-Min
Zhang
a,
Shu-Yu
Zhang
b and
Yong-Qiang
Tu
*ab aState Key Laboratory of Applied Organic Chemistry and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: tuyq@lzu.edu.cn; tuyq@sjtu.edu.cn
Received
30th August 2017
First published on 22nd January 2018
Abstract
Covering: 2011 to July 2017.
Spiroketal (spiroacetal), a common moiety in numerous natural products, drugs and functional molecules, has been a central topic in organic chemistry for a long time. Owing to their structural diversity, important bioactivity and functional irreplaceability, natural spiroketals have attracted the interest of natural product chemists, medical chemists, biological chemists, agricultural chemists, synthetic chemists, and chemical biologists. In this review, we focus on the overview of the isolation, bioactivity, biosynthesis and total synthesis of spiroketals from 2011 to July 2017.
Fu-Min Zhang
Fu-Min Zhang received his Ph.D. from Lanzhou University in 2006, working under the supervision of Prof. Yong-Qiang Tu. Subsequently, he worked as a postdoctoral fellow at the Mayo Clinic with Prof. Yuan-Ping Pang (2007–2008). He returned to the Lanzhou University as a lecturer and was then appointed Associate Professor in 2009, and became a full Professor in 2014. Currently, he is interested in the total synthesis of natural products.
Shu-Yu Zhang
Shu-Yu Zhang received his B.S. (2002) and Ph.D. (2010) in Organic Chemistry from Lanzhou University under the supervision of Prof. Y.-Q. Tu. Following postdoctoral studies at Chicago University (2010–11) working with Prof. C. He, and Penn State University (2011–14) working with Prof. G. Chen, he began his independent career at Shanghai Jiao Tong University in 2015. Currently, his research is mainly focused on transition metal catalyzed C–H activation reactions and their application in the synthesis of natural products.
Yong-Qiang Tu
Yong-Qiang Tu received his B.S. and M.S. from Lanzhou University in 1982 and 1985, respectively. He obtained his Ph.D. in Organic Chemistry in 1989 from Lanzhou University under the supervision of Prof. Yao-Zu Chen. He spent three years (1993–95) as a postdoctoral fellow in W. Kitching's group at Queensland University, Australia. He became a full Professor at Lanzhou University in 1995, and was Director of the State Key Laboratory of Applied Organic Chemistry from 2001 to 2010. In 2009, he was elected an Academician of the Chinese Academy of Sciences.
1. Introduction
Spiroketal, also called spiroacetal, is a unique moiety that occurs in numerous natural products, drugs and functional molecules. Structurally, the spiroketal contains at least two oxacyclic rings, in which the oxygen atoms belonging to different rings share a common spiro-carbon atom. Spiroketal is designated as a substituted spirane base in systematic nomenclature or as an [m,n]-spiroketal in customary nomenclature. There are three common ring systems, 1,6-dioxaspiro[4.4]nonane (also [5,5]-spiroketal), 1,6-dioxaspiro[4.5]decane (also [5,6]-spiroketal), and 1,7-dioxaspiro[5.5]undecane (also [6,6]-spiroketal), while other ring systems are relatively seldom observed (Fig. 1).
Fig. 1 The common spiroketal ring systems.
Because of the inherent spiro-chiral centre in spiroketals, their diastereoisomers and enantiomers have special terms, and these issues have been extensively discussed in previously published reviews and books.1–6 In particular, the anomeric effect in the spiroketal system plays a critical role in its conformational analysis, its bioactive contributions and the design of total synthesis strategies. Generally, natural spiroketals have a stabilizing anomeric effect; however, exceptions have also been found in some spiroketal scaffolds due to the presence of substituents or an intramolecular H-bond.7
Due to their diverse ring structures and varied substituents, spiroketals exhibit wide and various bioactivities. For example, (R)-olean (1) is active in males, while (S)-olean (2) is active in females;8 avermectin A1 (3), along with its derivatives, is an important pesticide for the treatment of parasitic worms.9 Berkelic acid (4) shows broad bioactivities including the selective inhibition of OVCAR-3 (GI50 = 91 nM) and the pronounced inhibition of the matrix metalloproteinase MMP3 (GI50 = 1.8 μM).10 Leonuketal (5) exhibits vasorelaxant activity with EC50 = 2.32 μM,11 and ganoapplanin (6) displays inhibitory activity against T-type voltage-gated calcium channels.12 Interestingly, 21-hydroxy-oligomycin C (7) has proved to be an exceptionally potent inhibitor of K-Ras PM localization with an IC50 value of approximately 6 nM (Fig. 2).13
Fig. 2 Representative bioactive natural products containing the spiroketal moiety.
Although new chemical structures, the investigation of their corresponding activity, biosynthesis, and total synthesis of spiroketals have been reported in many articles and some reviews,14–16 no systematic collection and classification has yet been performed. In this review, we have systematically summarized the isolation, bioactivities and possible biosynthetic pathways of new natural spiroketals reported from 2011 to July 2017, and have highlighted recent representative examples of the biosynthesis and total synthesis of natural spiroketals during the same period.
The abovementioned spiroketal descriptions imply that each ring contains only one oxygen atom. Spiroketals having more than one oxygen atom17 or involving another heteroatom18 are omitted from this review.
Spiroketal skeletons also occur embedded in other structural moieties, including alkaloids,19 and steroids.20 Although these embedded spiroketals are structurally important units, they are also not included in this review.
2. Isolation, bioactivities and biosynthesis
The isolation of new spiroketals and the investigation of biological functions are still a growing field of natural product chemistry. In the past seven years, more than one hundred new spiroketals with unusual skeletons have been isolated from various sources including plants, animals, and microbes. In this section, we provide an overview of the isolation and bioactivities of “new” spiroketals according to a classification based on the size of the spiroketal ring, such as [5,5]-, [5,6]-, [6,6]-, and [5,7]-spiroketals. These spiroketals with different ring systems are tabulated below, and the tables include their names, source organisms, structural features, biological activities, and related references; they are then described individually according their chemical structures. Finally, the biosynthesis of some spiroketals is also discussed.21,22
2.1 [5,5]-Spiroketals
The newly isolated [5,5]-spiroketals are listed in Table 1, and their chemical structures are shown in Fig. 3.
Table 1[5,5]-Spiroketals isolated from natural sources
Fig. 3 The isolated new natural [5,5]-spiroketals.
2.1.1 Terpenes. Lanceolactone A (13), a bicyclic monoterpenoid bearing a butenolide, exhibited no significant effects on the growth of S. mitis, S. oralis, or S. sobrinus;27 its possible biosynthetic pathway is proposed in Scheme 1. The oxidation of nerolidol (13a) afforded an acid 13b, which was then cyclized to produce lanceolacetone A.
Scheme 1 The possible biosynthesis pathway for lanceolactone A.
Pleurospiroketals A–F (29–34) feature novel perhydrobenzannulated sesquiterpenes, and the position of the double bond and the absolute configuration of the spiroatom are the major differences.33,34 The assay of bioactivity against nitric oxide (NO) production in the lipopolysaccharide-activated macrophage cell RAW 264.7 revealed that pleurospiroketals A–C showed NO inhibitory activity with IC50 values of 6.8, 12.6, and 20.8 μM, respectively, and the preliminary structure–activity relationship analysis proved that the exocyclic double bond was beneficial for NO inhibitory activity. Additionally, pleurospiroketals A–C exhibited cytotoxicity against the HeLa cell line with IC50 values of 20.6, 32.8, and 18.8 μM, respectively. Compound 29a, derived from FPP, was proposed as the possible biosynthetic precursor of pleurospiroketals A–F (Scheme 2). It can be subsequently oxidized to introduce multiple oxygen-containing groups and to construct the multi-substituted furan 29b, then further transformed into pleurospiroketals A–F.
Scheme 2 The possible biosynthesis pathway of pleurospiroketals.
An assay of cytotoxic activities of epi-danshenspiroketallactone A (10) against five human cell lines (HCT-8, Bel-7402, BGC-823, A549, and A2780) revealed no significant activity.25 In the ongoing search for immunosuppressive agents from Chinese medicinal herbs, Yue and coworkers isolated phainanoids A–F (22–27), six structurally novel triterpenoids incorporating a unique [5,5]-spirocyclic system in 2015.31 Notably, phainanoid F (27) exhibited remarkable activities against the proliferation of T-cells (IC50 = 2.04 ± 0.01 nM) and B-cells (IC50 < 1.60 ± 0.01 nM). The unique structure and interesting immunosuppressive bioactivity of these spiroketals open a new window for the exploration of new lead compounds.
2.1.2 Polyphenols. Spiroketals bearing polyphenols are rare skeletons in isolated natural products. In 2012, Ito and co-workers reported two rare spiroketal-hexosefuranosides of acetophenone, upuborneols A (37) and B (38).37 Four years later, melicospiroketals A–E (14–18), five compounds with similar skeleton containing β–D-glucose moieties on their acetophenone rings, were isolated by Oh and coworkers.28 The major differences in these novel structures are in both the stereochemistry of the spiroatom and the various substituents on the tetrahydrofuran ring. In 2015, another flavonoid substituted analogue, pinnatifin E (28), was also identified by Li and coworkers.32
2.1.3 Polyketides. Opaliferin (19) features a novel C19 tetracyclic product containing a [5,5]-spiroketal fused with a cyclopentane connected to another tetrahydrofuran ring by an exo-double bond,29 and it shows weak cytotoxicity against the HSC-2, HeLa, and RERF-LC-KJ tumour cell lines. A plausible biosynthetic route to opaliferin is proposed in Scheme 3. Decarboxylation of the precursor (5S,6S,9R)-5,6,9-trihydroxy-3-oxodecanoic acid (19a) derived from the biotransformation of acetyl CoA and malonyl CoA followed by the addition of cephalosporolide B (19b) gave the intermediate (19c), which was cyclized through Claisen condensation to afford 2H-cyclopenta[b]oxepin (19d). The subsequent rearrangement, dehydration, and spiroketalization produced opaliferin.
Scheme 3 The possible biosynthesis pathway for opaliferin.
Spiroplakortone (35) features an unprecedented γ-spiroketal-γ-lactone moiety and it displays moderate cytotoxicity against the L5178Y cell line (IC50 = 37.5 μM).35 A plausible biosynthetic pathway of spiroplakortone was postulated (Scheme 4). An aldol reaction between β-ketoacid (35a) and α-ketoisocaproate (35b) derived from the biotransformation of leucine gave the adduct 35c, which was decarboxylated and then cyclized to afford spiroplakortone.
Scheme 4 The postulated biogenesis of spiroplakortone.
2.1.4 Benzoquinone. Capilloquinol (9) exhibited cytotoxicity against P-388 with an ED50 value of 3.8 μg mL−1.24 As depicted in Scheme 5, the author proposed a possible biosynthesis pathway for capilloquinol. The oxidation of a furan derivative (9a) afforded its lactone analogue (9b), which was subsequently cyclized to yield capilloquinol.
Scheme 5 The possible biosynthesis pathway for capilloquinol.
2.1.5 Miscellaneous. Aspersclerotiorone C (8) features a new spirofuranone-γ-butenolide;23 unfortunately, the trace amount limited further bioassays. To evaluate the bioactivities of (±)-japonones A (11) and B (12),26 four enantiomers, (+)-/(−)-japonones A (11a/11b) and (+)-/(−)-japonones B (12a/12b) were separated by a chiral preparative column, and the results indicated that (±)-japonones A and B exhibited no efficacy for the BACE1 inhibition and cytotoxicity assays (IC50 > 40 μM) and for the NO production inhibition assay (IC50 > 25 μM), while (+)-japonones A displayed potential anti-KSHV activity with lower toxicity and better selectivity due to both the key hydrophilic interaction with residues Lys 52, Asp 165, and Gly 167 and the hydrophobic interactions with Pro 56, Ile 101, and Va 199. Biosynthetically, acetate–acetate–propionate was first converted to the intermediate (11c) through methylation and decarboxylation, and 11c was geranylated and subsequently oxidized to afford the epoxide (11e). The proton-promoted cyclization of the resulting epoxide (11e) produced (±)-japonones A and B (Scheme 6).
Scheme 6 The possible biosynthesis pathway for japonones.
In 2015, Jung and colleagues described the isolation and identification of paeciloketal B (20) and 2-epi-paeciloketal B (21), which share a rare benzannulated [5,5]-spiroketal skeleton.30 The combination of the high-throughput screening system for polo box domain (PBD) inhibitors and the methodology of the fractionation of microbial metabolites with a spectral database is an alternative tool in the search for new natural products, and using this method, Osada and colleagues isolated a new 5,5-spiroacetal metabolite, trachyspic acid 19-butyl ester (36).36 Although this compound may be an artefact derived from trachyspic acid due to using butanol in the extraction process, it should be an attractive lead compound for polo-like kinase 1 (PlK1) inhibition.
2.2 [5,6]-Spiroketals
The newly isolated [5,6]-spiroketals are listed in Table 2, and their chemical structures are shown in Fig. 4 and 5.
Table 2[5,6]-Spiroketals isolated from natural sources
Fig. 4 The isolated new natural [5,6]-spiroketals.
Fig. 5 The isolated new natural [5,6]-spiroketals (continued).
2.2.1 Terpenes. In the ongoing research on Chinese herbal medicine for the treatment of inflammatory diseases, a new sesquiterpene spiroketal, phaeocaulisin A (56), was isolated by Qiu and co-workers.45 It showed strong inhibitory activity against NO production (IC50 = 1.5 μM). Two phaeocaulisin A analogues 5746 and 58,47 were also identified, respectively. Unfortunately, no related bioactivities were reported. Fourteen highly oxygenated norbisabolane sesquiterpenoid-type spiroketals, phyllaemblicins H1–H14 (60–73), were isolated by Zhang and coworkers.49 The major structural differences lay in the different sugar substituents. Phyllaemblicin H1 (60) exhibited moderate cytotoxicity against the human cancer cell lines A-549 (IC50 = 4.7 ± 0.7 μM) and SMMC-7721 (IC50 = 9.9 ± 1.3 μM). Phyllaemblicin H6 (65) displayed inhibitory activity against HSV-1, whereas phyllaemblicin H8 (67) and phyllaemblicin H12 (71) showed moderate anti-CVB3 cell activities.
2.2.2 Polyacetylenes. Polyacetylenes bearing a spiroketal skeleton constitute an important subunit that confers various bioactivities. In 2011, Shi et al. isolated two polyacetylene spiroketals, chrysindins C–D (44–45).40 The same year, six new diacetylenic spiroketals, lactiflodiynes A–F (47–52), were isolated by Ye and coworkers.42 Five years later, Luo and colleagues reported three new diacetylenic spiroacetal enol ethers, designated artemiselenols A–C (40–42).39 Three compounds were evaluated for neuroprotection activity and the inhibition of human monoamine oxidase (hMAOs) in vitro. Artemiselenols B (41) and C (42) exhibited good neuroprotection activity and inhibitory activity against hMAOs. The IC50 values of the former were measured to be 90 μM and 77 μM in hMAO-A and hMAO-B, respectively, and the IC50 values of the latter were 98 μM and 87 μM.
2.2.3 Polyketides. In 2014, Wakimoto and Abe isolated phosphocalyculin C (59), a pyrophosphate derivative of the known spiroketal calyculin C.48 The cytotoxicity investigation suggested that the cytotoxicity of phosphocalyculin C could be derived from the protoxin calyculin C.
Screening of the secondary metabolites produced by entomopathogenic fungi grown in the presence of epigenetic modifying agents is a possible method to find new secondary metabolite products. Using the abovementioned approach, Oshima isolated a novel spiroketal bearing an unprecedented tetracyclic ring system, named tenuipyrone (75).51 A plausible biosynthetic route is postulated (Scheme 7). Michael addition between 4-hydroxy-6-methyl-2-pyrone (75a) and cephalosporolide B (19b) would result in the enolated intermediate 75b. Claisen condensation, rearrangement, and spiroketalization would give tenuipyrone.
Scheme 7 The possible biosynthesis pathway of tenuipyrone.
2.2.4 Oxaphenalenone. Neonectrolide A (53), a new oxaphenalenone spiroketal, was isolated by Che et al.43 It showed moderate cytotoxicity against T24 (IC50 = 47 μM). Two possible biosynthetic precursors of neonectrolide A may be co-isolated: 3-dehydroxy-4-O-acetylcephalosporolide (53a) and corymbiferan lactone E (53b). The former was converted to a precursor (53c) through cycle-opening and oxidation, followed by the reaction with the hemiacetal (53d) derived from the latter to afford the ketone 53e. Intermolecular cyclization of the resulting ketone 53e gave neonectrolide A (Scheme 8).
Scheme 8 The possible biosynthesis pathway of neonectrolide A.
Very recently, Prado and coworkers isolated talaroketal A (74), a novel oxaphenalenone dimer.50 It features a rare benzannulated [5,6]-spiroketal and the dimeric bis(oxaphenalenone) skeleton, and it displayed modest antimicrobial activity against Staphylococcus aureus. The biosynthetic precursor of talaroketal A was proposed based on the co-isolation of duclauxin (74a), which possesses the same skeleton except for the spiroketal unit, and the substituted exo-enol (74d), derived from compound 74b, was proposed as another biosynthetic partner. The reaction of duclauxin 74a with enol 74d yielded the intermediate 74e, which was subsequently spiro-cyclized to produce talaroketal A (Scheme 9).
Scheme 9 The plausible biogenetic pathway of talaroketal A.
2.2.5 Macrolide. Acuminolide A (39) features a new 33-membered macrolide, two dihydropyrans, two tetrahydrofurans, one [5,6]-spiroketal consisting of a dihydroisobenzofuran and an octahydroisochromene, one epoxide, and a side chain containing a sulfated substituent.38 Interestingly, although acuminolide A was isolated from potent marine toxins, it did not show any cytotoxicity against four tested cancer cell lines; however, it displayed potent stimulation of actomyosin ATPase activity.
2.2.6 Miscellaneous. Very recently, Rukachaisirikul et al. isolated a γ-butenolide-type spiroketal, aspersclerotiorone F (43);23 however, no further bioactive investigation was reported, perhaps due to the limited amount of material isolated (1.5 mg).
Structurally, eleganketal A (46) is a rare naturally occurring spiroketal bearing a spiro[isobenzofuran-1,3′-isochroman] ring system.41
In 2015, paeciloketal A (54), a new benzannulated [5,6]-spiroketal bearing a sole chiral spiro-stereocentre, was isolated as a mixture of two enantiomers by Jung and colleagues.30 Interestingly, the authors found that a pair of enantiomers of paeciloketal A could interconvert when they were dissolved in methanol at room temperature, which indicated that the ketal formation was reversed. This case is a rare example of interconverting enantiomers among natural products. Paeciloketal A showed modest antibacterial activity (MIC = 40 μg mL−1) against the marine pathogen Vibrio ichthyoenteri.
Peniciketal C (55),44 a new [5,6]-spiroketal, along with two [6,6]-spiroketal analogues, peniciketals A (110) and B (111) were isolated. Peniciketal C (55) features a fully substituted phenyl ring bearing a rare [5,6]-spiroketal core and an oxa-bicyclo[3.3.1]nonane skeleton, and peniciketal C exhibited selective effects on HL-60 cells with an IC50 value of 4.5 μM. A plausible biogenetic pathway for peniciketal C was proposed (Scheme 10). The common precursor 55a, derived from the biotransformation of the acetyl malonyl and malonyl CoA, was methylated and cyclized to afford intermediate 55b. Compound 55b was further transformed into 55c, 55d, and 55e through different reaction pathways. Compound 55c produced the spiroketal 55f, which reacted with 55e to afford peniciketal C through electrophilic aromatic substitution and ketal formation. A similar approach was proposed for the biosynthesis of peniciketals A and B.
Scheme 10 The plausible biogenetic pathway for peniciketals A–C.
2.3 [6,6]-Spiroketals
The newly isolated [6,6]-spiroketals are listed in Table 3, and their chemical structures are shown in Fig. 6 and 7.
Table 3[6,6]-Spiroketals isolated from natural sources
Entries
Compound names/number
Isolation source
Structural elucidation
Structural classification
Bioactivity
Ref.
1
Alotaketals C, D and E (76–78)
Specimens of the sponge Phorbas sp.
HRMS (ESI), UV, 1D- and 2D-NMR
Sesterterpenoid
An activator of cAMP signaling in HEK293 cells; potent inhibitors of HIV
Fig. 6 The isolated new natural [6,6]-spiroketals.
Fig. 7 The isolated new natural [6,6]-spiroketals (continued).
2.3.1 Terpenes. In the search for potent inhibitors of HIV from natural sources, Andersen and coworkers isolated three sesterpenoid-type spiroketals, designated alotaketals C–E (76–78).52,53 Preliminary biological investigation showed that alotaketal C was an activator of cAMP signalling in HEK293 cells. In particular, in the extensive biological investigation of these compounds, alotaketal C was found to induce HIV proviral gene expression of J-Lat cells more intensely than prostratin, which is an attractive lead compound, albeit with higher toxicity than prostratin, while alotaketal D has the same biological function as prostratin. Further mechanistic investigation indicated that these compounds induce provirus expression through the PKC activation pathway, as does prostratin. These interesting activities and unique structure open a window for the further investigation of SAR, relative to the design and synthesis of derivatives as novel latency reversal agents for a “shock and kill” approach to alternative HIV-1 treatment. The polyene precursor 76a was cyclized to give the cyclohexene derivative 76b. It was oxidized at various allylic positions to afford the Michael acceptor 76c, which reacted with water, resulting in precursor 76d. Further cyclization and transformation gave alotaketal C (76) (Scheme 11). Additionally, its analogues, such as alotaketal D (77), alotaketal E (78) and phorbaketals, could be synthesized by a similar biotransformation approach.
Scheme 11 The possible biosynthesis pathway of alotaketals and phorbaketals.
In the investigation of biologically active metabolites from Red Sea marine organisms, Youssef and coworkers isolated four new spiroketals, i.e., didemnaketals D–G (83–86) in 2014.56,57 These new compounds are different from the previously reported didemnaketals A–C by Faulkner and coworkers,58 although the molecular skeleton was not obviously changed. In contrast to didemnaketal A and B,58 which exhibit anti-HIV activity, these compounds show no activity against HIV. However, didemnaketals D and E showed moderate activity against CDK5, CK1, DyrK1A, and GSK3 kinases and moderate antimicrobial activity against Staphylococcus aureus and Bacillus subtilis. Didemnaketal F displayed potent antimicrobial activity against E. coli and C. albicans and moderate activity against HeLa cells (IC50 = 49.9 μM), while didemnaketal G exhibited moderate activity against HeLa cells (IC50 = 14.0 μM).
In 2016, ganoapplanin (6) was isolated as a mixture of two enantiomers in unequal amounts and possesses an unprecedented dioxaspirocyclic skeleton containing a 6/6/6/6 tetracyclic system and an unusually flabellate tricycle motif.12 Biological activity studies showed that (±)-ganoapplanin (6), (+)-ganoapplanin (6a), and (−)-ganoapplanin (6b) exhibited moderate inhibitory activities against T-type voltage-gated calcium channels (TTCC) with IC values of 36.6 μM, 51.5 μM, and 56.4 μM, respectively. These findings suggested the strong possibility of a TTCC inhibitor from Ganoderma species. Its biosynthetic approach was shown in Scheme 12. Lingzhilactone (6c) was converted to its hemiacetal 6d, which was reacted with the gentisic acid derivative 6f to afford the spiroketal 6g. Subsequent diazotization reaction and Gomberg–Bachmann reaction gave the precursor 6l, and an intermolecular esterification of 6l furnished ganoapplanin.
Scheme 12 The possible biosynthesis pathway for ganoapplanin.
In the ongoing search for bioactive compounds from Korean water sponges, Shin and coworkers isolated three tetracyclic sesterterpenes spiroketals, i.e., gombaspiroketals A–C (87–89).59 They exhibited moderate cytotoxicity against the A549 and K562 cell lines, antibacterial activity against ATCC 6538p, ATCC 6633, ATCC 14028, ATCC 35270, NBRC 3851, and NBRC 12708, and weak inhibition of Na+/K+–ATPase and isocitrate lyase.
Leonuketal (5), a complex spiroketal diterpenoid, was isolated by Peng et al. in 2015.11 It features an unprecedented tetracyclic skeleton incorporating a bridged spiroketal moiety fused with a ketal-γ-lactone unit. It displayed significant vasorelaxant activity (EC50 = 2.32 μM) against KCl-induced contraction of the rat aorta. A possible biosynthesis pathway of leonuketal was proposed (Scheme 13). Water would react with an intermediate (5a) to give compound 5b, in which the double bond in the six-membered cycle could undergo selective cleavage to give triketone 5c. The spiroketalization and then selective reduction ketone of 5f could afford leonuketal.
Scheme 13 The possible biosynthesis pathway of leonuketal.
Phorbaketals D–K (114–121) and phorbaketals L–N (122–124) were isolated by Kang and coworkers in 2013 and in 2014, respectively.69,70 Phorbaketals E (115), F (116), H (118), and I (119) exhibited weak cytotoxicity against the A498 human renal cancer cell line, while phorbaketal N (124) showed potent cytotoxicity against human pancreas cancer cells.
2.3.2 Polyketides. In 2017, limaol (96), which features a 47-carbon skeleton bearing multiple hydroxyl and alkenyl groups and containing tetrahydropyran and octahydrospiro[pyran-2,2′-pyrano[3,2-b]pyran] cyclic moieties, was reported.62 In a bioassay investigation, limaol showed moderate cytotoxicity against the HepG2, HCT-116 and Neuro2a cancer cell lines.
Six rare reveromycin class spiroketals were isolated by Capon et al. using HPLC-DAD-MS and HPLC-DAD-SPE-NMR methodology, and named reveromycins H–M (125–130).71 Reveromycins K–M showed moderate antifungal activities.
In the search for the biosynthetic gene cluster of reveromycin A using reveromycin T as an intermediate, two new derivatives of reveromycin T, reveromycin T 1-methyl ester (131) and 1-ethyl ester (132), were obtained through adding alcohol to the culture broth of Streptomyces reveromyceticus SN-593.72 This alcohol-added fermentation may be a potent approach to obtain new metabolites of active products containing an acid group.
Sargassopenillines A–G (133–139), were isolated in 2014.73 Two years later, sargassopenilline H (140) was also ecludicated.74 Bioactivity investigation showed that sargassopenilline A (133) possessed radical scavenging activity against DPPH (IC50 = 100 μM), sargassopenilline C (135) inhibited the transcriptional activity of the oncogenic nuclear factor AP-1 (IC50 = 15 μM), and sargassopenilline E (137) exhibited cytotoxicity against splenocytes (IC50 = 38 μM) and radical scavenging activity against DPPH (IC50 = 30 μM). Sargassopenilline H (140) in non-cytotoxic concentrations inhibited the colony formation of the RPMI-7951 and MDA-MB-231 cell lines.
Three new [6,6]-spiroketal polyketides, named streptospirodienoic acids A (153) and B (154) and streptospirodienoate A (155), were isolated.77 Notably, the authors excluded the possibility that streptospirodienoate A, a methyl esterified derivative of streptospirodienoic acid A (153) at the terminal acid, was an artefact. However, streptospirodienoate A (155) showed moderate cytotoxic activity against HCT-116 cells (IC50 = 5.2 μM), while streptospirodienoic acids A and B showed no activity.
Ten new spirangiens, designated spirangiens G–P (141–150), were isolated by Kalesse and coworkers in 2015.75 Most of them showed significant cytotoxicity against the KB-3-1 cell line and against untransformed cells (L-929) in the relatively low nanomolar range.
In the process of isolating structurally novel metabolites and investigating their bioactivities based on the spectral database of the fraction library method, Osada and coworkers identified two new [6,6]-spiroacetal polyketides, spirotoamides A (151) and B (152).76 Based on the biosynthetic approaches to tautomycin and avermectin and some experimental results, a possible pathway was proposed (Scheme 14). Dihydroxyl ketone precursor (151a) was first converted into a thermodynamically and electrostatically preferred spiroketal (151b), which was transformed to spirotoamides A and B through amidation with carboxamide synthase and selective hydroxylation by P450.
Scheme 14 The possible biosynthesis route of spirotoamides A and B.
In the search for bioactive natural products from Thai microorganisms, Sadorn et al. isolated xerucitrinic acids A (161) and B (162).80 The preliminary bioassay indicated that xerucitrinic acid A exhibited moderate anti-Bacillus cereus activity (MIC 12.5 μg mL−1). Based on the citrinin moiety produced in various fungal genera, a plausible biosynthetic approach was proposed (Scheme 15). 1,6-Conjugate addition between citrinin (161a) and another precursor aedoxcitrinin (161b) gave the adduct 161c, which was selectively reduced and cyclized to afford xerucitrinic acids A and B.
Scheme 15 The possible biosynthesis pathway of xerucitrinic acids A and B.
In 2011, Ishibashi reported four new pyranonaphthoquinones, named (+)-griseusin E (90), (+)-methyl ester of 4′-deacetyl-griseusin B (91), (+)-4′-deacetyl-griseusin A (92) and (+)-4′-deacetyl-griseusin B (93).60 Four natural products showed significant ability to overcome tumour necrosis factor-related apoptosis-inducing ligand resistance in human gastric adenocarcinoma cell lines. One year later, two analogues, named griseusin F (94) and G (95), were isolated by Wen and Zhu.61 Both compounds feature a previously undescribed C23 polyketide skeleton. Griseusins F and G exhibited strong cytotoxicity against the B16, MDA-MB435S, CFPAC-1, ACHN, and HCT-116 human cancer cell lines (IC50 0.37–0.82 μM) and antibacterial activity against Staphylococcus aureus ATCC 29213 with MIC 0.91 and 0.80 μg mL−1, respectively.
As a new member of the griseusin family, yoropyrazone (163), was isolated by Ishibashi.81 In contrast to other griseusins, yorophenazone bears a usual naphthoiminoquinone, a naphthoquinone and an amide chain containing a vicinal diol, for which stereochemistries have not been determined. It is the first naturally occurring microbial naphthopyridazone. An investigation of the TRAIL resistance-overcoming activity of yoropyrazone in human gastric adenocarcinoma (AGS) cell lines showed moderate cytotoxic activity.
2.3.3 Macrolide. Two aplysiatoxin analogues, 3-methoxyaplysiatoxin (79) and 3-methoxydebromoaplysiatoxin (80) were isolated by Chu et al.54 The latter displayed significant anti-CHIKV activity with an EC50 value of 2.7 μM and a selectivity index of 9.2, and this is the first report of antiviral activity of an aplysiatoxin-type compound. Interestingly, the absence of bromine in 3-methoxydebromoaplysiatoxin (80) is a key factor for its antiviral activity, and the antiviral mechanism is likely the inhibition of CHIKV replication cycle.
Very recently, Sun reported the isolation and structural elucidation of two new avemectin analogues, 81 and 82;55 bioassay revealed that they exhibited moderate cytotoxicities in vitro against MG-63, B16, and Saos-2 cell lines.
Ras protein is important in cell growth, proliferation and differentiation, and inhibition of K-Ras plasma membrane localization would be an effective approach in the treatment of cancer. Therefore, the discovery and development of new chemical scaffolds to mislocalize oncogenic mutant K-Ras remain a challenge. Very recently, Capon and colleagues isolated two new 26-membered macrocyclic lactone oligomycin derivatives, 21-hydroxy-oligomycin C (7) and 40-hydroxy-oligomycin B (106).13 Preliminary bioactivity screening results confirmed that 21-hydroxy-oligomycin C and 40-hydroxy-oligomycin B are potent inhibitors of K-Ras and exhibit moderate cytotoxicity to colon cancer cell line SW620. These results pave the way for the development of new probes to treat K-Ras-related cancers.
Screening of the fermentation extract of strain NPS702 revealed nine new analogues of maclafungin, neomaclafungins A–I (97–105), which possess a 26-membered macrolide.63 The presence of alkane or alkanol branches on the [6,6]-spiroketal skeleton are the major differences in their chemical structures. Neomaclafungins A–I showed strong antifungal activity against Trichophyton mentagrophytes (ATCC 9533) with MIC values between 1 and 3 μg mL−1in vitro.
Gene replacement of polyketide synthase (PKS) could serve as an alternative approach to obtaining some biologically important natural products. Using this method, Zheng and coworkers discovered two new avemectin analogues, named tenvermectins A (156) and B (157).78 They showed better bioactivities against Tetranychus cinnabarinus and Bursaphelenchus xylophilus than avermectin, ivermectin, and milbemycin, respectively.
2.3.4 Miscellaneous. Peneciraistins A–B (107–108), two rare benzannulate [6,6]-spiroketals, along with peneciraistin C (109), were isolated by Liu and coworkers.64 The preliminary bioassay of peneciraistins A and B showed radical scavenging activities against DPPH (1,1-diphenyl-2-picrylhydrazyl) (IC50 = 38.9 and 42.7 μM, respectively). Peneciraistin C showed significant cytotoxicity against the A549 and MCF-7-60 cell lines (IC50 = 3.2, 7.6 μM, respectively). Notably, the extensive investigation of the anticancer activity of peneciraistin C showed that it could induce lung cell death through apoptotic and autophagic means; therefore, peneciraistin C could be a potential lead compound relevant to lung cancer.65 Later, Sobolevskaya's assessment of the bioactivity revealed that peneciraistin C inhibited the growth of developing oocytes in the sea urchin Strongylocentrotus intermedius (IC50 = 0.8 μM).66
In an ongoing effort to find structurally diverse natural products with interesting bioactivity, Zhu and coworkers isolated peniciketals A–B (110–111), two new [6,6]-spiroketals bearing a benzo-fused 2,8-dioxabicyclo[3.3.1]nonane moiety.44 Peniciketals A and B showed moderate cytotoxicity against HL-60 cells (IC50 = 3.2 and 6.7 μM, respectively). Their possible biosynthesis route is shown in Scheme 10.
In 2016, Zhang and coworkers reported the isolation of penicophenone A (112).67 A preliminary 11β-HSD1 assay of this compound was carried out, but unfortunately, no activity was observed. Its biosynthesis was proposed to proceed through a similar pathway to that of peniphenone A (see below).
Peniphenone A (113), bearing a rare benzannulated [6,6]-spiroketal, was isolated.68 Based on the co-isolated compound 2,4-dihydroxy-3,5-dimethylacetophenone (113a), a possible biosynthetic pathway was proposed (Scheme 16). 3,6-Dimethyl-4-hydroxy-2-pyrone derivative (113b), derived from a biotransformation of acetyl CoA, reacted with ortho-quinone methide 113d from 2,4-dihydroxy-3,5-dimethylacetophenone (113c) to afford the intermediate 113e, which further transformed to produce peniphenone A. A similar transformation could explain the possible biosynthesis of its analogues, such as penicophenone A and sargassopenillines A–G.
Scheme 16 The possible biosynthesis route of (±)-peniphenone A, penicophenone A, and sargassopenillines.
In 2011, Che and coworkers isolated three benzannulated spiroketals, virgatolides A–C (158–160).79 These novel compounds feature a 3,4,5,6-tetrahydrospiro[chroman-2,2′-pyran] core. Remarkably, virgatolide A contains two γ-lactone units, one spiralled with the spiroketal skeleton and the other fused with the substituted benzyl ring. Notably, virgatolide A is the first example of the unique benzannulated [6,6]-spiroketal bearing γ-lactone skeleton. The preliminary bioactivity investigation indicated that virgatolides A, B, and C exhibited modest cytotoxicity against HeLa cells with IC50 values of 19.0, 22.5 and 20.6 μM, respectively. Based on the co-isolated compounds pestaphthalide A and B, the biosynthetic pathway to these spiroketals was also proposed (Scheme 17). 3,6-Dimethyl-4-hydroxy-2-pyrone (158a) was reacted with demethyl pestaphthalide A (158c) and B (158d) to produce the spiroketals 158e and 158f, respectively. Subsequent cascade transformations of two intermediates yielded virgatolides B and C, while virgatolide A was derived from virgatolide B through oxidation/nucleophilic reaction/lactonization/reduction processes.
Scheme 17 The possible biosynthetic approach for virgatilodes A–C.
2.4 [m,n]-Spiroketals
The newly isolated [m,n]-spiroketals are listed in Table 4, and their chemical structures are shown in Fig. 8.
Table 4m,n-Spiroketals isolated from natural sources
Entries
Compound names/number
Isolation source
Structural elucidation
Structural classification
Bioactivity
Ref.
1
Ramariolide A (164)
The coral mushroom Ramaria cystidiophora
ESI-QIT-MS, HRMS (ESI), 1D- and 2D-NMR, XRD, Mosher's method
Ramariolide A (164), containing an unusual spiro-oxiranebutenolide moiety, was isolated.82 It displayed antimicrobial activity against Mycobacterium smegmatis and Mycobacterium tuberculosis in vitro. Along with ramariolide A, ramariolide B (165) was obtained by the same group.82 Structurally, it is the second natural product isolated that bears a spirooxetanebutenolide.
Tagitinins G (166) and H (167) were isolated by Sun and coworkers in 2012.83 Tagitinin G exhibited remarkable anti-hyperglycaemic activity by glucose uptake in 3T3-L1 adipocytes without significant toxic effects at 10 μg mL−1 concentration.
In the process of the structure elucidation of natural products using HPLC-SPE-NMR, which is superior to the classical preparative-scale fractionation of extracts originating from natural sources in some cases, Jaroszewski et al. isolated two metabolites, i.e., pestalospiranes A (168) and B (169).84 These compounds feature a rare benzo-[c]-oxepin skeleton, and their highly symmetric chemical structures were assigned by a combination of NMR spectroscopy and ECD spectroscopy.
2.5 Biosynthesis of spiroketals
2.5.1 Avermectins and analogues. To solve the mystery of the spiroketal formation process in the biosynthesis of avermectins, Liu and colleagues first revealed that AveC has dual functions in this important transformation based on numerous experimental results (Scheme 18).85 One is to stereospecifically catalyze spiroketalization of dihydroxyketone polyketide intermediate 170c, and the other is a regiospecific dehydration to produce precursor 170f. Therefore, structurally diverse avermectins could be easily obtained.
Scheme 18 Biosynthesis of avermectins.
In 2014, Liu and co-workers reported nine avermectin analogues (174–182), which were all derived from a common biosynthetic intermediate 171, from the triple-mutant S. avermectinius strain VL1004 (Scheme 19).22 Interestingly, the absolute configuration of spiroatom (C-21) in some analogues was S, which is different from the previously isolated natural avermectins (i.e. R configuration at C-21). The C-21 carbons of these analogues are in the S configuration, indicating they were produced via different biosynthetic pathways and have different bioactivities than those of the C-21 R-type natural products. Moreover, these bioactivity differences indicate that the furan core and the sugar moiety in the avermactins have an important role in the activity. One year later, the same group reported three new derivatives (183–185) of 1,19-seco-avermactin from the ΔaveCDE mutant S. avermetinius strain, which were deduced from the compounds 173 and 180, respectively (Scheme 19).86
Scheme 19 Biosynthesis of analogues of avermectin.
2.5.2 Olean and analogues. In 2011, De Voss proposed a biosynthetic approach towards some minor C12 and C13 spiroketals that had been identified as the volatile emissions of female Bactrocrea tryoni.21 First, the deuterium-labelled 2,6-, 3,7-, and 4,8-dioxygenated precursors and some deuterium-labelled monooxygenated precursors were prepared, and then these compounds were administered to female B. tryoni. Gas chromatograms of the solid-phase microextraction (SPME) analyses of the volatiles from B. tryoni were used to identify possible intermediates. Based on these results, a possible pathway was proposed. The fatty acid 186/187 was converted into diketone 192/193 either through four subsequent oxidation/decarboxylation steps or through two oxidation/decarboxylation/oxidation cycles, and then they were selectively reduced to the hydroxyketones 192a/193a. Finally, P450-catalysed hydroxylation of ketone at the 2- and 3-positions provided spiroketals 194 and 195, respectively, while the 2-hydroxylation of ketone 193b afforded (E,E)-196 (Scheme 20). Additionally, the biosynthesis of a spiroketal bearing a branched-chain was also proposed.
Scheme 20 Biosynthesis of olean analogues.
Three years later, based on the extensive experimental results, De Voss and colleagues proposed the possible biosynthetic pathway of olean and its derivatives in Bactrocera cacuminata.87 Starting from a fatty acid equivalent 196a, an enzyme-mediated C–H bond oxidation process at specific positions yielded a trioxygenated intermediate 196b with a vicinal diol moiety, which could be converted to key synthetic precursor 196c through oxidative C–C bond cleavage. This compound was transformed to olean through subsequent reduction, selective oxidation and cyclization, and the oxidation of the resulted olean afforded its derivatives (Scheme 21). Therefore, considering previously reported results, the complete biosynthetic pathway of olean and its spiroacetal derivatives was well-established. Notably, this oxidative transformation involving P450 was originally reported in insects.
Scheme 21 Biosynthesis of olean and its derivatives.
2.5.3 Reveromycin A. In 2011, Osada and coworkers discovered that RevG and RevJ are two key synthetic enzymes for the biosynthesis of reveromycin A (197) (Scheme 22).88 The former, a dihydroketone synthase, was involved in the dehydrogenation of the triol intermediate to yield the cyclization precursor keto-diol; the latter, a new spiroketal synthase, catalyzed the stereo-specific cyclization process to afford the natural S-configuration reveromycin A. This speculation was further verified by the corresponding nonenzymatic transformation of keto-diol in the presence of acid to produce both native S-configuration compound 197c and non-native 197d in a 3:2 ratio.
Scheme 22 Biosynthesis of reveromycin A.
2.5.4 Salinomycin. Very recently, Bai and coworkers reported that SlnM, a methyltransferase-like enzyme, has abilities to form the [6,6]-spiroketal core and to eliminate the allylic OH group at C-19 to introduce a double bond at C-18 and C-19 in the biosynthesis of salinomycin (198) (Scheme 23).89 Based on the experimental results and computational modeling, S-adenosylmethionine (SAM, 199) or sinefungin (200) was speculated to play the role of assistant by both stabilizing the SlnM's active conformation and enhancing the acidity.
Scheme 23 Biosynthesis of salinomycin.
3. Total syntheses of natural spiroketals
The total synthesis of natural spiroketals has been a promising field for synthetic chemists because these heterocyclic compounds possess unique structures and interesting biological activities, albeit with only small amounts, even trace amounts, isolated from nature. Consequently, access to numerous natural spiroketals has mainly relied on total synthesis via different strategies. In this section, some representative examples have been provided and correspondingly categorized into six subclasses. This review aims to emphasize the key steps for the total syntheses of spiroketals, especially synthetic methods for the construction of the spiroketal scaffold, and therefore, other chemical transformations will be discussed only briefly. Because some elegant syntheses of natural subclasses of spiroketal products have been recently reviewed by Brimble,90 Tong,91,92 Pettus,93 and Paterson,94 these previously reviewed synthetic approaches will not be introduced. Additionally, both the fragmentary synthetic studies on natural spiroketals and the preparation of natural spiroketal analogues will be neglected in this review.
3.1 Synthesis based on the classical cyclization of a dihydroxyketone or its equivalent to the key spiroketal moiety
3.1.1 Attenol A. In 2013, Yadav and coworkers developed a new TosMIC-derived spiroketalization to achieve the total synthesis of attenol A (201).95 Starting from iodide 202, two alkylations of TosMIC furnished the entire carbon skeleton (204), which was cyclized to provide the spiroketal precursor in 90% yield. Further removal of the Bn protecting group resulted in attenol A (Scheme 24).
Scheme 24 Total synthesis of attenol A by Yadav.
3.1.2 Proposed didemnaketal A. Didemnaketal A was isolated from an ascidian Didemnum sp. by Faulkner's group in 1991.58 The scarce natural source, rare structure and significant HIV-1 inhibition (IC50 = 2 μM) of didemnaketal A has attracted the interest of synthetic chemists. In 2012, the first total synthesis of the proposed didemnaketal A (205) was achieved by Tu's group (Scheme 25).96 Dihydroxyl ketone 206 was treated with the reagent NH4HF2, resulting in the desired the spiro-moiety compound 207 in 86% yield, by using an “internal” protection strategy of the keto-diol. More importantly, the MOM and ketal groups in compound 207 were retained in this transformation, and this key transformation is beneficial to the subsequent installation of various ester groups in the target molecule. TBS-protection of the secondary alcohol followed by the Suzuki coupling reaction gave the desired product 208 and simultaneously installed the chiral methyl group at the C-10 position in the side chain. The asymmetric dihydroxylation of 208 both introduced the C-7 and C-8 hydroxyl groups and self-protected the C-7 hydroxyl group as a lactone (209). Subsequent protection, reduction and Wittig reaction with ylide (212) provided compound 213 containing the total carbon skeleton of didemnaketal A. Selective repeated deprotections and esterification manipulations completed the total synthesis of the proposed didemnaketal A.
Scheme 25 Total synthesis of the proposed didemnaketal A by Tu.
3.1.3 EBC-23. In 2008, the first total synthesis of EBC-23 (214) was reported by Williams' group, and the chemical structure of EBC-23 was confirmed.97 Three years later, Yamamoto described an efficient synthesis of EBC-23 based on the development of a supersilyl-directed aldol reaction (Scheme 26).98 Starting from tetradecanal (215), ketone 216 was obtained through two aldol reactions, while aldehyde 217 was prepared from 3-hydroxy-1,4-pentadiene. With these two reaction partners in hand, the efficient condensation of 216 and 217 was tested. After a large number of attempts, compound 218 was obtained in 63% yield with high diastereoselectivity. Inspired by Williams' methods, alcohol 218 was subjected to a one-pot acylation/ring-closing metathesis/deprotection process, resulting in a mixture of anomers (219). This mixture was reacted with DDQ to afford EBC-23 with high enantioselectivity through deprotection/spiroketalization.
Scheme 26 Total synthesis of EBC-23 by Yamamoto.
3.1.4 Laidlomycin sodium salt. Very recently, Kang et al. reported the asymmetric total synthesis of laidlomycin sodium salt (220) with the longest linear sequence of 33 steps.99 Coupling aldehyde 221a and phosphonate 221b afforded an intermediate, which was subsequently desilylated, oxidized, and methylated to furnish the total carbon skeleton enone 222. Hydrogenation of the double bond and hydrogenolysis of the benzylidene and benzyl groups afforded the 1:1 mixture of spirocyclic products, which were isomerized to furnish spiroketal 223 with the expected stereochemistry in 79% yield over two steps. The removal of the protected groups completed the total synthesis of laidlomycin sodium salt (Scheme 27).
Scheme 27 Total synthesis of laidlomycin sodium salt by Kang.
3.1.5 Lysidicin A. The first total synthesis of (±)-lysidicin A (224) was effectively achieved in 15 steps with 3.5% overall yield.100 In the presence of water, Me3Al could effectively catalyse the expected Claisen rearrangement reaction at room temperature, resulting in the product 226 with excellent yield. Next, the acetylation of phenolic hydroxyl groups followed by the cleavage of the double bond by ozonolysis afforded the precursor 226a, which was deprotected and cyclized under acidic conditions to give the spiroketal 227 with 81% yield over two steps. Protection of the resulting phenolic hydroxyl group and installation of three isovaleryl groups on the aryl ring using Friedel–Crafts acylation promoted by AgOTf provided an efficient approach to obtaining two regioisomeric derivatives (228a and 228b) of lysidicin A. Both isomers were then separately subjected to the acid-catalysed isomerization of the spiro[furan–furofuran] ring system, resulting in (±)-lysidicin A in 73% and 63% yield, respectively (Scheme 28).
Scheme 28 Total synthesis of (±)-lysidicin A by Watanabe.
3.1.6 (−)-Marinisporolide C. The first total synthesis of (−)-marinisporolide C (229) was reported by the Dias group.101 The synthetic approach features highly stereoselective aldol reactions for the formation of five C–C bonds, the construction of six corresponding stereogenic centres and the cyclization of the macrocyclic ring by an intramolecular Horner–Wadsworth–Emmons olefination. Coupling sulfone segment 230 with aldehyde 231 under Julia–Kocienski olefination conditions afforded the desired compound 232 with high diastereoselectivity. The selective removal of the PMB group in compound 232 was followed by the enlargement of the carbon chain using the CM reaction and Takai reaction to provide iodide, which was further esterified to give the precursor 233. Classical HF acid promoted spiroketalization was performed to give a mixture spiroketal, followed by the protection of the released OH group to give 234 with 87% yield over two steps. Stille coupling with stannane segment 235 afforded the allylic alcohol, which was oxidized to furnish the aldehyde 236. Finally, under the Masamune–Roush conditions, the macrocyclization was completed using intramolecular Horner–Wadsworth–Emmons olefination to afford the macrolactone intermediate, which was deprotected using HF·pyridine and then treated with HCl to complete the total synthesis of the (−)-marinisporolide C (Scheme 29).
Scheme 29 Total synthesis of (−)-marinisporolide C by Dias.
3.1.7 (+)-Peniphenone A. The nine-step synthesis of (+)-peniphenone A (133) was completed by George and coworkers.102 The treatment of TBS ether 237 with LiTMP in the presence of HMPA generated the chiral (Z)-enolate intermediate 238, which underwent the Michael reaction with the o-quinone methide 239 generated in situ to afford the adduct 240. Tandem double desilylation/spiroketalization was completed under acidic conditions to furnish 242. Due to the effect of hydrogen bonding with the adjacent ketone at the aryl ring, the hydroxyl group at C-1 was less reactive than that at C-5, and the spiroketalization of diol 241 gave the desired regioselective product 242 in 23% total yield from 237. Moreover, the formation of a new spirocentre is also highly diastereoselective, as three chiral methyl substituents in the product are all in equatorial positions. Notably, the desired spiroketal with two stereocentres was efficiently prepared in two steps from the linear material 237, albeit in modest yield. Finally, the oxidation of 242 afforded (+)-peniphenone A (Scheme 30).
Scheme 30 Total synthesis of (+)-peniphenone A by George.
3.1.8 Virgatolide B. Virgatolide B (159) was synthesized for the first time by Brimble and coworkers in 2013.103 The Suzuki coupling of 243 with the aryl bromide 244 provided the desired amide 245, and subsequent removal of chiral template was followed by asymmetric dihydroxylation and then selective iodination to afford iodide 247. Subsequently, phthalide construction using Pd-catalysed carboalkoxylation and the key Mukayama aldol reaction using a temporary protection strategy afforded the hydroxyl ketone 248 with excellent diastereoselectivity. Finally, removal of three hydroxyl protection groups followed by treatment with a catalytic amount of CSA afforded the target molecule in 55% yield over two steps. Notably, another regioisomer of the product was not observed, which may be due to the presence of the intermolecular hydrogen bond between the phenolic hydroxyl group and carbonyl group of the phthalide core, decreasing the reactivity of the cyclic precursor (Scheme 31).
Scheme 31 Total synthesis of virgatolide B by Brimble.
3.2 Based on transition metal-promoted transformation to obtain key spiroketal moiety
3.2.1 Ascospiroketals A and B. Ascospiroketal A (249) was isolated from A. Isalicorniae by König and coworkers in 2007, and it represents a rare [5,5]-spiroacetal-containing family.104 However, the relative stereochemistry of its side chain and the absolute stereochemistry within the tricyclic core of the ascospiroketals remained unclear. In 2015, Britton and coworkers described an efficient approach to synthesizing ascospiroketal A and further confirmed its stereochemistry.105 The tricyclic core (252a/252b) of ascospiroketal A was conveniently constructed by a tandem silver(I)-promoted cascade cyclization developed by the author's group in 82% yield, albeit with low selectivity. However, the undesired isomer 252b could be easily transformed into the expected isomer 252a by the epimerization of the spiroketal centre. The conversion of the primary alcohol 252a into its corresponding carboxyl acid 253 was then achieved through a two-step oxidation, and vinyl iodide 254, a Sonogashira coupling partner in the next transformation, was obtained by NIS-promoted silicon–iodine exchange. Because the absolute configuration at C-15, C-2′ and C-3′ in natural ascospiroketal A was not determined in the original literature, four possibly coupled homopropargyl esters were prepared and reacted separately with iodide 254. After Lindlar reduction of the coupling enyne and careful comparison of the spectral data of the four synthetic isomers of ascospiroketal A with the data of the natural product, the absolute configuration for natural ascospiroketal A was completely clarified (Scheme 32).
Scheme 32 Total synthesis of ascospiroketal A by Britton.
3.2.2 EBC-23. In 2016, a gold-catalyzed spiroketalization toward EBC-23 (214) was reported by Yadav (Scheme 33).106 The coupling of terminal alkyne 255 with aldehyde 256 provided a mixture of propargylic alcohol (257a and 257b), and 257b could be easily transformed into 257a through the Mitsunobu reaction. Protection of the resulting secondary alcohol and selective removal of the TES group afforded the precursor 258, and Au-catalysed cyclization provided the spiroketone 259 in 62% yield under the revised Trost procedure. Involvement of the hydroxyl group at the C-5 position through a 5-exo-dig cyclization afforded the enol ester 258a, which was reacted with methanol to furnish ketol 258b. The hydroxyl group at the C-13 position attacked the oxocarbenium ion 258c to provide the desired spiroketal skeleton 259. Finally, the construction of a six-membered lactone completed the total synthesis of EBC-23 (Scheme 33).
Scheme 33 Total synthesis of EBC-23 by Yadav.
3.2.3 (E)-2-Ethyl-1,7-dioxaspiro[5.5]undecane. (E)-2-Ethyl-1,7-dioxaspiro[5.5]undecane (260) was reported previously as a component of the cephalic secretions of a kleptoparasitic bee, and its first total synthesis was reported by the De Voss group.107 Very recently, Carreira and coworkers described a simple and efficient synthesis of this natural product based on their newly developed novel Ir-catalysed enantio- and diastereo-selective spiroketalization process.108 A racemic hydroxylketone bearing allylic ester (261) was treated with chiral Ir catalyst and Zn(OTf)2 to afford the spiroketal 262 with 98% ee, 93% yield, and >25:1 dr through ring-chain tautomerism/mutarotation approach. Hydrogenation of the resulting product gave the target molecule 260 in 90% yield (Scheme 34).
Scheme 34 Synthesis of (E)-2-ethyl-1,7-dioxaspiro[5.5]undecane by Carreira.
3.2.4 α-Levantanolide and α-levantenolide. Recently, Dai and coworkers successively synthesized two labdanolic diterpene spiroketals, α-levantanolide (263) and α-levantenolide (264),109 based on a catalytic carbonylative spirolactonization method in two and four steps, respectively. The commercially available (3aR)-(+)-sclareolide (265) was first converted to cyclopropanol 266, which was subjected to optimal conditions to result in the natural α-levantanolide (263) with 30% yield and two isomers of the lactone (267a/267b) in 23% yield. The introduction of a CC double bond into the lactone ring was achieved by the treatment of a mixture of α-levantanolide and other isomers with phenylselenyl chloride, followed by oxidation with H2O2 to provide α-levantenolide (264) as a single product in 46% yield in two steps. The structure of α-levantenolide was further confirmed by X-ray diffraction (Scheme 35).
Scheme 35 Total synthesis of α-levantanolide and α-levantenolide by Dai.
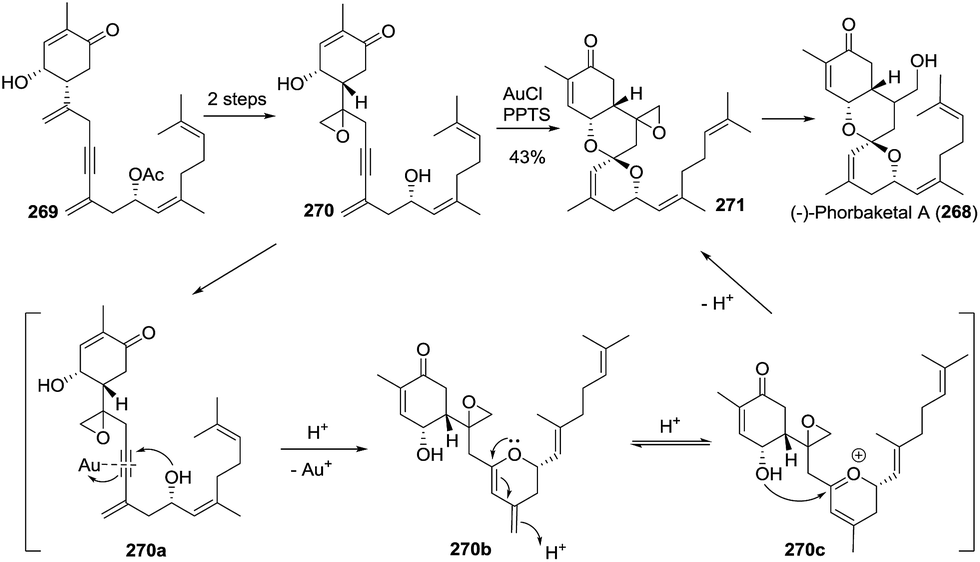
3.2.5 (−)-Phorbaketal A. Very recently, Lee and coworkers described a convergent asymmetric total synthesis of (−)-phorbaketal A (268) through the Au-catalysed intramolecular spiroketalization reaction.110 Starting from alkyne 269, hydroxyl group directed epoxidation and deprotection of the Ac group gave the diol 270, and subsequent Au-catalysed spiroketalization under acidic conditions furnished the compound 271 in 43% yield. These mild reaction conditions did not impact the sensitive functional groups such as epoxy, double bonds, and conjugated ketone. The hydroxyl group in the linear region attacked the Au-activated alkyne 270a through an endo-cyclization, which favored the hydroxyl group on the six-membered ring. Acid-catalysed isomerization of diene 270b afforded the oxocarbenium intermediate 270c, which was trapped by the hydroxyl group to provide the desired precursor 271. The opening of epoxide 271 by the promotion of BF3·Et2O afforded (−)-phorbaketal A (Scheme 36).
Scheme 36 Total synthesis of (−)-phorbaketal A by Lee.
3.2.6 Schindilactone A. In 2011, the first total synthesis of schindilactone A (272), which represents a class of biologically important, structurally sophisticated and synthetically challenging nortriterpenoids, was achieved by Yang and coworkers.111 Hemiketal 273 was transformed to enyne 274, which was subjected to the optimal Pauson–Khand reaction conditions in the presence of the complex of tetramethyl thiourea (TMTU) and Co2(CO)8, resulting in the spiroketone 275 in 74% yield. The ring-opening of spiroketal 275 and some subsequent chemical conversions afforded allylic alcohol 276, which was re-spiroketalized through thiourea/Pd-catalysed carbonylative annulation developed by this group to give intermediate 277 in 78% yield. The introduction of a methyl group, construction of the A ring by Dieckmann condensation and deprotection completed the total synthesis of (±)-schindilactone A in a longest linear sequence of 29 steps (Scheme 37).
Scheme 37 Total synthesis of (±)-schindilactone A by Yang.
3.3 Based on organocatalysed transformation to obtain key spiroketal moiety
The organocatalysed asymmetric synthesis of spiroketals, especially for the corresponding synthetic precursors without active groups to interact with the reaction catalyst, has been a great challenge and remains an unclear topic in organic synthesis.112 Delightfully, this tricky issue was solved by List and co-worker in 2012. They applied the exceptionally bulky chiral imidodiphosphoric acid as an organic catalyst to induce allylic ether to form planar oxocarbenium ions and then interact with an intermediate through a rigid counter ion pair approach, resulting in the highly stereoselective construction of spiroketals.8
3.3.1 Chalcogran. In 2015, Matsubara et al. discovered a novel tandem intramolecular hemiacetalization/oxy-Michael addition promoted by a bifunctional aminothiourea catalyst, resulting in a chiral spiroketal skeleton with high enantioselectivity.113 Based on this novel method, 2S,5S-chalcogran (278), a pheromone, was conveniently asymmetrically synthesized in three steps from 279 (Scheme 38).
Scheme 38 Total synthesis of 2S,5S-chalcogran by Matsubara.
3.3.2 Olean. Olean, one of the simplest [6,6]-spiroketals isolated from the olive fruit fly, exhibits remarkably selective activity for the different sexes. Based on the abovementioned strategy, an enantiomeric pair of oleans was prepared by List from the same starting material (282).8 In contrast to the previous approach involving either chiral starting materials and reagents or chiral separation of a racemic mixture, the current approach features high enantioselectivity and excellent yield. Importantly, other analogues of olean were also prepared with a similar approach using different substrates (Scheme 39).
Scheme 39 Total synthesis of (R)- and (S)-olean by List.
3.4 Based on the cycloaddition reaction to obtain the key spiroketal moiety
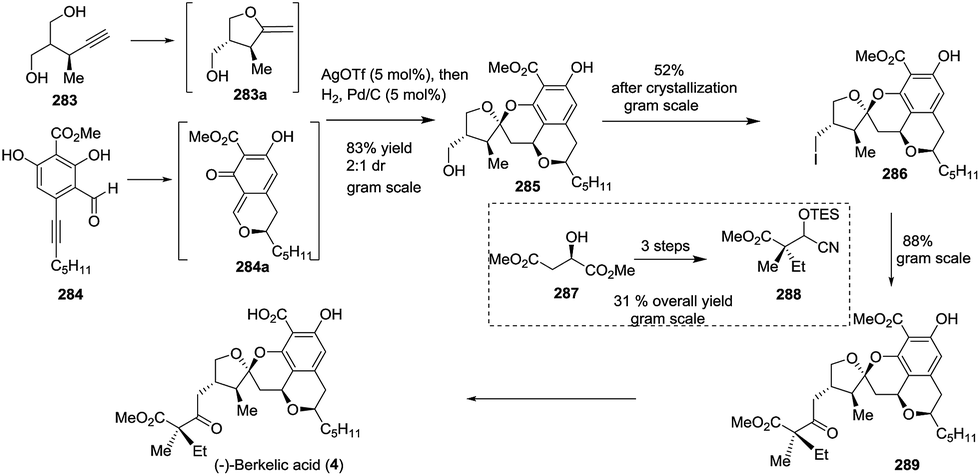
3.4.1 (−)-Berkelic acid. (−)-Berkelic acid (4), a biologically active and architecturally unique secondary metabolite, was isolated in 2006 by Stierle and coworkers. Although several synthetic approaches have been reported, scalable synthesis remains challenge. In 2012, a practical and scalable route to (−)-berkelic acid was achieved by Fañanás and coworkers.114 This novel approach featured a new silver-catalysed reaction that allowed the construction of a spiroketal core bearing five chiral centres in just one step. Notably, the approach to synthesizing (−)-berkelic acid was protection-free and was conducted on the gram scale, except for the last step. The formation of an exocyclic enol ether (283a) and an o-quinonemethide (284a) in situ was followed by the formal cycloaddition reaction between two intermediates promoted by AgOTf. Hydrogenation of the resulting carbon–carbon double bond afforded the mother core of the natural product, 285, with 83% yield and 2:1 dr value, containing four rings with five stereocentres, in only one step, and produced 2.4 g of the isolated product. To introduce the lateral chain and avoid a tedious oxidation process, the new umpolung synthesis strategy was applied. The treatment of the primary iodide 286, which was obtained from alcohol 285 in gram scale, with cyanohydrin 288 afforded the precursor 289, of which the benzylic ester was removed to complete the total synthesis of (−)-berkelic acid in a seven-step linear sequence (Scheme 40).
Scheme 40 Total synthesis of (−)-berkelic acid by Fañanás.
3.4.2 Bistramide A. In 2014, Floreancig and coworkers described the shortest reported approach to synthesizing bistramide A (290) through a one-pot hetero-Diels–Alder reaction/oxidative cleavage of carbon–hydrogen bond/spiroketalization.115 Olefin 291 was first transformed to yield diene 292, which was then reacted with aldehyde 293 in the presence of Jacobsen's catalyst 294 to afford the adduct 292a, which was directly treated with DDQ without purification and then with PTS to provide the spiroketal compound 295 as a single stereoisomer. Enlargement of the side chain and the installation of some stereocentres gave azide, which was reduced to a primary amine 296 and subsequently condensed with another partner 297 to afford bistramide A. Notably, a longest linear sequence of 14 steps sequence was needed to achieve the target molecule from the commercially available materials (Scheme 41).
Scheme 41 Total synthesis of bistramide A by Floreancig.
3.5 Based on the rearrangement reaction to obtain the key spiroketal moiety
3.5.1 Cereoanhydride. Cereoanhydride (298) was isolated from green algae as a new secondary metabolite; its original chemical structure (299),116 bearing an unusual seven-membered cyclic anhydride and a fused tetrahydrofuran hemiacetal moiety, was assigned by König and coworkers in 2012. Its structure was revised by Hu and coworkers based on the total synthesis of its spiroketal isomer 298.117 Starting from the commercially available compound 300, two selective additions and the subsequent protection of the resulting tertiary alcohol to its acetal derivative gave cyclobutanone 301. The introduction of an Ac group efficiently inhibited the oxidation of ring-expanded phenol product and increased the yield of the product 302 in the cascade ring expansion/hemiketalization process. The hemiketal 302 was twice reduced and then oxidized with CAN to furnish the diketone 303. The total synthesis of cereoanhydride was completed by the Baeyer–Villiger oxidation of ketone 303, resulting in the anhydride 304. A subsequent hydration process proceeded through the proposed structure 304 as an intermediate under acidic conditions to afford cereoanhydride 298 in 30% yield over two steps. The synthetic product 298 was further verified by X-ray analysis, resulting in the unequivocal reassignment of the natural product (Scheme 42).
Scheme 42 Total synthesis of cereoanhydride by Hu.
3.6 Based on other novel processes to obtain the key spiroketal moiety
3.6.1 (−)-Alotaketal A. (−)-Alotaketals A–D and phorbaketals A–K, structurally novel spiroketal sesterterpenoid natural products isolated from different sources, exhibited broad-spectrum bioactivities. Therefore, total synthesis of these subunit products attracted the interest of synthetic chemists.91,92
The first total synthesis of (−)-alotaketal A (306) was reported by Yang and coworkers in 2012.118 The coupling of lactone 307 (prepared from (R)-carvone) and allylic iodide 308 (prepared from ethyl acetoacetate) was carried out under Barbier conditions to afford the desired hemiacetal 309, which was desilylated using TBAF and then cyclized using PPTS to give the expected spiroketal 311 as a sole product with 40% yield over three steps. The author speculated that the electron-withdrawing inductive effect of PMB at the C-22 position is a key factor. The oxidation of allylic alcohol 311 followed by the removal of the PMB group from the compound furnished alotaketal A (Scheme 43a).
Scheme 43 Three approaches to (−)-alotaketals and (−)-phorbaketal A.
In the same year, Dalby and coworkers described a convergent approach towards (−)-alotaketal A.119 The key LiDBB-mediated coupling of the chiral lactone 312 with allyl bromide 313 afforded the hemiacetal 314, which was subjected to HF·pyridine buffered solution to give the spiroketone 315 with 55% yield, and the spirocentre was efficiently constructed through the anomerically stabilized configuration. The subsequent selective removal of the TIPS group/oxidation/selective reduction completed the total synthesis of (−)-alotaketal A (Scheme 43b).
Very recently, Tong and coworkers described a novel synthetic strategy for the collective synthesis of (−)-alotaketals A–D and (−)-phorbaketal A.120 The sequential treatment of 316 with HF-pyridine and DMDO afforded the epoxide 317, which was subjected to TfOH to promote the designed cascade reaction of vinyl epoxy δ-ketoalcohols, resulting in the desired tricyclic spiroketal 318 in 56% yield on a 4.5 g scale, along with the minor isomer. Interestingly, the isomerization product of the exo-double bond was not observed. The adjustment of the oxidation state and the installation of some functional groups afforded the common intermediate 319, which was transformed to (−)-alotaketals A (306), B (320), C (94), D (95) and (−)-phorbaketal A (243) based on different transformations (Scheme 43c).
3.6.2 Bistramide A. In 2012, Goekjian and coworkers reported an approach toward the total synthesis of bistramide A.121 Julia coupling of lactone 321 with sulfone 322 and subsequent elimination afforded enol ether 323, which was cyclized to furnish spiroketal 324 in 69% yield over two steps. Amine 325 was prepared through multi-step transformation from 324, followed by deprotection of the Fmoc group and subsequent condensation with activated ester 326 to afford bistramide A (Scheme 44).
Scheme 44 Total synthesis of bistramide A by Goekjian.
3.6.3 Didemnaketal B. In 2013, the Fuwa group described the total synthesis of the proposed didemnaketal B (327), further confirming that the structure of didemnaketals needed to be revised.122 One year later, the revised structure of didemnaketal B (328) was synthesized by the same group.123 Coupling iodide 329 and ester 330 afforded the intermediate 331, and deprotection of the silyl ethers within 331 with TBAF resulted in the dihydroxy enol ether, which was treated with PPTS under thermodynamic conditions to afford the desired spiroacetal 332 in 80% yield (two steps) as the sole isolable product (dr > 20:1). The enlargement of a side chain, installation of ester, deprotection and oxidation gave the C1–C21 aldehyde segment 333, which was coupled with the C22–C28 side chain under Nozaki–Hiyama–Kishi (NHK) reaction conditions to complete the total synthesis of the proposed didemnaketal B, 327. Unfortunately, the spectral data of the synthetic didemnaketal B did not match the spectral data of the reported didemnaketal B, 328. After careful examination of the original literature and comparison of the spectral data of the key intermediate to elucidate the chemical structure of didemnaketal B, the author proposed that the absolute configuration of the C10–C20 spiroketal core might be erroneously assigned. Therefore, starting from ent-329 and ent-330, the natural didemnaketal B was synthesized by following a similar approach toward the proposed didemnaketal B, resulting in the real structure of the didemnaketal B (Scheme 45).
Scheme 45 Total synthesis of didemnaketal B by Fuwa.
3.6.4 Dinemasone A. In 2012, a convenient synthesis of dinemasone A (335) was reported by Sharma.124 Alkyne 336 was treated with BuLi and then reacted with aldehyde 337 to afford the propargylic alcohol, which was oxidized to give the ynone 338. Double oxa-Michael addition under acidic conditions afforded the monoanomeric spiroketal 339 with 79% yield through the 6-endo-dig approach. The formation of the axial-equatorial isomer 339 is favoured over that of the axial–axial isomer 340 because the former avoids the steric interaction between an axial hydrogen and a hydrogen on a methyl group. The monoanomeric spiroketal 339 was then transformed into the more stable double anomeric spiroketal 340 with 35% yield, followed by hydrogenation to give dinemasone A (Scheme 46).
Scheme 46 Total synthesis of dinemasone A by Sharma.
3.6.5 Griseusin A, 4′-deacetyl-griseusin A, and griseusin C. Very recently, Thorson and coworkers reported the first enantioselective total synthesis of three griseusin family products in a concise and divergent approach.125 The compound 342 was prepared from commercially available 1,5-dihydroxynaphthalene (341), which was then subjected to revised Sharpless conditions, resulting in the desired product 343. The screening of hydroxyl-oriented olefination conditions found that compound 345 and compound 346 were obtained in almost equivalent yield; fortunately, 346 could be easily transformed into the desired product 345 through two-step manipulation. Subsequently, the designed tandem reaction was investigated. The removal of TBS ether and treatment of the resulting intermediate 347 with DMDO and TfOH in DCM efficiently afforded the common precursor 348 in 80% yield and >49:1 dr through TfOH-controlled cascade si-facial selective epoxidation/cyclization. The precursor 348 was concisely transformed into the natural griseusin A (349), 4′-deacetyl-griseusin A (113), and griseusin C (350) in a 1–3 step chemical transformation (Scheme 47).
Scheme 47 Total synthesis of griseusin A, 4′-deacetyl-griseusin A, and griseusin C by Thorson.
3.6.6 (−)-Halichondrin C. In 2012, Kishi and coworkers completed the first total synthesis of (−)-halichondrin C (351) based on the iterant catalytic asymmetric NHK reaction for the construction of the carbon skeleton of the target molecule.126 In these procedures, the authors found that the sulfonamide ligand could improve the efficiency of the NHK reaction. The NHK coupling product of 352 and 353 was oxidized to afford the enone 354, which was then subjected to TBAF, DDQ and PPTS, resulting in the removal of five TBS groups, the formation of the hemiketal group, and spiroketalization through intermolecular oxa-Michael addition to construct the [6,6]-spiroketal core 355. The deprotection of the MPM group and cyclization by PPTS were then performed to construct another [5,5]-spiroketal core in the target molecule, producing the precursor 356 in 30% yield over 3 steps. The removal of the allylic group of precursor 356 afforded the natural product (−)-halichondrin C (Scheme 48).
Scheme 48 Total synthesis of natural (−)-halichondrin C by Kishi.
3.6.7 Pestalospiranes A and B. Based on the bioinspired approach proposed by Jaroszewski et al., Brimble and coworkers completed the first asymmetric total synthesis of two enantiomers of pestalospirane B (194) using a tandem dimerization/spiroketalization reaction as the key step.127 Starting from phenylacetylene derivative 357, the reaction with Weinreb amide and complete hydrogenation of the acetylene bond of the resulting ynone gave ketone 358, followed by acid-catalysed cyclization to furnish the chiral alcohol 359. Subsequent oxidation of compound 359 afforded the ketone 360; unfortunately, the stereocentre in compound 360 was lost in the oxidative process. An enantioselective reduction of ketone 360 was therefore investigated, resulting in a mixture of the expected precursor 361 and another byproduct 362. This mixture was directly subjected to trifluoroacetic acid in DCM without purification and thereby afforded (+)-pestalospirane B, (+)-pestalospirane A and another unnatural isomer, albeit with low ee of 20%. Further optimization of the enantioselective reduction was achieved using the combination of (R)-Me-CBS and BH3·diethylaniline, resulting in the precursor 363, which was cyclized to afford (−)-pestalospirane B with 97% ee and 49% yield. The absolute stereochemistry of natural pestalospirane B was confirmed by spectral analysis as well as the total synthesis of another enantiomer and X-ray structure of its p-bromobenzoyl derivative 364. Therefore, the originally assigned absolute configuration of pestalospirane B was revised. Notably, pestalospirane A (193), an isomer of pestalospirane B, was also synthesized for the first time, albeit with low yield (Scheme 49).
Scheme 49 Total synthesis of (−)-pestalospirane B by Brimble.
3.6.8 Ramariolides A and B. In 2017, Nanda and coworkers described the first asymmetric total synthesis of ramariolide A (189).128 The key reaction features bimetallic cascade cyclization and Z–E isomerization. Starting from the chiral alkynol 365, which was obtained through the combination of the CAL-B enzyme and Ru catalyst from its racemic form in two steps, the Pd/Cu catalysed tandem Sonogashira-type cross-coupling reaction/5-exo-dig cyclization reaction with (Z)-3-bromoacrylic acid (366) afforded the unprecedented isomerization of γ-(Z)-butenolide 367. The transformation of γ-(Z)-butenolide into the synthetically required, but thermodynamically less stable γ-(E)-butenolide 369 was achieved by the Ru-catalysed CM reaction with olefin 368. Finally, the epoxidation of TBS ether, followed by the removal of the TBS group, completed the first asymmetric total synthesis of ramariolide A. Notably, this synthetic approach is protection-group free and leads to diversity (Scheme 50a).
Scheme 50 Total synthetic route toward ramariolides A and B.
At the same time, the first total synthesis of ramariolides A (189) and B (190) was reported by Sieber and coworkers.129 The commercially available fatty acid 371 was first transformed into the corresponding α-bromoketone 372 through three consecutive conversions, and the subsequent Wittig reaction with maleic anhydride afforded the (E)-butenolide moiety 373 as a major isomer, which was reduced by NaBH4 to yield the allylic alcohol 375. The subsequent Sharpless epoxidation accomplished the total synthesis of ramariolide A with 35% yield and a modest 55% ee. In parallel, the oxidation of the allylic alcohol 376 derived from a minor (Z)-butenolide moiety 374 with mCPBA yielded the syn-configuration α-hydroxy epoxide 377, and the subsequent inversion of configuration of the hydroxyl group and then CSA-promoted cyclization completed the first total synthesis of ramariolide B with low 25% yield (Scheme 50b).
4. Summary and perspectives
From the simple insect pheromone olean to the sophisticated anticancer agent spongistatin, from pesticide avermectins to T-type voltage-gated calcium channel inhibitor ganoapplanin, natural spiroketals isolated from various sources including plants, animals, and microbes have shown broad and interesting bioactivities, providing great possibilities for drug discovery and design, structure optimization, and functional reassignment, which are important topics in scientific research. As such, spiroketals have attracted substantial attention from the relevant chemists, and amazing achievements have consequently been made due to the relentless endeavours of researchers in recent years. These achievements include the following: (1) new chemical structures have been discovered based on bioactivity-oriented isolation or the combination of multiple methods and techniques, such as LS-MS-NMR, HPLC-DAD-MS and HPLC-DAD-SPE-NMR; (2) the novel bioactivities of natural spiroketals have been explored; (3) numerous convenient and efficient synthetic routes toward natural spiroketals have been reported from synthetic communities; (4) the biosynthesis of spiroketals has been a blooming topic, especially polyketide-related spiroketals, which have been extensively investigated by many biologists and chemists.
Nevertheless, the investigation of natural spiroketals remains in its infancy because many spiroketal-related issues that need to be solved are still being investigated. (1) Although novel chemical structures have been discovered, the isolation of only trace amounts and the scarce resources in nature have limited widespread bioassays and other functional studies. Therefore, how to efficiently and practically obtain highly bioactive spiroketals is a central issue that needs to be solved, but it is a great challenge. Total synthesis and biosynthesis may be two preferred approaches. Total synthesis could solve the problem of scarce spiroketals by chemical transformation, and would also provide plentiful analogues of natural spiroketals. At the same time, biosynthesis provides the simplest and the most efficient approach to the synthesis of natural products in some cases, and it is of great benefit to reveal the mystery of the biotransformation of spiroketals. These syntheses will facilitate bioassays, drug design, SAR studies, and discovery of new lead compounds. Gratifyingly, some new synthetic techniques have also been applied in the total synthesis of sophisticated spiroketal natural products. For example, the total syntheses of spiranien A and spiranien A methyl ester were achieved by flow chemistry technologies.130 (2) The bioactivity model of spiroketals needs to be further clarified, because a deep understanding of the origin of bioactivity could provide a platform for the design of potential lead compounds and drug candidates for the treatment of a variety of human diseases. (3) Structurally, due to the presence of two oxygen atoms interacting with a transition metal ion such as Fe, Zn, or Cu, even Li, K, or Na, spiroketals could be used as ideal probes for protein interaction and the investigation of other biological process, providing many chances for chemical biology.131,132 (4) The easy epimerization of the spiro-atom stereocentre is another challenge for drug metabolism, drug development and the design of total synthetic strategies. (5) The biosynthesis of spiroketals is still in its infancy, since only a few types of spiroketals have been explored. For example, polyketide cyclization and P450 function in the formation of spiroketals, and numerous spiroketal biotransformations remain unknown.
The spiroketals have therefore provided wonderful opportunities for further scientific research, and we believe that the related investigations will lead to the discovery and development of novel functional spiroketal molecules. These can be widely applied in medicinal chemistry, material chemistry, chemical biology, and other fields, resulting in a number of achievements in the near future.
5. Conflicts of interest
There are no conflicts to declare.
6. Acknowledgements
We gratefully acknowledge financial support from the NSFC (Grant Nos. 21772076, 21272097, 21290181, and 21372104) and PCSIRT of MOE (No. IRT-15R28).
7. Notes and references
R. G. Carter and D. L. Kuiper, Science of Synthesis, Stereoselective Synthesis, ed. J. G. De Vries, G. A. Molander and P. A. Evans, 2011, vol. 2, pp. 863–914 Search PubMed.
J. A. Palmes and A. Aponick, Synthesis, 2012, 44, 3699–3721 CrossRefCAS.
R. Quach, D. P. Furkert and M. A. Brimble, Org. Biomol. Chem., 2017, 15, 3098–3104 CAS.
S. V. Ley, L. G. Milroy and R. M. Myers, Science of Synthesis, 2007, vol. 29, pp. 613–689 Search PubMed.
M. A. Brimble and L. A. Stubbing, Synthesis of Saturated Oxygenated Heterocycles I, Topics in Heterocyclic Chemistry, ed. J. Cossy, Springer-Verlag, Berlin Heidelberg, 2014, vol. 35, pp. 189–267 Search PubMed.
F. Perron and K. F. Albizati, Chem. Rev., 1989, 89, 1617–1661 CrossRefCAS.
J. E. Aho, P. M. Pihko and T. K. Rissa, Chem. Rev., 2005, 105, 4406–4440 CrossRefCASPubMed.
I. Čorić and B. List, Nature, 2012, 483, 315–319 CrossRefPubMed.
W. C. Campbell, Angew. Chem., Int. Ed., 2016, 55, 10184–10189 CrossRefCASPubMed.
A. A. Stierle, D. B. Stierle and K. Kelly, J. Org. Chem., 2006, 71, 5357–5360 CrossRefCASPubMed.
L. Xiong, Q.-M. Zhou, Y. Zou, M.-H. Chen, L. Guo, G.-Y. Hu, Z.-H. Liu and C. Peng, Org. Lett., 2015, 17, 6238–6241 CrossRefCASPubMed.
L. Li, H. Li, X.-R. Peng, B. Hou, M.-Y. Yu, J.-R. Dong, X.-N. Li, L. Zhou, J. Yang and M.-H. Qiu, Org. Lett., 2016, 18, 6078–6081 CrossRefCASPubMed.
A. A. Salim, L. Tan, X.-C. Huang, K.-J. Cho, E. Lacey, J. F. Hancock and R. J. Capon, Org. Biomol. Chem., 2016, 14, 711–715 CAS.
D. J. Atkinson and M. A. Brimble, Nat. Prod. Rep., 2015, 32, 811–840 RSC.
Q. Zheng, Z. Tian and W. Liu, Curr. Opin. Chem. Biol., 2016, 31, 95–102 CrossRefCASPubMed.
Y. K. Booth, W. Kitching and J. J. De Voss, Nat. Prod. Rep., 2009, 26, 490–525 RSC.
L.-Y. Wang, J.-J. Qin, Z.-H. Chen, Y. Zhou, W. Tang, J.-P. Zuo and W.-M. Zhao, J. Nat. Prod., 2017, 80, 1102–1109 CrossRefCASPubMed.
M. Li, J. Xiong, Y. Huang, L.-J. Wang, Y. Tang, G.-X. Yang, X.-H. Liu, B.-G. Wei, H. Fan, Y. Zhao, W.-Z. Zhai and J.-F. Hu, Tetrahedron, 2015, 71, 5285–5295 CrossRefCAS.
W.-W. Jiang, Y.-C. Liu, Z.-J. Zhang, Y.-C. Liu, J. He, J. Su, X. Cheng, L.-Y. Peng, L.-D. Shao, X.-D. Wu, J.-H. Yang and Q.-S. Zhao, Fitoterapia, 2016, 109, 155–161 CrossRefCASPubMed.
L. Wang, X.-L. Jiang, W.-M. Zhang, F. Li, A.-A. Khan, X. Liu, K. Yu and M.-K. Wang, Phytochemistry, 2017, 136, 125–132 CrossRefCASPubMed.
Y. K. Booth, W. Kitching and J. J. De Voss, ChemBioChem, 2011, 12, 155–172 CrossRefCASPubMed.
P. Sun, Q. Zhao, H. Zhang, J. Wu and W. Liu, ChemBioChem, 2014, 15, 660–664 CrossRefCASPubMed.
P. Phainuphong, V. Rukachaisirikul, K. Tadpetch, Y. Sukpondma, S. Saithong, S. Phongpaichit, S. Preedanon and J. Sakayaroj, Phytochemistry, 2017, 137, 165–173 CrossRefCASPubMed.
S.-Y. Cheng, K.-J. Huang, S.-K. Wang and C.-Y. Duh, Mar. Drugs, 2011, 9, 1469–1476 CrossRefCASPubMed.
D.-W. Zhang, X. Liu, D. Xie, R. Chen, X.-Y. Tao, J.-H. Zou and J. Dai, Chem. Pharm. Bull., 2013, 61, 576–580 CrossRefCASPubMed.
L. Hu, H. Zhu, L. Li, J. Huang, W. Sun, J. Liu, H. Li, Z. Luo, J. Wang, Y. Xue, Y. Zhang and Y. Zhang, Sci. Rep., 2016, 6, 27588 CrossRefCASPubMed.
M. Kubo, Y. Nishikawa, K. Harada, M. Oda, J.-M. Huang, H. Domon, Y. Terao and Y. Fukuyama, J. Nat. Prod., 2015, 78, 1466–1469 CrossRefCASPubMed.
N. Nguyen, T. Ha, S. Choi, S. Eum, C. H. Lee, T. T. Buch and W. K. Oh, Phytochemistry, 2016, 130, 291–300 CrossRefCASPubMed.
A. Grudniewska, S. Hayashi, M. Shimizu, M. Kato, M. Suenaga, H. Imagawa, T. Ito, Y. Asakawa, S. Ban, T. Kumada, T. Hashimoto and A. Umeyama, Org. Lett., 2014, 16, 4695–4697 CrossRefCASPubMed.
H. Wang, J. Hong, J. Yin, H. R. Moon, Y. Liu, X. Wei, D.-C. Oh and J. H. Jung, J. Nat. Prod., 2015, 78, 2832–2836 CrossRefCASPubMed.
Y.-Y. Fan, H. Zhang, Y. Zhou, H.-B. Liu, W. Tang, B. Zhou, J.-P. Zuo and J.-M. Yue, J. Am. Chem. Soc., 2015, 137, 138–141 CrossRefCASPubMed.
Y.-L. Zhang, L.-L. Jiang, T.-S. Xiao, S.-Q. Chen and Y.-B. Li, J. Asian Nat. Prod. Res., 2015, 17, 717–723 CrossRefCASPubMed.
S.-J. Wang, L. Bao, J.-J. Han, Q.-X. Wang, X.-L. Yang, H.-A. Wen, L.-D. Guo, S.-J. Li, F. Zhao and H.-W. Liu, J. Nat. Prod., 2013, 76, 45–50 CrossRefCASPubMed.
Q.-Q. Tao, K. Ma, L. Bao, K. Wang, J.-J. Han, W.-Z. Wang, J.-X. Zhang, C.-Y. Huang and H.-W. Liu, Planta Med., 2016, 82, 639–644 CrossRefCASPubMed.
G. Chianese, B.-B. Gu, F. Yang, W.-H. Jiao, Y.-W. Guo, H.-W. Lin and O. Taglialatela-Scafati, RSC Adv., 2015, 5, 63372–63376 RSC.
T. Nogawa, N. Ogita, Y. Futamura, S. Negishi, N. Watanabe and H. Osada, J. Antibiot., 2017, 70, 705–707 CrossRefCASPubMed.
T. Ito, H. Ito, M. Oyama, T. Tanaka, D. Darnaedi and M. Iinuma, Phytochem. Lett., 2012, 5, 325–328 CrossRefCAS.
B. S. Hwang, H. S. Kim, W. Yih, E. J. Jeong and J.-R. Rho, Org. Lett., 2014, 16, 5362–5365 CrossRefCASPubMed.
K. D. G. Wang, J. Wang, S.-S. Xie, Z.-R. Li, L.-Y. Kong and J. Luo, Tetrahedron Lett., 2016, 57, 32–34 CrossRefCAS.
L.-L. Liu, R. Wang and Y.-P. Shi, Planta Med., 2011, 77, 1806–1810 CrossRefCASPubMed.
Y. Luan, H. Wei, Z. Zhang, Q. Che, Y. Liu, T. Zhu, A. Mándi, T. Kurtán, Q. Gu and D. Li, J. Nat. Prod., 2014, 77, 1718–1723 CrossRefCASPubMed.
L. Ma, F. Ge, C.-P. Tang, C.-Q. Ke, X.-Q. Li, A. Althammer and Y. Ye, Tetrahedron, 2011, 67, 3533–3539 CrossRefCAS.
J. Ren, F. Zhang, X. Liu, L. Li, G. Liu, X. Liu and Y. Che, Org. Lett., 2012, 14, 6226–6229 CrossRefCASPubMed.
Y. Liu, J. Ma, Q. Zhao, C. Liao, L. Ding, L. Chen, F. Zhao and F. Qiu, J. Nat. Prod., 2013, 76, 1150–1156 CrossRefCASPubMed.
C. N. Diep, E. G. Lyakhova, D. V. Berdyshev, A. I. Kalinovsky, V. A. Tu, N. X. Cuong, N. H. Nam, C. V. Minh and V. A. Stonik, Tetrahedron Lett., 2015, 56, 7001–7004 CrossRefCAS.
M. G. Phan, T. T. N. Tran, T. S. Phan, K. Matsunami and H. Otsuka, Phytochem. Lett., 2014, 9, 137–140 CrossRefCAS.
Y. Egami, T. Wakimoto and I. Abe, Bioorg. Med. Chem. Lett., 2014, 24, 5150–5153 CrossRefCASPubMed.
J.-J. Lv, S. Yu, Y. Xin, R.-R. Cheng, H.-T. Zhu, D. Wang, C.-R. Yang, M. Xu and Y.-J. Zhang, Phytochemistry, 2015, 117, 123–134 CrossRefCASPubMed.
Y. Zang, G. Genta-Jouve, P. Retailleau, A. Escargueil, S. Mann, B. Nay and S. Prado, Org. Biomol. Chem., 2016, 14, 2691–2697 CAS.
T. Asai, Y.-M. Chung, H. Sakurai, T. Ozeki, F.-R. Chang, K. Yamashita and Y. Oshima, Org. Lett., 2012, 14, 513–515 CrossRefCASPubMed.
J. Daoust, M. Chen, M. Wang, D. E. Williams, M. A. G. Chavez, Y. A. Wang, C. E. Merchant, A. Fontana, T. J. Kieffer and R. J. Andersen, J. Org. Chem., 2013, 78, 8267–8273 CrossRefCASPubMed.
M. Wang, I. Tietjen, M. Chen, D. E. Williams, J. Daoust, M. A. Brockman and R. J. Andersen, J. Org. Chem., 2016, 81, 11324–11334 CrossRefCASPubMed.
D. K. Gupta, P. Kaur, S. T. Leong, L. T. Tan, M. R. Prinsep and J. J. H. Chu, Mar. Drugs, 2014, 12, 115–127 CrossRefPubMed.
Y. Zhang, C. Zhang, K. Wang, G. Chen and P. Sun, Chem. Biodiversity, 2017, 14, e1700054 Search PubMed.
G. A. Mohamed, S. R. M. Ibrahim, J. M. Badr and D. T. A. Youssef, Tetrahedron, 2014, 70, 35–40 CrossRefCAS.
L. A. Shaala, D. T. A. Youssef, S. R. M. Ibrahim, G. A. Mohamed, J. M. Badr, A. L. Risinger and S. L. Mooberry, Mar. Drugs, 2014, 12, 5021–5034 CrossRefCASPubMed.
B. C. M. Potts, D. J. Faulkner, J. A. Chan, G. C. Simolike, P. Offen, M. E. Hemling and T. A. Francis, J. Am. Chem. Soc., 1991, 113, 6321–6322 CrossRefCAS.
J.-K. Woo, C.-K. Kim, S.-H. Kim, H. Kim, D.-C. Oh, K.-B. Oh and J. Shin, Org. Lett., 2014, 16, 2826–2829 CrossRefCASPubMed.
M. S. Abdelfattah, T. Kazufumi and M. Ishibashi, J. Antibiot., 2011, 64, 729–734 CrossRefCASPubMed.
Z.-G. Ding, J.-Y. Zhao, M.-G. Li, R. Huang, Q.-M. Li, X.-L. Cui, H.-J. Zhu and M.-L. Wen, J. Nat. Prod., 2012, 75, 1994–1998 CrossRefCASPubMed.
A. R. Yang, S. Lee, Y. D. Yoo, H. S. Kim, E. J. Jeong and J.-R. Rho, J. Nat. Prod., 2017, 80, 1688–1692 CrossRefCASPubMed.
S. Sato, F. Iwata, S. Yamada and M. Katayama, J. Nat. Prod., 2012, 75, 1974–1982 CrossRefCASPubMed.
L.-Y. Ma, W.-Z. Liu, L. Shen, Y.-L. Huang, X.-G. Rong, Y.-Y. Xu and X.-D. Gao, Tetrahedron, 2012, 68, 2276–2282 CrossRefCAS.
X. Pan, D. Liu, J. Wang, X. Zhang, M. Yan, D. Zhang, J. Zhang and W. Liu, Cancer Sci., 2013, 104, 1476–1482 CrossRefCASPubMed.
M. P. Sobolevskaya, O. I. Zhuravleva, E. V. Leshchenko, S. S. Afiyatullov, Y. V. Khudyakova, N. Y. Kim, N. N. Kirichuk and S. A. Dyshlovoy, Chem. Nat. Compd., 2014, 50, 1122–1124 CrossRefCAS.
W. Sun, X. Chen, Q. Tong, H. Zhu, Y. He, L. Lei, Y. Xue, G. Yao, Z. Luo, J. Wang, H. Li and Y. Zhang, Sci. Rep., 2016, 6, 26418 CrossRefCASPubMed.
H. Li, J. Jiang, Z. Liu, S. Lin, G. Xia, X. Xia, B. Ding, L. He, Y. Lu and Z. She, J. Nat. Prod., 2014, 77, 800–806 CrossRefCASPubMed.
W. Wang, B. Mun, Y. Lee, M. V. Reddy, Y. Park, J. Lee, H. Kim, D. Hahn, J. Chin, M. Ekins, S.-J. Nam and H. Kang, J. Nat. Prod., 2013, 76, 170–177 CrossRefCASPubMed.
Y. Lee, W. Wang, H. Kim, A. G. Giri, D. H. Won, D. Hahn, K. R. Baek, J. Lee, I. Yang, H. Choi, S.-J. Nam and H. Kang, Bioorg. Med. Chem. Lett., 2014, 24, 4095–4098 CrossRefCASPubMed.
L. Fremlin, M. Farrugia, A. M. Piggott, Z. Khalil, E. Lacey and R. J. Capon, Org. Biomol. Chem., 2011, 9, 1201–1211 CAS.
T. Nogawa, S. Takahashi, Y. Sekiyama, H. Takagi, M. Uramoto, H. Koshino, M. Kawatani, T. Shimizu and H. Osada, J. Antibiot., 2013, 66, 247–250 CrossRefCASPubMed.
O. I. Zhuravleva, M. P. Sobolevskaya, S. S. Afiyatullov, N. N. Kirichuk, V. A. Denisenko, P. S. Dmitrenok, E. A. Yurchenko and S. A. Dyshlovoy, Mar. Drugs, 2014, 12, 5930–5943 CrossRefCASPubMed.
O. I. Zhuravleva, M. P. Sobolevskaya, V. A. Denisenko, N. N. Kirichuk, S. A. Dyshlovoy and S. S. Afiyatullov, Nat. Prod. Commun., 2016, 11, 207–210 Search PubMed.
N. Bruns, W. Collisi, S. Bernecker, M. Stadler, C. Richter, H. Schwalbe and M. Kalesse, Eur. J. Org. Chem., 2015, 847–857 CrossRefCAS.
T. Nogawa, S. Takahashi, A. Okano, M. Kawatani, M. Uramoto, T. Saito and H. Osada, J. Antibiot., 2012, 65, 123–128 CrossRefCASPubMed.
Y.-P. Chen, Q. Liu, H. Gao, H.-P. Lin, H.-Y. Tian, K. Hong, J. Li, R.-W. Jiang, X.-S. Yao and J.-S. Tang, RSC Adv., 2014, 4, 63324–63327 RSC.
J. Huang, A.-L. Chen, H. Zhang, Z. Yu, M.-H. Li, N. Li, J.-T. Lin, H. Bai, J.-D. Wang and Y.-G. Zheng, Appl. Environ. Microbiol., 2015, 81, 5326–5334 CrossRefCASPubMed.
J. Li, L. Li, Y. Si, X. Jiang, L. Guo and Y. Che, Org. Lett., 2011, 13, 2670–2673 CrossRefCASPubMed.
K. Sadorn, S. Saepua, N. Boonyuen, P. Laksanacharoen, P. Rachtawee and P. Pittayakhajonwut, RSC Adv., 2016, 6, 94510–94523 RSC.
M. S. Abdelfattah, K. Toume and M. Ishibashi, J. Antibiot., 2012, 65, 245–248 CrossRefCASPubMed.
R. M. Centko, S. Ramón-García, T. Taylor, B. O. Patrick, C. J. Thompson, V. P. Miao and R. J. Andersen, J. Nat. Prod., 2012, 75, 2178–2182 CrossRefCASPubMed.
G. Zhao, X. Li, W. Chen, Z. Xi and L. Sun, Fitoterapia, 2012, 83, 1590–1597 CrossRefCASPubMed.
J. R. Kesting, L. Olsen, D. Staerk, M. V. Tejesvi, K. R. Kini, H. S. Prakash and J. W. Jaroszewski, J. Nat. Prod., 2011, 74, 2206–2215 CrossRefCASPubMed.
P. Sun, Q. Zhao, F. Yu, H. Zhang, Z. Wu, Y. Wang, Y. Wang, Q. Zhang and W. Liu, J. Am. Chem. Soc., 2013, 135, 1540–1548 CrossRefCASPubMed.
P. Sun, Q. Zhao, Z. Wu, W. Zhang and W. Liu, J. Nat. Prod., 2015, 78, 301–305 CrossRefCASPubMed.
A. A. Singh, J. A. Rowley, B. D. Schwartz, W. Kitching and J. J. De Voss, J. Org. Chem., 2014, 79, 7799–7821 CrossRefCASPubMed.
S. Takahashi, A. Toyoda, Y. Sekiyama, H. Takagi, T. Nogawa, M. Uramoto, R. Suzuki, H. Koshino, T. Kumano, S. Panthee, T. Dairi, J. Ishikawa, H. Ikeda, Y. Sakaki and H. Osada, Nat. Chem. Biol., 2011, 7, 461–468 CrossRefCASPubMed.
C. Jiang, Z. Qi, Q. Kang, J. Liu, M. Jiang and L. Bai, Angew. Chem., Int. Ed., 2015, 54, 9097–9100 CrossRefCASPubMed.
R. Quach, D. F. Chorley and M. A. Brimble, Org. Biomol. Chem., 2014, 12, 7423–7432 CAS.
H. Yao, J. Wang and R. Tong, Chem. Rec., 2017, 17, 1109–1123 CrossRefCASPubMed.
L. Song, H. Yao, Y. Dai, M. Wu and R. Tong, Tetrahedron Lett., 2016, 57, 4257–4263 CrossRefCAS.
J. C. Green, G. L. Burnett IV and T. R. R. Pettus, Pure Appl. Chem., 2012, 84, 1621–1631 CrossRefCASPubMed.
I. Paterson, S. M. Dalby and P. Maltas, Isr. J. Chem., 2011, 51, 406–419 CrossRefCAS.
J. S. Yadav, P. A. N. Reddy, Y. J. Reddy, S. Meraj and A. R. Prasad, Eur. J. Org. Chem., 2013, 6317–6324 CrossRefCAS.
F.-M. Zhang, L. Peng, H. Li, A.-J. Ma, J.-B. Peng, J.-J. Guo, D. Yang, S.-H. Hou, Y.-Q. Tu and W. Kitching, Angew. Chem., Int. Ed., 2012, 51, 10846–10850 CrossRefCASPubMed.
L. Dong, V. A. Gordon, R. L. Grange, J. Johns, P. G. Parsons, A. Porzelle, P. Reddell, H. Schill and C. M. Williams, J. Am. Chem. Soc., 2008, 130, 15262–15263 CrossRefCASPubMed.
B. J. Albert, Y. Yamaoka and H. Yamamoto, Angew. Chem., Int. Ed., 2011, 50, 2610–2612 CrossRefCASPubMed.
W. Lee, S. Kang, B. Jung, H.-S. Lee and S. H. Kang, Chem. Commun., 2016, 52, 3536–3539 RSC.
Y. Ogura, K. Ishigami and H. Watanabe, Tetrahedron, 2012, 68, 1723–1728 CrossRefCAS.
L. C. Dias and E. C. de Lucca Jr, J. Org. Chem., 2017, 82, 3019–3045 CrossRefCASPubMed.
J. T. J. Spence and J. H. George, Org. Lett., 2015, 17, 5970–5973 CrossRefCASPubMed.
P. A. Hume, D. P. Furkert and M. A. Brimble, Org. Lett., 2013, 15, 4588–4591 CrossRefCASPubMed.
S. F. Seibert, A. Krick, E. Eguereva, S. Kehraus and G. M. König, Org. Lett., 2007, 9, 239–242 CrossRefCASPubMed.
S. Chang, S. Hur and R. Britton, Angew. Chem., Int. Ed., 2015, 54, 211–214 CrossRefCASPubMed.
D. V. Reddy, G. Sabitha, T. P. Rao and J. S. Yadav, Org. Lett., 2016, 18, 4202–4205 CrossRefCASPubMed.
J. E. Stok, C.-S. Lang, B. D. Schwartz, M. T. Fletcher, W. Kitching and J. J. De Voss, Org. Lett., 2001, 3, 397–400 CrossRefCASPubMed.
J. Y. Hamilton, S. L. Rössler and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 8082–8085 CrossRefCASPubMed.
D. C. Davis, K. L. Walker, C. Hu, R. N. Zare, R. M. Waymouth and M. Dai, J. Am. Chem. Soc., 2016, 138, 10693–10699 CrossRefCASPubMed.
S. Joung, R. Kim and H.-Y. Lee, Org. Lett., 2017, 19, 3903–3906 CrossRefCASPubMed.
Q. Xiao, W.-W. Ren, Z.-X. Chen, T.-W. Sun, Y. Li, Q.-D. Ye, J.-X. Gong, F.-K. Meng, L. You, Y.-F. Liu, M.-Z. Zhao, L.-M. Xu, Z.-H. Shan, Y. Shi, Y.-F. Tang, J.-H. Chen and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 7373–7377 CrossRefCASPubMed.
M. Wilsdorf and H.-U. Reissig, Angew. Chem., Int. Ed., 2012, 51, 9486–9488 CrossRefCASPubMed.
N. Yoneda, Y. Fukata, K. Asano and S. Matsubara, Angew. Chem., Int. Ed., 2015, 54, 15497–15500 CrossRefCASPubMed.
F. J. Fañanás, A. Mendoza, T. Arto, B. Temelli and F. Rodríguez, Angew. Chem., Int. Ed., 2012, 51, 4930–4933 CrossRefPubMed.
X. Han and P. E. Floreancig, Angew. Chem., Int. Ed., 2014, 53, 11075–11078 CrossRefCASPubMed.
M. F. Elsebai, M. Nazir, S. Kehraus, E. Egereva, K. N. Ioset, L. Marcourt, D. Jeannerat, M. Gütschow, J.-L. Wolfender and G. M. König, Eur. J. Org. Chem., 2012, 6197–6203 CrossRefCAS.
Z. Ren, Y. Hao and X. Hu, Org. Lett., 2016, 18, 4958–4961 CrossRefCASPubMed.
J. Huang, J. R. Yang, J. Zhang and J. Yang, J. Am. Chem. Soc., 2012, 134, 8806–8809 CrossRefCASPubMed.
M. Xuan, I. Paterson and S. M. Dalby, Org. Lett., 2012, 14, 5492–5495 CrossRefCASPubMed.
H. Cheng, Z. Zhang, H. Yao, W. Zhang, J. Yu and R. Tong, Angew. Chem., Int. Ed., 2017, 56, 9096–9100 CrossRefCASPubMed.
L. Tomas, G. Boije af Gennäs, M. A. Hiebel, P. Hampson, D. Gueyrard, B. Pelotier, J. Yli-Kauhaluoma, O. Piva, J. M. Lord and P. G. Goekjian, Chem.–Eur. J., 2012, 18, 7452–7466 CrossRefCASPubMed.
H. Fuwa, K. Sekine and M. Sasaki, Org. Lett., 2013, 15, 3970–3973 CrossRefCASPubMed.
H. Fuwa, T. Muto, K. Sekine and M. Sasaki, Chem.–Eur. J., 2014, 20, 1848–1860 CrossRefCASPubMed.
G. V. M. Sharma, G. Srikanth and P. P. Reddy, Org. Biomol. Chem., 2012, 10, 8119–8124 CAS.
Y. Zhang, Q. Ye, X. Wang, Q.-B. She and J. S. Thorson, Angew. Chem., Int. Ed., 2015, 54, 11219–11222 CrossRefCASPubMed.
A. Yamamoto, A. Ueda, P. Brémond, P. S. Tiseni and Y. Kishi, J. Am. Chem. Soc., 2012, 134, 893–896 CrossRefCASPubMed.
S. Badrinarayanan, C. J. Squire, J. Sperry and M. A. Brimble, Org. Lett., 2017, 19, 3414–3417 CrossRefCASPubMed.
P. Pal and S. Nanda, Org. Lett., 2017, 19, 1164–1167 CrossRefCASPubMed.
J. Lehmann, J. Richers, A. Pöthig and S. A. Sieber, Chem. Commun., 2017, 53, 107–110 RSC.
S. Newton, C. F. Carter, C. M. Pearson, L. de, C. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915–4920 CrossRefCASPubMed.
M. Scheepstra, S. A. Andrei, M. Y. Unver, A. K. H. Hirsch, S. Leysen, C. Ottmann, L. Brunsveld and L.-G. Milroy, Angew. Chem., Int. Ed., 2017, 56, 5480–5484 CrossRefCASPubMed.
B. Borgström, X. Huang, C. Hegardt, S. Oredsson and D. Strand, Chem.–Eur. J., 2017, 23, 2077–2083 CrossRefPubMed.

 a,
Shu-Yu
Zhang
a,
Shu-Yu
Zhang











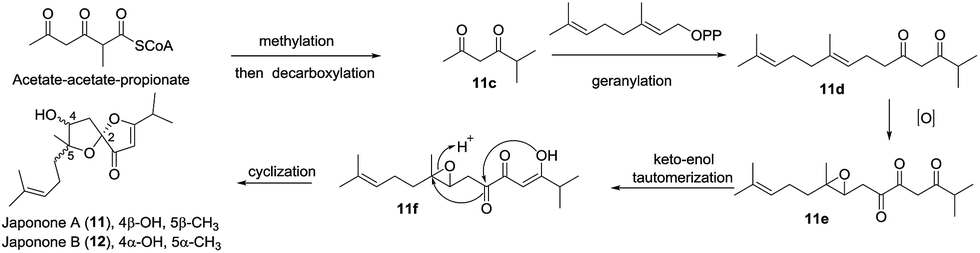


![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 and 58,47 were also identified, respectively. Unfortunately, no related bioactivities were reported. Fourteen highly oxygenated norbisabolane sesquiterpenoid-type spiroketals, phyllaemblicins H1–H14 (60–73), were isolated by Zhang and coworkers.49 The major structural differences lay in the different sugar substituents. Phyllaemblicin H1 (60) exhibited moderate cytotoxicity against the human cancer cell lines A-549 (IC50 = 4.7 ± 0.7 μM) and SMMC-7721 (IC50 = 9.9 ± 1.3 μM). Phyllaemblicin H6 (65) displayed inhibitory activity against HSV-1, whereas phyllaemblicin H8 (67) and phyllaemblicin H12 (71) showed moderate anti-CVB3 cell activities.
46 and 58,47 were also identified, respectively. Unfortunately, no related bioactivities were reported. Fourteen highly oxygenated norbisabolane sesquiterpenoid-type spiroketals, phyllaemblicins H1–H14 (60–73), were isolated by Zhang and coworkers.49 The major structural differences lay in the different sugar substituents. Phyllaemblicin H1 (60) exhibited moderate cytotoxicity against the human cancer cell lines A-549 (IC50 = 4.7 ± 0.7 μM) and SMMC-7721 (IC50 = 9.9 ± 1.3 μM). Phyllaemblicin H6 (65) displayed inhibitory activity against HSV-1, whereas phyllaemblicin H8 (67) and phyllaemblicin H12 (71) showed moderate anti-CVB3 cell activities.
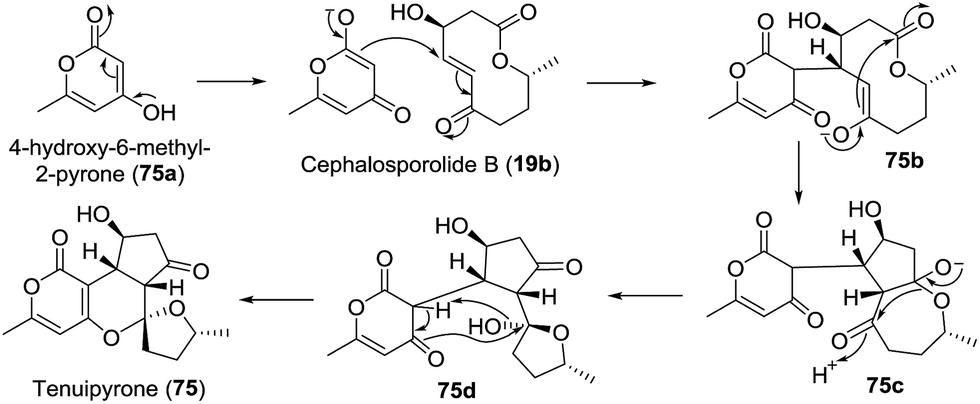












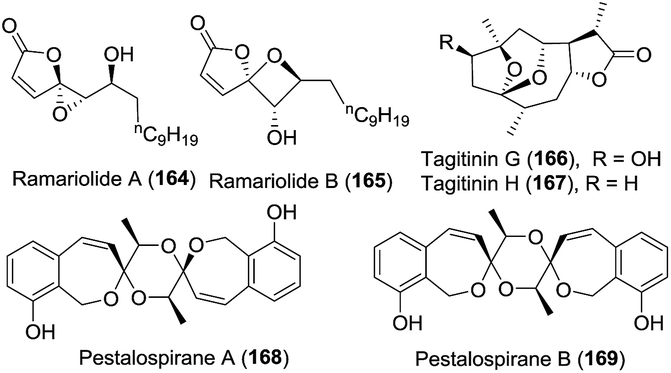



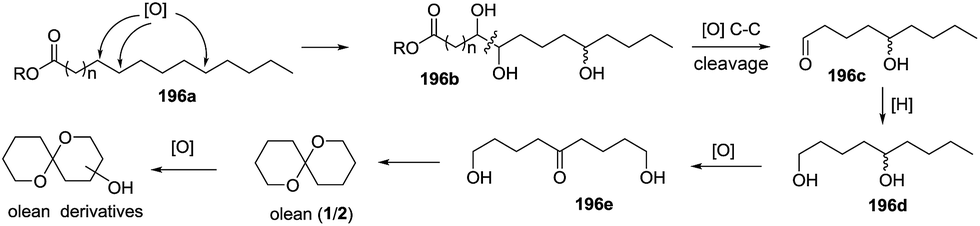

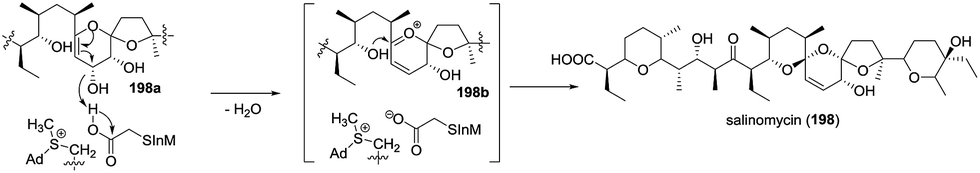


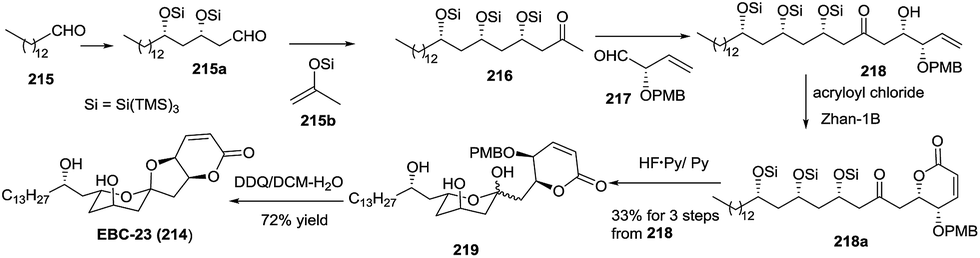
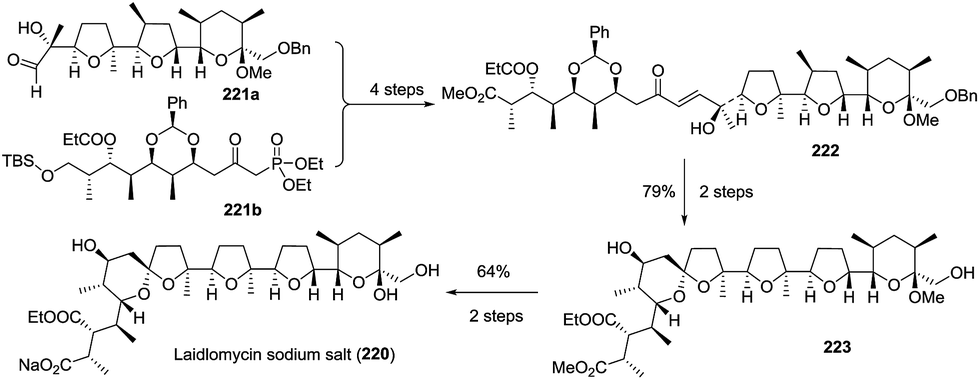







![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond into the lactone ring was achieved by the treatment of a mixture of α-levantanolide and other isomers with phenylselenyl chloride, followed by oxidation with H2O2 to provide α-levantenolide (264) as a single product in 46% yield in two steps. The structure of α-levantenolide was further confirmed by X-ray diffraction (Scheme 35).
C double bond into the lactone ring was achieved by the treatment of a mixture of α-levantanolide and other isomers with phenylselenyl chloride, followed by oxidation with H2O2 to provide α-levantenolide (264) as a single product in 46% yield in two steps. The structure of α-levantenolide was further confirmed by X-ray diffraction (Scheme 35).