
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVarious types of non-covalent interactions contributing towards crystal packing of halogenated diphospha-dicarbaborane with an open pentagonal belt†
Josef
Holub
a,
Zdeňka
Růžičková
b,
Pavel
Hobza
 *c,
Jindřich
Fanfrlík
*c,
Drahomír
Hnyk
*a and
Aleš
Růžička
*b
*c,
Jindřich
Fanfrlík
*c,
Drahomír
Hnyk
*a and
Aleš
Růžička
*b
aInstitute of Inorganic Chemistry of the Czech Academy of Sciences, 250 68 Husinec-Řež, Czech Republic. E-mail: hnyk@iic.cas.cz
bDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 53210 Pardubice, Czech Republic. E-mail: ales.ruzicka@upce.cz
cInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nam. 2, 166 10 Prague 6, Czech Republic. E-mail: hobza@uochb.cas.cz; fanfrlik@uochb.cas.cz
First published on 29th March 2018
Abstract
We have prepared 3-Cl-10-I-nido-7,8,9,11-P2C2B7H7 by the reaction of 3-Cl-nido-7,8,9,11-P2C2B7H8 with I2 in the presence of AlCl3. The product was obtained in a yield of 49% and was characterized by NMR spectroscopy and X-ray crystallography. Quantum chemical calculations have demonstrated that the crystal structure is stabilized by hydrogen, dihydrogen and pnictogen bonds.
Innerly and/or outerly substituted boron cluster hydrides (heteroboranes) of various architectures are known for various types of non-covalent intermolecular interactions and thus serve as good models for the evaluation and mutual comparison of various types of hydrogen, dihydrogen (diH), halogen (X), chalcogen, pnictogen (Pn) or even tetrel bonds.1,2 The neutral nido-B7P2[C(R)]2H7 cage with an open P2CBC pentagonal belt can be formally obtained from nido-B11H114− (nido-B11H14− exists)3 by replacing four BH vertices with two tetrel and Pn vertices. Miscellaneous dicarba dipnictogen nido eleven-vertex heteroboranes have been prepared by reacting arachno-4,5-C2B7H13 with PCl3, AsCl3 and SbCl3.4–6 The crystal structures of 3-Cl-nido-7,8,9,11-P2C2B7H8 (1), 3-I-nido-7,8,9,11-As2C2B7H8 (2) and nido-7,8,9,11-Sb2C2B7H9 (3) compounds are already known.4–6 The crystal packing of 3 is governed by Pn-bonds.6 In contrast to 3, it has not been possible to record the single-crystal X-ray data of the parent nido-7,8,9,11-P2C2B7H9 and nido-7,8,9,11-As2C2B7H9 in sufficient quality. Interestingly, it is feasible to determine the structure of single-crystal halogenated compounds 1 and 2, revealing a supramolecular architecture mainly resulting from extensive Pn-bonding.4–6 In order to proceed in the examination of the non-covalent interactions of nido-heteroboranes, we attempted to iodinate 1 (Fig. 1) by the reaction with elemental iodine in the presence of aluminum trichloride (details in ESI†). The resulting 3-Cl-10-I-nido-7,8,9,11-P2C2B7H7 (4) was obtained in a moderate yield of 49% and was characterized in solution in terms of multinuclear NMR spectroscopy (Table 1).
 | ||
| Fig. 1 A simplified reaction scheme. | ||
| B(1) | B(3) | B(2,4) | B(10)b | B(5,6) | |
|---|---|---|---|---|---|
| a All in ppm with respect to BF3·OEt2. Atomic numbering is shown in Fig. 2. b The discrepancy between the calculated and experimental chemical shift results from the neglect of the effect of spin–orbit (SO) coupling on the overall shielding, which is not taken into account in the GIAO-MP2 scheme. However, it has been shown that due to iodine being terminally bonded to a boron atom within a neutral boron cluster, SO coupling increases the contribution of this B atom towards overall shielding by about 20 ppm,7,8 which would considerably improve the fit in the present case. | |||||
| GIAO | −29.3 | 2.5 | −2.0 | 5.9 | 3.5 |
| Exp. | −31.2 | −0.7 | −1.9 | −14.7 | 0.3 |
In the crystal structure of 4 (Fig. 2), four molecules have been found in the unit cell. The structure of the cage of 4 is essentially the same as found for 14 except for the very slight shortening of the B(10)–C(9/11) bonds inside the open P2CBC pentagonal belt caused by the substitution of the B(10) atom by the heavy iodine. In fact, there are only two examples of a similar structural pattern in the Cambridge Structural Database, i.e. dicarba-closo-dodecaborane-1,2-diyl-1,2-bis(diisopropylamino)diphosphines,9 but these are heavily distorted with no option of a serious structural comparison.
 | ||
| Fig. 2 The molecular structure (ORTEP 50% probability level) and atomic numbering of 4. The selected interatomic distances [Å] are: I1–B10 2.184(2), Cl1–B3 1.786(2), B2–C11 1.730(3), B2–P7 2.053(2), B3–P8 2.1117(19), B3–P7 2.113(2), B4–C9 1.732(2), B4–P8 2.0516(18), B5–C9 1.715(3), B6–C11 1.715(2), C9–B10 1.608(2), C9–P8 1.8681(17), C9–H9 0.91(3), B10–C11 1.604(3), C11–P7 1.8706(19) and P8–P7 2.1970(6). For all the distances and other structural characteristics, see Table S2 (ESI†). | ||
To better understand non-covalent interactions of 4, we computed its ESP potential (Fig. 3) and compared it with other recently studied analogous compounds, 1–3. Compound 1 has only one highly positive σ-hole located on the P vertices. The other expected σ-holes of 1 could not be defined on the P vertices due to the more positive neighboring CH vertices. The Sb and As vertices of 2 and 3 have more positive ESP surfaces than the CH vertices. Both molecules thus have three well-defined σ-holes on the Pn atoms. From this point of view, the ESP surface of 4 is thus analogous to 2 and 3. Compound 4 has three σ-holes located on P atoms with the same magnitude (VS,max) of 31.5 kcal mol−1. The P vertices of 4 have even more positive ESP values than the CH vertices. The ESP surface on top of the CH vertices has the value of 28.2 kcal mol−1. Besides the σ-holes located on P atoms, the 4 compound also has σ-holes on the X atoms. While the I atom has a positive σ-hole with the VS,max value of 6.8 kcal mol−1, the Cl atom only has a relative σ-hole (i.e. the top of the Cl atom is less negative than its belt but the VS,max value is negative). The 4 compound can thus form Pn-, H- and X-bonds, the last of which are, however, less probable due to the lower VS,max values on the X atoms (Table 2). The most negative ESP surface of 4 is on the negative belt on the I and Cl atoms (the magnitudes of −15.1 and −12.3 kcal mol−1, respectively).
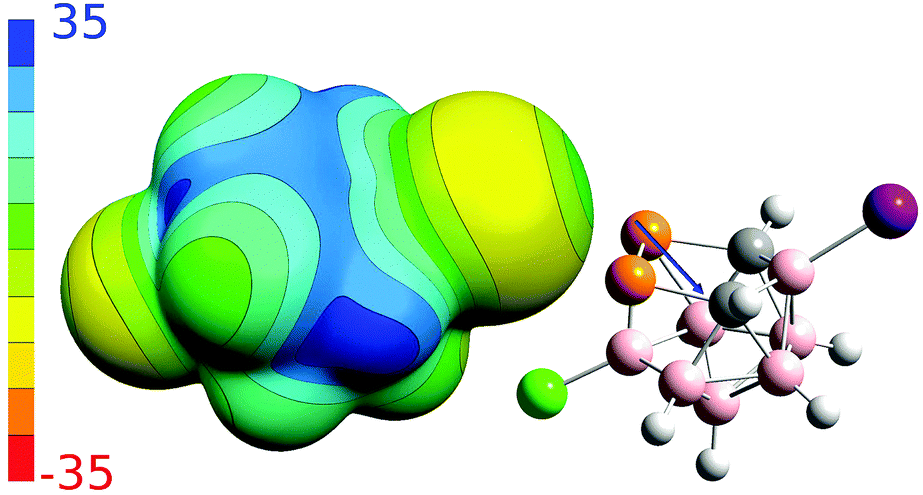 | ||
| Fig. 3 The computed electrostatic potential (ESP) surface for compound 4. The color range of the ESP in kcal mol−1. The direction of the dipole moment is shown as an arrow derived at the HF/cc-pVTZ level. | ||
Finally, we have studied pairwise interactions in the crystal structure of 4 and compared them with those of 1, 2 and 3. Highly-accurate MP2/CBS interaction energy (ΔE) values are summarized in Table 3 (see also Fig. 4). The A⋯B motif is the most stable motif of 4. The motif can be described as two symmetrical P2⋯I Pn-bonds. Since the Pn-bond is quite long (3.8 Å), it could also be seen as a nonspecific dispersion contact with an optimal dipole–dipole arrangement. The ΔE value of −9.5 kcal mol−1 is very large, considerably more negative than the most negative bonding motifs of 1 and 2 (the ΔE of about −6.0 kcal mol−1), which also had two Pn-bonds. However, considering that the A⋯B motif of 4 has two Pn-bonds, they are weaker than the Pn-bonds of 3. Compound 3 has a binding motif that is exclusively formed by a single Pn-bond and has the ΔE value of −6.5 kcal mol−1.6
| Interaction | MP2.5/CCSD(T) | |
|---|---|---|
| 3-Cl-nido-7,8,9,11-P2C2B7H8 (1) | ||
| A⋯B | 2× Pn-bond | −5.936/−6.106 |
| 3-I-nido-7,8,9,11-As2C2B7H8 (2) | ||
| A⋯B | 2× Pn-bond | −5.986 |
| A⋯C | 2× Pn-bond | −5.676 |
| nido-7,8,9,11-Sb2C2B7H9 (3) | ||
| A⋯B | 2× Pn-bond, HH-bond | −6.806/−7.056 |
| A⋯C | Single Pn-bond | −6.226/−6.466 |
| 3-Cl-10-I-nido-7,8,9,11-P2C2B7H7 (4) | ||
| A⋯B | 2× Pn-bond | −9.5 |
| A⋯C | 2× H-bond | −6.5 |
| A⋯D | diH and H-bonds | −6.1 |
| A⋯E | Stacking | −4.8 |
 | ||
| Fig. 4 The most significant interaction motifs from the crystal structure of 3-Cl-10-I-nido-7,8,9,11-P2C2B7H7 (4). The distances are in Å. The positions of the H atoms have been optimized at the DFT-D3/BLYP/DZVP level.10 | ||
The A⋯C motif, formed by two symmetrical C–H⋯I–B H-bonds, has ΔE of −6.5 kcal mol−1. The H-bond is again rather long (3.2 Å). The third most stable motif is A⋯D with ΔE of −6.1 kcal mol−1. It is formed by the C–H⋯H–B dihydrogen bond and the C–H⋯I–B H-bond. The C–H⋯H–B diH-bond is the only contact in the crystal structure of 4 with the length below the sum of van der Waals radii (2.9 Å between the H and the center of the B–H bond; H⋯H and H⋯B distances of 2.3 and 3.0 Å, respectively). The A⋯E motif also has all contacts much higher than the sum of the van der Waals radii (e.g. the P2⋯Cl Pn bond of 3.9 Å).
In summary, compound 4 (3-Cl-10-I-nido-7,8,9,11-P2C2B7H7) was prepared by reacting 3-Cl-nido-7,8,9,11-P2C2B7H8 with elemental iodine in the presence of aluminum trichloride. Its molecular structure has been characterized by NMR spectroscopy, X-ray crystallography and ab initio computations. Apart from B(10) there is a good accord between the experimental and computed 11B chemical shifts. It is the spin–orbit coupling that is responsible for such a disagreement related to B(10). The combination of Pn-, diH and H-bonds turned out to be a driving force for the crystal packing as revealed by quantum chemical computations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the research project RVO 61388963 of the Czech Academy of Sciences. We acknowledge the financial support of the Czech Science Foundation (17-08045S).Notes and references
- (a) G. Cavall, P. Metrangolo, R. Milani, T. Pilati, A. Primagi, G. Ransani and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef PubMed; (b) S. Scheiner, Acc. Chem. Res., 2013, 46, 280 CrossRef CAS PubMed; (c) A. Bundhun, P. Ramasami, J. S. Murray and P. Politzer, J. Mol. Model., 2013, 19, 2739 CrossRef CAS PubMed; (d) A. Bauza, T. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef CAS PubMed.
- R. Sedlak, J. Fanfrlík, A. Pecina, D. Hnyk, P. Hobza and M. Lepšík, Boron – the Fifth Element, in Challenges and Advances in Computational Chemistry and Physics, ed. D. Hnyk and M. McKee, Springer, Heidelberg, New York, Dordrecht and London, ch. 9, vol. 20, 2015 Search PubMed.
- For the structure of B11H14− with three hydrogen bridges, see: J. Fritchie, Inorg. Chem., 1967, 6, 1199–1203 CrossRef.
- J. Holub, T. Jelínek, D. Hnyk, Z. Plzák, I. Císařová, M. Bakardjiev and B. Štíbr, Chem. – Eur. J., 2001, 7, 1546 CrossRef CAS.
- L. Mikulášek, B. Grüner, I. Císařová and B. Štíbr, Dalton Trans., 2003, 1332 RSC.
- J. Holub, P. Melichar, Z. Růžičková, J. Vrána, D. A. Wann, J. Fanfrlík, D. Hnyk and A. Růžička, Dalton Trans., 2017, 46, 13714 RSC.
- J. Macháček, J. Plešek, J. Holub, D. Hnyk, V. Všetečka, I. Císařová, M. Kaupp and B. Štíbr, Dalton Trans., 2006, 1024 RSC.
- B. Štíbr, J. Holub, M. Bakardjiev, P. D. Lane, M. L. McKee, D. A. Wann and D. Hnyk, Inorg. Chem., 2016, 56, 852 CrossRef PubMed.
- A. Kreienbrink, M. B. Sarosi, E. G. Rys, P. Lonnecke and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2011, 50, 4701 CrossRef CAS PubMed.
- J. Hostaš and J. Řezáč, J. Chem. Theory Comput., 2017, 13, 3575 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1819146. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj00450a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |