
Preparation of metal oxide supported catalysts and their utilization for understanding the effect of a support on the catalytic activity†
Jhumur
Seth
,
Prashant
Dubey‡
,
Vijay R.
Chaudhari
* and
Bhagavatula L. V.
Prasad
 *
*
Physical and Material Chemistry Division, National Chemical Laboratory (CSIR-NCL), Dr Homi Bhabha Road, Pune 411008, India. E-mail: pl.bhagavatula@ncl.res.in; v.chaudhari@ncl.res.in
First published on 20th November 2017
Abstract
A convenient way of anchoring transition metal nanoparticles (palladium, platinum, rhodium and ruthenium) onto metal oxide supports (magnesium oxide and zirconium oxide) by means of a modified sol–gel technique is demonstrated. Use of toluene dispersed, ligand protected pre-synthesized nanoparticles during sol–gel synthesis delivered size-controlled, spatially distributed, well-adhered transition metal nanoparticles (MNPs) on metal oxide supports. The catalytic activities of these supported nanoparticles were tested for the p-nitro phenol reduction reaction. It was observed that the reaction kinetics were crucially dependent on the catalyst support and MNP size. The influence of the magnesium oxide and zirconium oxide supports towards the catalytic performance of the anchored transition MNPs was probed using cyclic voltammetry and the differences in the same were attributed to the support-induced modification in the electronic properties of the MNPs. Our results indicated that magnesium oxide is a better support than zirconium oxide.
Introduction
The field of heterogeneous catalysis is seeing a great surge with possible applications in many fields that include energy, materials, medicine and environmental remediation.1,2 Today, most of the chemical processes and chemical industries focus on supported nanomaterials/nanoparticles as catalysts due to the convenience in recycling and the high turnover numbers.3–7 The catalytic activity, being a surface phenomenon, is sensitive to the nature of active surface sites which in turn is governed by the particle size, shape and composition of the material concerned.8,9 Recent advances in chemical syntheses provide access to nanoparticles with well-defined structures that have revitalized the interest in their usage as heterogeneous catalysts, especially those that are supported on an oxide surface.10,11 The main challenge one faces en route is to retain the particle composition and structure on the support avoiding the anchoring site dependence.12–14 Generally, impregnation and precipitation methods are adopted to prepare the supported nanoparticles. The impregnation method involves selective adhesion of precursors (metal ions) on the existing support followed by their conversion to the desired composition (metal/metal oxide/sulfide etc.) through appropriate chemical reactions.10 Alternatively, one can use the precipitation technique, which is broadly categorized into two routes – (i) co-precipitation, i.e. simultaneous synthesis of the support and metal nanoparticles, and (ii) deposition precipitation, which involves in situ preparation and deposition of nanoparticles on pre-formed support materials. However, all these methods suffer from one disadvantage or another, like aggregation of particles on the support, burial of the particles into the support matrix and therefore their non-availability for catalytic conversions or the lack of control over the particle size and shape, which is very crucial for the catalytic performance of the material.In this context, we envisaged the sol–gel synthesis of metal oxide supports in the presence of pre-synthesized nanoparticles where nanoparticles are kept away from the hydrolysis and the condensation process as an efficient strategy to maintain the physico-chemical nature of the supported nanoparticles and achieve the desired material architecture. Moreover, in the procedure for oxide preparation if there is no necessity of a separate step for the generation of anchoring sites such a procedure would be versatile and material independent.
Our search for such a versatile method led us to the strategy proposed by Klaubunde and co-workers15,16 wherein it was revealed that a sol–gel process carried out in the presence of a spectator non-polar solvent like toluene affords nanocrystalline metal oxides with high surface area. We then envisioned that if we used ligand-protected MNPs dispersed in toluene instead of pure toluene during the sol–gel process we could get access to oxide-supported MNPs.17 Accordingly, herein we illustrate the deposition of a wide range of nanomaterials on two different metal oxide supports, namely, magnesium oxide (MgO) and zirconium oxide (ZrO2) using this modified sol–gel method. MgO and ZrO2 are distinctly different from each other as the former is more basic in nature than the latter18 and hence we reckoned that we could systematically study the influence of the support on the MNP catalysts and gain useful insights. Pt, Pd, Rh and Ru MNPs were chosen as examples as they have potential applications in many catalytic processes. Furthermore, the size effect of the anchored nanoparticle catalysts was also investigated by depositing two different sized nanoparticles for each metal. The differences in catalytic activity of the obtained supported MNPs were resolved, in terms of support and size, through kinetic investigations using the convenient and standard sodium borohydride mediated p-nitro phenol reduction.19 Interesting trends observed in terms of the rates of reactions were rationalized based on the size of the metal particles as well as the chemical/electronic nature of the support. The origin of the dissimilar catalytic activities was delineated using simple cyclic voltammetric (CV) investigations. The CV measurements suggested that the observed enhanced activity is due to the modulation of the redox properties of the metal in the presence of the support. We hasten to add here that the catalytic hydrogenation using hydrogen gas would be more reliable if someone is interested in organic transformations rather than in understanding the kinetics. Presented below are the details of the investigation.
Experimental procedure
Chemicals
Magnesium methoxide in methanol (6–10 weight%) solution and zirconium isopropoxide in isopropanol (40 weight%) solution, palladium acetate dimer, platinum chloride, rhodium acetate dimer, ruthenium chloride, sodium borohydride, didodecyldimethylammonium bromide (DDAB) and dodecanethiol were purchased from Sigma-Aldrich. Methanol, iso-propanol and toluene were obtained from Merck chemicals.Preparation of transition metal nanoparticles (Pd, Pt, Rh, Ru)
All the transition MNPs were prepared by the chemical reduction method and their size distribution was controlled by traditional digestive ripening (DR) and modified digestive ripening (mDR) methods, as reported by our group.20 Briefly, dodecanethiol capped transition MNPs were prepared by dissolving a known amount of metal precursor (22 mg palladium acetate, 26.5 mg platinum(II) chloride, 44.2 mg rhodium acetate dimer, and 20.7 mg ruthenium(III) chloride were used as a precursor for the synthesis of Pd, Pt, Rh and Ru NPs, respectively) in 10 mL toluene with the aid of DDAB. The final concentration of the respective metal ion in toluene was 0.01 M. These were reduced by the addition of aqueous NaBH4 solution (50 μL of 9.4 M) under vigorous stirring followed by the addition of dodecanethiol (719 μL, 0.3 M) and stirring for one more hour. The product obtained at the end of this procedure is referred to as as-prepared nanoparticles.The size distribution (monodispersity) of the as-prepared nanomaterials was controlled through the DR and mDR methods. During DR, the as-prepared nanoparticle sol was purified by adding ethanol as a co-solvent and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 minutes. This process was repeated thrice and the final product was dispersed in 10 mL toluene. An extra amount of dodecanethiol (719 μL) was added to the nanoparticle dispersion and refluxed at 120 °C for an hour under argon atmosphere. Finally, the ligand protected particles were obtained as a black sticky material by repetitive ethanol washing and dried under vacuum. These particles get easily dispersed in non-polar solvents. MNPs obtained after the DR treatment are named Pd-DR, Pt-DR, Rh-DR and Ru-DR. Alternatively, the size distribution was also controlled through mDR. The mDR procedure is roughly similar to the DR procedure. The main difference is that in the case of mDR the as-prepared nanoparticles were directly subjected to reflux without any purification treatments such as washing and centrifugation. Nanoparticles obtained after mDR treatment are named Pd-mDR, Pt-mDR, Rh-mDR and Ru-mDR.
000 rpm for 20 minutes. This process was repeated thrice and the final product was dispersed in 10 mL toluene. An extra amount of dodecanethiol (719 μL) was added to the nanoparticle dispersion and refluxed at 120 °C for an hour under argon atmosphere. Finally, the ligand protected particles were obtained as a black sticky material by repetitive ethanol washing and dried under vacuum. These particles get easily dispersed in non-polar solvents. MNPs obtained after the DR treatment are named Pd-DR, Pt-DR, Rh-DR and Ru-DR. Alternatively, the size distribution was also controlled through mDR. The mDR procedure is roughly similar to the DR procedure. The main difference is that in the case of mDR the as-prepared nanoparticles were directly subjected to reflux without any purification treatments such as washing and centrifugation. Nanoparticles obtained after mDR treatment are named Pd-mDR, Pt-mDR, Rh-mDR and Ru-mDR.
Synthesis of transition metal nanoparticles anchored on MgO and ZrO2 supports
Mg(OMe)2 in methanol (1.6 mL) and 0.4 mL of nanoparticle dispersion in toluene were taken in vessel 1. In vessel 2, 0.5 mL of methanol, 1.5 mL of nanoparticle dispersion in toluene and 36 μL of de-ionized water were taken. Both these solutions were thoroughly sonicated and mixed with each other which resulted in the formation of an instantaneous viscous gel. This gel was left undisturbed at room temperature for slow evaporation of the solvent. After 7 days, the gel was dried in a vacuum oven at 60 °C until a constant weight was obtained. Finally, the dried gel was annealed under hydrogen gas flow at 550 °C in a furnace for 1 hour. The material thus obtained was cooled to room temperature slowly and ground in a mortar and pestle to obtain a fine powder.ZrO2-supported nanoparticles were prepared by taking 0.9 mL of zirconium isopropoxide in isopropanol (70 weight%), 0.4 mL of nanoparticle sol dispersed in toluene and 0.7 mL of isopropanol in vessel 1 and 0.5 mL of methanol, 1.5 mL of nanoparticle sol dispersed in toluene and 72 μL of de-ionized water in vessel 2. Both these solutions were thoroughly sonicated and mixed with each other which led to the formation of an instantaneous viscous gel. This gel was dried in a similar way as adopted for MgO. Different nanoparticles anchored on MgO and ZrO2 supports are named M-DR-MO and M-mDR-MO (M: Pd, Pt, Rh and Ru and MO: MgO and ZrO2). Details of the abbreviated nomenclature are given in Table 1.
| Transition metals (TM) | Method of NP synthesisa | Support materials | Abbreviated name |
|---|---|---|---|
| a mDR = modified digestive ripening method; DR = digestive ripening method. | |||
| Pd | mDR | MgO | Pd-mDR-MgO |
| Pd | DR | MgO | Pd-DR-MgO |
| Pd | mDR | ZrO2 | Pd-mDR-ZrO2 |
| Pd | DR | ZrO2 | Pd-DR-ZrO2 |
| Pt | mDR | MgO | Pt-mDR-MgO |
| Pt | DR | MgO | Pt-DR-MgO |
| Pt | mDR | ZrO2 | Pt-mDR-ZrO2 |
| Pt | DR | ZrO2 | Pt-DR-ZrO2 |
| Rh | mDR | MgO | Rh-mDR-MgO |
| Rh | DR | MgO | Rh-DR-MgO |
| Rh | mDR | ZrO2 | Rh-mDR-ZrO2 |
| Rh | DR | ZrO2 | Rh-DR-ZrO2 |
| Ru | mDR | MgO | Ru-mDR-MgO |
| Ru | DR | MgO | Ru-DR-MgO |
| Ru | mDR | ZrO2 | Ru-mDR-ZrO2 |
| Ru | DR | ZrO2 | Ru-DR-ZrO2 |
For comparative catalytic experiments a sample christened as Pd-DR-MgO-un-annealed was also prepared by anchoring Pd-DR particles having a thiol-protecting layer on MgO by a similar process as mentioned above but without the annealing step (for the detailed experimental procedure please refer to ESI,† S1).
p-Nitro phenol reduction reaction
The catalytic activities of the MgO- and ZrO2-anchored MNPs were evaluated through the sodium borohydride-assisted p-nitro phenol (PNP) reduction reaction in an aqueous medium. Typically, 5 mg of solid catalyst was dispersed in 10 mL of distilled water. 600 μL of the freshly prepared 0.01 M PNP was added to the catalyst dispersion so that the final concentration of PNP is 10−4 M. Finally, 500 μL of 0.5 M freshly prepared sodium borohydride solution was added and the kinetics of the reaction were monitored by recording UV-Vis spectra by withdrawing aliquots of samples at regular time intervals. The order of the reaction was determined by fitting the concentration (obtained from time-dependent absorbance) into different rate law equations. The best fitted equation was considered as the order of reaction for a particular catalyst.Comparative experiments were also performed wherein the reduction was carried out in a chloroform–water two-phase system (for the detailed experimental procedure please refer to ESI,† S2).
Characterization
The crystallographic phase was judged by powder X-ray diffraction (PXRD) recorded on a X'pert Pro model PANalytical diffractometer from Philips PANalytical instruments operated at a voltage of 40 kV and a current of 30 mA with CuKα (1.5418 Å) radiation and at different scan rates depending upon the sample. The exact morphology of the catalyst was observed by Transmission Electron Microscope (TEM) images recorded using an FEI model TECNAI G2 F20 instrument operating at an accelerating voltage of 200 kV. The samples were prepared by dispersing them in ethanol and drop casting them onto a 200 mesh carbon-coated copper grid (Ted Pella, Inc.). Kinetic investigations of the catalytic reactions were carried out spectro-photometrically using a Varian make CARY 300 UV-Visible spectro-photometer. Cyclic voltammograms (CVs) were recorded in a 0.5 M H2SO4 electrolytic medium using a CHI 660E (compliance voltage: ±13 V) electrochemical work station and a standard three-electrode system. A known amount of nanomaterial dispersion was drop cast onto a 3 mm diameter glassy carbon electrode and allowed to dry overnight. The total nanomaterial loading on the electrode surface was kept the same with respect to their mass. These deposited electrodes were used as working electrodes. Pt wire and Ag/AgCl (3 M) were used as the counter and reference electrode, respectively.Results and discussion
Klaubunde et al. prepared nano-MgO and other oxides by a modified sol–gel process using toluene as a spectator solvent. This procedure is known to result in nanocrystalline oxide materials possessing high surface area.15,16 Recently, we synthesized transition MNPs (Pd, Pt, Rh and Ru) dispersed in toluene with controlled size and size dispersity.20 In the work presented here, we used these nanoparticle dispersions in toluene during the sol–gel process instead of pure toluene as proposed by Klabunde et al., and believed that this would lead to a better deposition of MNPs on the oxide surface. As-prepared M-mDR-MgO, M-DR-MgO, M-mDR-ZrO2 and M-DR-ZrO2 (M: Pd, Pt, Rh and Ru) samples were characterized by PXRD. The representative PXRD patterns of Pd and Pt nanoparticles anchored on MgO and ZrO2 are depicted in Fig. 1. | ||
| Fig. 1 XRD pattern for (A) Pd, and (B) Pt nanoparticles prepared by traditional and modified digestive ripening and anchored on MgO and ZrO2 supports. Diffraction peaks are marked by symbols as # – Pd and Pt, * – MgO, Δ – monoclinic ZrO2 and Φ – tetragonal ZrO2. | ||
For the PXRD plots of the Rh and Ru-based samples please see Fig. S1 in the ESI.† In all cases peaks corresponding to metal reflections (marked as # in the figure) could be clearly seen. For Pd, Pt and Rh, the diffraction patterns match with the metallic cubic crystallographic phase, whereas Ru displayed a hexagonal phase. Diffraction lines marked as * are characteristics of cubic phase MgO (JCPDS No. 78-0430). The diffraction patterns for the ZrO2-supported nanoparticles displayed characteristic lines for mixed phase ZrO2. Lines marked with Δ correspond to monoclinic ZrO2 (JCPDS No. 89-9066) and those marked with Φ match with the tetragonal phase (JCPDS No. 89-9068). The relative intensities of both the lines indicate that the monoclinic ZrO2 phase is the dominant one. The feeble diffraction pattern observed for the metals is attributed to both the smaller particle sizes and their extended spatial distribution on the support. Thus, the PXRD data clearly endorse the presence of the respective metals along with either a cubic phase of MgO or a mixed phase of ZrO2.
The exact morphology and nature of the samples were analyzed by TEM. The representative TEM images and the corresponding size distribution histograms for the Pd and Pt nanoparticles anchored on MgO and ZrO2 are shown in Fig. 2 and 3, respectively.
 | ||
| Fig. 2 Upper panel: TEM images for (A) Pd-mDR-MgO, (B) Pd-DR-MgO, (C) Pd-mDR-ZrO2 and (D) Pd-DR-ZrO2 particles; lower panel: particle size distribution histogram for (E) Pd-mDR-MgO, (F) Pd-DR-MgO, (G) Pd-mDR-ZrO2 and (H) Pd-DR-ZrO2 particles. | ||
 | ||
| Fig. 3 Upper panel: TEM images for (A) Pt-mDR-MgO, (B) Pt-DR-MgO, (C) Pt-mDR-ZrO2 and (D) Pt-DR-ZrO2 particles; lower panel: particle size distribution histogram for (E) Pt-mDR-MgO, (F) Pt-DR-MgO, (G) Pt-mDR-ZrO2 and (H) Pt-DR-ZrO2 particles. | ||
In the images the presence of small sized transition metal nanoparticles having darker contrast anchored to the support (MgO or ZrO2) is very evident. These are attributed to the MNPs. It may be noticed that the metal nanoparticles are well dispersed on the metal oxide support. The corresponding particle size distribution histograms suggest the average sizes of ca. 2.2 nm and 5 nm for Pd and 3.3 nm and 5.9 nm for Pt. Though a few bigger particles are also observed (especially in case of ZrO2 support), it was gratifying to see that most of them retained their original sizes obtained after the DR and mDR procedures20 and were also found to be well separated from each other.
The mean sizes obtained from the TEM images and the crystallite sizes calculated from PXRD, using the Scherrer equation, for the supported particles are presented in Table 2. It may be noticed that there is good agreement between these two independent measurements which again indicates that the MNPs have not undergone any aggregation-induced growth during the deposition or annealing, exemplifying the utility of this method. Hence, based on the above results, the deposition of Pd, Pt, Rh and Ru nanoparticles on the MgO and ZrO2 supports is confirmed. The retention of the MNP sizes and their well-dispersed nature on the metal oxide support is attributed to the presence of toluene during the sol–gel process as proposed by Klabunde and co-workers.16
| Catalyst | Mean size (TEM) (nm) | Crystallite size (nm) | Catalyst | Mean size (TEM) (nm) | Crystallite size (nm) |
|---|---|---|---|---|---|
| Pd-mDR-MgO | 2.2 | 2.0 | Pt-mDR-MgO | 3.3 | 3.0 |
| Pd- DR-MgO | 5.4 | 5.1 | Pt-DR-MgO | 5.4 | 5.2 |
| Pd-mDR-ZrO | 2.4 | 2.1 | Pt-mDR-ZrO | 3.3 | 3.0 |
| Pd-DR-ZrO | 5.0 | 4.7 | Pt-DR-ZrO | 5.9 | 5.6 |
We reckon that the hydrophobic nature of toluene in which the MNPs are dispersed leads to the confinement of MNPs at the polar–non polar interface.16 This ensures that during the solvent evaporation process they get deposited on the metal oxide surface. Significantly, the present synthetic strategy does not involve the control of any critical solution parameters (viz. pH, concentration etc.) that are necessary for the surface confinement of nanoparticles.10 Thus, this method is more versatile and material independent and hence can be extended to any type of nanomaterial that one desires to load as long as the same can be obtained as a dispersion in a non-polar solvent. Moreover, the spatial distribution of the deposited particles without any morphology change is assured as they do not participate in the sol–gel process.
Having established the generality of the procedure to prepare different supported MNPs, we then proceeded to investigate their catalytic activity using the well-known borohydride-assisted reduction of PNP. PNP produces phenolate ions in a basic medium which gives a characteristic peak at ∼400 nm in the UV-Vis spectrum. PNP reduction results in the formation of p-amino phenol (PAP) which has a characteristic peak at ∼290 nm. Therefore, the progress of PNP reduction and kinetics of the reaction can be easily followed spectro-photometrically by monitoring the absorbance at ∼400 nm with respect to reaction time.21 Representative graphs for the time-dependent evolution of the UV-Vis spectra for the above reaction in the presence of two different palladium nanoparticles anchored on magnesium oxide and zirconia supports are depicted in Fig. 4A–D. For the UV-Vis spectra of the PNP reduction reaction catalyzed by Pt, Rh, and Ru DR and mDR nanoparticles deposited on MgO and ZrO2 supports, please refer to Fig. S4–S6 in the ESI.† As can be seen, the peak at 400 nm corresponding to phenolate ions gradually decreases along with a concomitant increase in absorbance at 290 nm. It may be noticed that the reaction gets completed within 15 minutes, 4 minutes, 15 minutes and 6 minutes when Pd-MgO-mDR, Pd-MgO-DR, Pd-ZrO2-mDR and Pd-ZrO2-DR are used as catalysts, respectively. The Pt, Rh and Ru nanoparticles prepared by the DR and mDR methods and anchored on MgO and ZrO2 supports showed similar catalytic activity and trends with slight variation in reaction time.
 | ||
| Fig. 4 UV-vis spectra for the PNP reduction reaction catalyzed by (A) Pd-mDR-MgO, (B) Pd-DR-MgO, (C) Pd-mDR-ZrO2 and (D) Pd-DR-ZrO2 catalysts. | ||
The overall general trend observed in the catalytic activity of different samples studied here can be summarized as M-DR-MgO > M-mDR-MgO > M-DR-ZrO2 > M-mDR-ZrO2 (where M: Pd, Pt, Rh and Ru). Most importantly, the extent of PNP reduction is higher for supported MNPs than unsupported MNPs and the effect is more pronounced for the MgO support. Compared to ZrO2 a clear isosbestic point is observed for the MgO support suggesting that PAP is the only product formed.22 The diffused isosbestic point observed in the case of ZrO2-supported MNPs could be due to the modification in the reaction mechanism (vide infra). Interestingly, there is no induction period observed for all the particles which indicates that the surface-assisted reduction reaction starts immediately and hence the catalyst is reasonably active. No PNP reduction was observed when a metal oxide without deposited nanoparticles was used as a catalyst which clearly emphasized the role of the deposited MNPs in the overall catalytic activity (Fig. S7 in the ESI†). It is important to notice here that the said reaction is completed using supported MNPs at significantly lower metal loading (just 4–6%) suggesting higher catalytic activities of the metal oxide supported particles compared with the respective unsupported particles which can also be evidenced by the turn over frequency (TOF) numbers (Table 3). The product formation rate in turn was determined based on decrease in absorbance at 400 nm, where the surface atom density of the metal catalyst was determined using the electrochemical technique i.e. based on charges under the oxide reduction peak (for Pd) and the hydrogen desorption peak (for Pt and Rh) (for detailed information please refer to ESI,† S4).23 To determine the TOF for unsupported particles we have used the data from ref. 20 where PNP reduction was carried out using the unsupported-ligand protected particles. It is observed that the TOF of the supported nanoparticles is 4–5 orders of magnitude higher than that of the unsupported ones. To rationalize the support contribution towards the improved catalytic activity of the supported particles, we have synthesized MgO-supported Pd-DR nanoparticles (Pd-DR-MgO-un-annealed) by a similar process where the thiol-protecting layer on the particle surface was retained by avoiding the annealing step (which removes the protecting thiol layer) and tested the same for PNP reduction (refer to Fig. S8 and Table S2, ESI†). It is observed that the TOF and rate constant for Pd-DR-MgO-un-annealed (k = 0.13 min−1 and TOF = 116) are four orders of magnitude higher than those of the unsupported nanoparticles but lower than those of Pd-DR-MgO. It may be noticed that the rate constant and TOF numbers both in the case of Pd-DR-MgO-un-annealed and Pd-DR-MgO are of similar orders of magnitude. The slightly higher values observed for Pd-DR-MgO could be due to the presence of a free metal surface devoid of any ligand protection. This clearly highlights that more than the presence or absence of ligands on the MNPs, the support surface plays a major role towards the enhanced catalytic activity.24 The ligand-free metal surface in Pd-DR-MgO enhances this effect even further.
| Sr. no. | Catalyst | n | K | TOFa |
|---|---|---|---|---|
| a M M−1 min−1. b Data taken from ref. 20. | ||||
| Pd-mDR | Pd-mDRb | 1 | 1.006 min−1 | 0.069 |
| Pd-mDR-MgO | 1 | 0.18 min−1 | 38.75 | |
| Pd-mDR-ZrO2 | 1 | 0.19 min−1 | 32.30 | |
| Pd-DR | Pd-DRb | 1 | 8.61 × 10−3 min−1 | 2.7 × 10−3 |
| Pd-DR-MgO | 1 | 0.86 min−1 | 464.68 | |
| Pd-DR-ZrO2 | 1 | 0.55 min−1 | 309.8 | |
| Pt-mDR | Pt-mDRb | 1 | 6.35 × 10−3 min−1 | 7.6 × 10−4 |
| Pt-mDR-MgO | 1 | 0.33 min−1 | 1.34 | |
| Pt-mDR-ZrO2 | 2 | 0.04 M−1 min−1 | 0.32 | |
| Pt-DR | Pt-DRb | 1 | 4.65 × 10−3 min-1 | 7.7 × 10−4 |
| Pt-DR-MgO | 1 | 0.37 min−1 | 1.96 | |
| Pt-DR-ZrO2 | 2 | 0.2 M−1 min−1 | 0.8 | |
| Rh-mDR | Rh-mDRb | 1 | 57.0 × 10−3 min−1 | 5.33 × 10−3 |
| Rh-mDR-MgO | 0 | 0.14 M min−1 | 11.09 | |
| Rh-mDR-ZrO2 | 1 | 0.23 min−1 | 6.8 | |
| Rh-DR | Rh-DRb | 1 | 6.49 × 10−3 min−1 | 2.29 × 10−3 |
| Rh-DR-MgO | 0 | 0.19 M min−1 | 23.56 | |
| Rh-DR-ZrO2 | 1 | 0.29 min−1 | 12.5 | |
| Ru-mDR | Ru-mDRb | 1 | 5.4 × 10−3 min−1 | — |
| Ru-mDR-MgO | 0 | 0.04 M min−1 | — | |
| Ru-mDR-ZrO2 | 2 | 0.05 M−1 min−1 | — | |
| Ru-DR | Ru-DRb | 1 | 1.96 × 10−3 min−1 | — |
| Ru-DR-MgO | 0 | 0.06 M min−1 | — | |
| Ru-DR-ZrO2 | 2 | 0.04 M−1 min−1 | — | |
The kinetics of the catalytic reaction were followed for all the samples by fitting the UV-Vis data into various rate law equations and the rate constants were determined based on best fitting of data into the corresponding rate law equation. Fig. 5 depicts the fitting results for Pd and Pt nanoparticles anchored on MgO and ZrO2 supports. The fitting results for Rh and Ru nanoparticles are depicted as Fig. S9 in the ESI.†
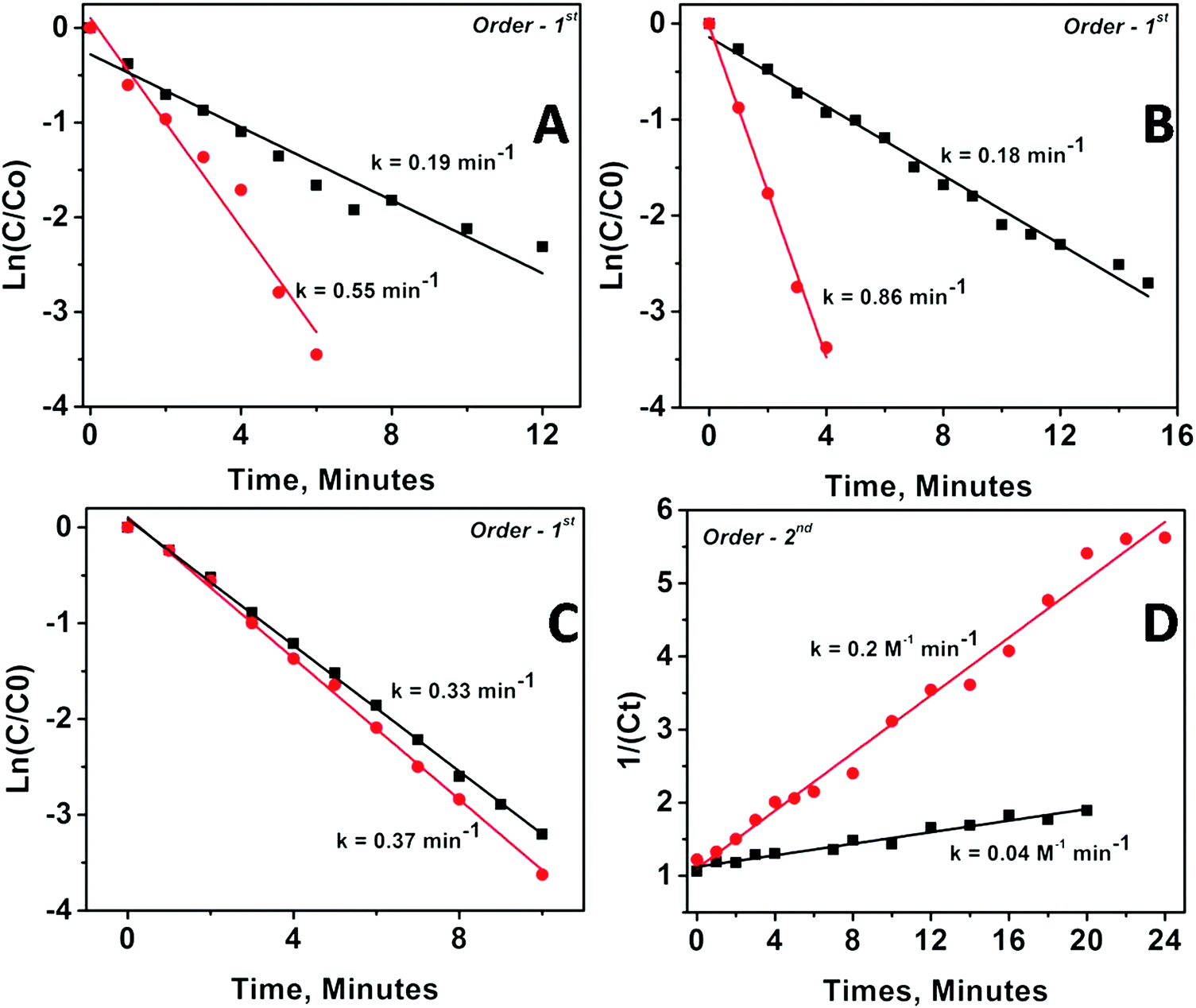 | ||
| Fig. 5 Fitting of the reaction kinetics of p-nitro phenol reduction with different rate law equations. In the presence of (A) Pd-mDR-MgO (black), and Pd-DR-MgO (red), (B) Pd-mDR-ZrO2 (black) and Pd-DR-ZrO2 (red) catalysts; (C) Pt-mDR-MgO (black), and Pt-DR-MgO (red), and (D) Pt-mDR-ZrO2 (black) and Pt-DR-ZrO2 (red) catalysts. | ||
The corresponding rate constants and order of reaction determined are given in Table 3. The values presented clearly indicate the modifications in kinetics of catalyzed PNP reduction in terms of rate constants and/or orders of reaction. Briefly, (1) the catalytic activity of all the supported nanoparticles is higher than the catalytic activity of the same un-supported nanoparticles; (2) MgO-supported particles show higher catalytic activity than ZrO2-supported particles; (3) metal oxide supported bigger particles (obtained from DR) show higher rate constant than smaller ones (obtained via mDR); and (4) ZrO2-supported particles always followed higher order kinetics as compared with their MgO-supported counterparts (except in the case of Pd nanoparticles, where the order of the reaction for MgO-supported and ZrO2-supported particles is the same albeit with different rate constants). It may be worth reemphasizing here that as compared with the unsupported particles, where the smaller sized ones are found to be more active than the bigger ones, the trend is reversed when the same particles are anchored to the oxide supports.
This turn-around and change in catalytic activity of the same material after supporting it on a metal oxide substrate demanded a detailed investigation of the role of the support and the same was attempted using X-ray photoelectron spectroscopy (XPS) using Pd as a representative case; depicted as Fig. S10 in ESI.† A sharp Pd 3d doublet was observed for unsupported particles with a peak spacing of 5.3 eV. Very interestingly, the XPS spectra of the same particles on the MgO support showed very broad peaks. Such broadening has been ascribed to the MgO charging effect25 and has been suggested to be indicative of the interaction between Pd and MgO. Thus, the XPS results indicate that even when metal particles are anchored on oxide supports their electronic nature gets altered. To probe this aspect in a more detailed way we carried out cyclic voltammetric studies on these materials. Fig. 6 depicts the representative example of CVs recorded using the unsupported and MgO-supported Pd nanoparticles prepared by the DR and mDR methods. It may be noted that the CVs displayed here are for the MNPs whose ligand protection was removed as the CVs recorded using unsupported nanoparticles having thiol protecting layers did not show any redox characteristics perhaps due to the insulating nature of the thiol layer that prevents electron transfer from the electrode to the nanoparticles. During electrochemical cycling, Pd and Pt undergo electrochemical oxide formation in an anodic scan and their subsequent reduction in a cathodic scan. The latter process is sensitive and informative to understand the nature of the metal surface. Hence, we have concentrated on the characteristic reduction peak for PdO located in the cathodic scan.26 In the case of unsupported nanoparticles, the PdO reduction peak appeared at 312 mV and 130 mV for Pd-mDR and Pd-DR, respectively. The negative shift in reduction peak for Pd-DR suggests the sluggish nature of the redox process in this case as compared with Pd-mDR. However, for MgO-supported particles, these peaks shifted to 378 mV and 400 mV respectively for Pd-mDR-MgO and Pd-DR-MgO. Moreover, complementary anodic peaks are seen to be developed at 867 mV and 767 mV for Pd-mDR-MgO and Pd-DR-MgO. The positive shift in reduction potential along with the improved reversibility clearly indicates the enhanced activity of MgO-supported particles. The higher activity of bigger supported particles (Pd-DR-MgO) as compared with smaller supported particles (Pd-mDR-MgO) could be due to positive reduction potential as well as faster electron transfer kinetics (smaller ΔV).27 Therefore, based on CV investigations the higher activity of the supported particles could be ascribed to the modification in the redox potential of MNPs and enhanced electron transfer kinetics stimulated by the MgO support. Such modifications in electronic nature have been explained earlier based on the Schottky metal insulator semiconductor (SMIS) concept.28 Surprisingly, we also observed a similar phenomenon though in this case no insulating layer between the MNPs and support is expected to be present. As our system would not be a true case to be explained by the SMIS effect, we attribute the observed modifications in redox behaviour to the basic nature of MgO which could push the electron density towards the metal particles, thereby uplifting its Fermi level and hence improving its catalytic activity.29
 | ||
| Fig. 6 CVs recorded for the (A) unsupported and (B) MgO-supported Pd nanoparticles prepared by traditional digestive ripening (DR) and modified digestive ripening (mDR). The material was deposited on a glassy carbon electrode and CVs were recorded in 0.5 M H2SO4 solution. Scan rate 50 mV s−1. | ||
To support this contention we carried out the CV measurements on ZrO2-supported MNPs as well. Fig. S11 (ESI†) depicts the CVs recorded using Pt-DR nanoparticles anchored on MgO and ZrO2 supports. It can clearly be seen from the figure that the reduction potential of MgO-supported Pt nanoparticles (430 mV) is more positive compared to that of ZrO2 (161 mV) supported particles and this is responsible for the higher catalytic activity of the former system, namely Pt-DR-MgO. The pronounced modification of the catalytic activity with MgO-supported particles can be explained on the basis of the more basic nature of MgO as compared to ZrO2.
Comparative kinetic investigations of all the supported transition MNPs depict the variation in the orders of reaction. In particular, the order of reaction for Pt, Rh and Ru nanoparticles anchored on ZrO2 is higher than that for the MgO support. The increase in the order of reaction for the ZrO2 support can again be explained on the basis of its less basic character as compared with the MgO. ZrO2 possess higher oxy-philic groups that are acidic in nature because of which its electron-donating tendency decreases.30 PNP possesses a nitrogen functionality that has a lone pair of electrons which can interact strongly with oxy-philic groups present on ZrO2. Hence, the overall reaction kinetics is now governed by surface-confined reactants and their adsorption/desorption rate. Owing to the interaction between PNP and ZrO2 their rate of desorption is slower and hence all the active sites get saturated, thereby influencing the overall reaction mechanism and shifting it to higher order.31,32 Interestingly, for Ru supported on ZrO2, the order of reaction increases from zero order to second order which we attributed to the less contribution from surface active sites.33 Modification in the order of reaction of PNP reduction due to an environmental effect has been reported earlier.32 In fact, the absence of a clear isosbestic point for ZrO2-supported nanoparticles (except Pd) supports the modification in the reaction mechanism. Hence, based on the comparative kinetic and CV investigations, MgO can be regarded as a better support for the improvement in catalytic activity of MNPs towards PNP reduction.
Conclusion
We have successfully demonstrated the anchoring of pre-synthesized, size-controlled Pd, Pt, Rh and Ru nanoparticles on the MgO and ZrO2 supports through a modified sol–gel method. Use of a nanoparticle dispersion in toluene is responsible for the unique architecture of the supported nanoparticles obtained. The simplicity of the synthetic technique makes it a more versatile method for anchoring a wide range of nanomaterials on suitable metal oxide supports. The catalytic activity of the supported nanoparticles was investigated through the p-nitro phenol reduction reaction where improved activity of the supported particles is observed. Furthermore, MgO is found to be an efficient support as compared to ZrO2. Support-dependent catalytic activity and modification in the reaction mechanism were evaluated through cyclic voltammetric investigations which suggest that support-induced modification in the electronic nature of the particles is responsible for the observed performances.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JS thanks the University Grant Commission for the fellowship. BLVP and VRC thank the Council of Scientific and Industrial Research for the financial support through the project CSC0134. PD thanks Indian Science Academies for the faculty summer fellowship which enabled him to visit CSIR-NCL and carry out this work.References
- S. E. Lohse and C. J. Murphy, J. Am. Chem. Soc., 2012, 134, 15607–15620 CrossRef CAS PubMed.
- L. Dolatyari, M. R. Yaftian and S. Rostamnia, J. Environ. Manage., 2016, 169, 8–17 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- J. A. Schwarz, C. Contescu and A. Contescu, Chem. Rev., 1995, 95, 477–510 CrossRef CAS.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- P. Munnik, P. E. de Jongh and K. P. de Jong, J. Am. Chem. Soc., 2014, 136, 7333–7340 CrossRef CAS PubMed.
- K. Morgan, R. Burch, M. Daous, J. J. Delgado, A. Goguet, C. Hardacre, L. A. Petrov and D. W. Rooney, Catal. Sci. Technol., 2014, 4, 729–737 CAS.
- C. Amiens, D. Ciuculescu-Pradinesa and K. Philippot, Coord. Chem. Rev., 2016, 308, 409–432 CrossRef CAS.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- N. J. S. Costa and L. M. Rossi, Nanoscale, 2012, 4, 5826–5834 RSC.
- Y. Wang, J. Ren, K. Deng, L. Gui and Y. Tang, Chem. Mater., 2000, 12, 1622–1627 CrossRef CAS.
- W. Li, C. Liang, W. Zhou, J. Qiu, Z. Zhou, G. Sun and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292–6299 CrossRef CAS.
- R. M. Rioux, H. Song, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, J. Phys. Chem. B, 2005, 109, 2192–2202 CrossRef CAS PubMed.
- K. T. Ranjit and K. J. Klabunde, Chem. Mater., 2005, 17, 65–73 CrossRef CAS.
- Y. Diao, W. P. Walawender, C. M. Sorensen, K. J. Klabunde and T. Ricke, Chem. Mater., 2002, 14, 362–368 CrossRef CAS.
- J. Seth, D. Nepak, V. R. Chaudhari and B. L. V. Prasad, Appl. Surf. Sci., 2017, 418, 87–91 CrossRef CAS.
- J.-M. Arce-Ramos, L. C. Grabow, B. E. Handy and M.-G. Cárdenas-Galindo, J. Phys. Chem. C, 2015, 119, 15150–15159 CAS.
- D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed.
- J. Seth and B. L. V. Prasad, Nano Res., 2016, 9, 2007–2017 CrossRef CAS.
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2010, 26, 2885–2893 CrossRef CAS PubMed.
- A. Fedorczyk, J. Ratajczak, O. Kuzmych and M. Skompska, J. Solid State Electrochem., 2015, 19, 2849–2858 CrossRef CAS.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CrossRef CAS.
- Bokhimi, A. Aceves, O. Novaro, T. Lopez and R. Gomez, J. Phys. Chem., 1995, 99, 14403–14406 CrossRef CAS.
- D. J. O’Connor, B. A. Sexton and R. St. C. Smart, Surface Analysis Methods in Materials Science, 2nd edn, Springer, 2003, pp. 405–432 Search PubMed.
- L.-L. Fang, Q. Tao, M.-F. Li, L.-W. Liao, D. Chen and Y.-X. Chen, Chin. J. Chem. Phys., 2010, 23, 543–548 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, John Wiley & Sons, Inc., 2001, 2016 Search PubMed.
- I. A. Digdaya, G. W. P. Adhyaksa, B. J. Trześniewski, E. C. Garnett and W. A. Smith, Nat. Commun., 2017, 8, 15968 CrossRef CAS PubMed.
- N. Mahata, K. V. Raghavan, V. Vishwanathan, C. Park and M. A. Keane, Phys. Chem. Chem. Phys., 2001, 3, 2712–2719 RSC.
- P. M. de Souza, R. C. Rabelo-Neto, L. E. P. Borges, G. Jacobs, B. H. Davis, U. M. Graham, D. E. Resasco and F. B. Noronha, ACS Catalysis, 2015, 5, 7385–7398 CrossRef CAS.
- Z. D. Pozun, S. E. Rodenbusch, E. Keller, K. Tran, W. Tang, K. J. Stevenson and G. Henkelman, J. Phys. Chem. C, 2013, 117, 7598–7604 CAS.
- M. Li and G. Chen, Nanoscale, 2013, 5, 11919–11927 RSC.
- C. Ou, S. Zhang, J. Liu, J. Shen, Y. Liu, X. Sun, J. Li and L. Wang, Phys. Chem. Chem. Phys., 2015, 17, 22072–22078 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray diffractograms for Rh and Ru nanoparticles prepared by digestive and modified digestive ripening and anchored on MgO and ZrO2 supports (Fig. S1), TEM images and corresponding histograms for Rh and Ru nanoparticles prepared by digestive and modified digestive ripening and anchored on MgO and ZrO2 supports (Fig. S2 and S3), and time dependent UV-Vis spectra for p-nitro phenol catalyzed by Pt, Rh, and Ru nanoparticles prepared by digestive and modified digestive ripening and anchored on MgO and ZrO2 supports (Fig. S4–S6). See DOI: 10.1039/c7nj03753h |
| ‡ Permanent address: Centre of Material Sciences, Institute of Interdisciplinary Studies, University of Allahabad, Allahabad-211002, Uttar Pradesh, India. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |