
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrooxidative CH/PH functionalization as a novel way to synthesize benzo[b]phosphole oxides mediated by catalytic amounts of silver acetate
V. V.
Khrizanforova
 *,
K. V.
Kholin
,
M. N.
Khrizanforov
,
M. K.
Kadirov
and
Yu. H.
Budnikova
*,
K. V.
Kholin
,
M. N.
Khrizanforov
,
M. K.
Kadirov
and
Yu. H.
Budnikova
A.E. Arbuzov Institute of Organic and Physical Chemistry Russian Academy of Sciences, Kazan Scientific Center, Arbuzov str. 8, 420088 Kazan, Russian Federation. E-mail: khrizanforovavera@yandex.ru; Fax: (+7) 8432752253
First published on 27th November 2017
Abstract
Electrochemical joint oxidation of diphenylphosphine oxide with acetylenes in the presence of silver acetate (10%) as a catalyst yields benzo[b]phosphole oxide derivatives under mild conditions at room temperature without additional oxidants or initiators and with yields up to 100%. The redox properties of the key intermediate of the catalytic cycle have been investigated by cyclic voltammetry and EPR spectroscopy. The Ph2P(O)Ag intermediate oxidizes easier (+0.05 V vs. Fc+/Fc) than all reaction precursors to form the phosphinyl radical Ph2P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)˙, fixed as a spin-adduct by ESR. Thus, a radical mechanism type through the Ag+/Ag0 couple was developed for electrocatalytic conditions.
O)˙, fixed as a spin-adduct by ESR. Thus, a radical mechanism type through the Ag+/Ag0 couple was developed for electrocatalytic conditions.
Introduction
Phosphorus-containing heterocycles have attracted significant interest from synthetic chemists over the last few years because of their wide application in organic synthesis, medicinal chemistry, and materials science.1–5 Among them, benzo[b]phosphole oxide derivatives, phosphorus-containing π-conjugated compounds, have attracted significant attention as promising organic optoelectronic materials due to their unique physical and photoelectric properties.6–14 This explains the urgent need to develop new effective, atom-economical catalytic routes for their synthesis.Since 2013, various one-stage methods for the synthesis of benzo[b]phosphole oxide derivatives 3 through dehydrogenative annulation (Scheme 1) have been demonstrated.15–20 The disadvantages of all the described methods are requirement of high temperature and excessive use of oxidants such as Mn(OAc)3 or AgOAc (4 equiv.), as reported by Satoh,15 Ag2O (2 equiv.) or Pd(OAc)2 with three-fold excess of Ag salts and an equivalent amount of metal nitrates, as reported by Duan,16 AgOAc (2 equiv.), as reported by Ackermann,17 and TBHP (2 equiv.) with CuSO418 or K2S2O8 (5 equiv.), as reported by Zhao.19 Metal derivatives (2% CuSO4,18 5% Ag2O,16 2.5% Cp*RhCl216 or Pd(OAc)216) in catalytic amounts perform well only under conditions of excess various oxidants, with the product yield dependent on the nature of the latter (therefore, for each condition, a screening is required), as well as at elevated temperatures and often in excess of diphenylphosphine oxide.15,16,18 Low temperature for benzo[b]phosphole oxide preparation was used only in some instances. Only Lakhdar synthesized benzo[b]phosphole oxides under photocatalytic conditions using Eosin Y as the catalyst and N-ethoxy-2-methylpyridinium tetrafluoroborate as an oxidant at a lower temperature, 35 °C,20 and Satoh used 4 equiv. of Mn(OAc)315 at room temperature, but the yields were lower than those obtained at 100 °C. The use of additional reagents (oxidants and bases), typical for traditional reactions, leads to an increase in the production cost, complicates the separation, and increases waste.
 | ||
| Scheme 1 Synthesis of benzophosphole oxide derivatives. | ||
The mechanism of most Ag-catalyzed reactions is proposed to be radical type based on the absence of the target product when some equivalent of TEMPO is present in the reaction mixture. However, the failure of the reaction in the presence of TEMPO can be explained by the fact that the latter is easily oxidized by the applied oxidant, for example, K2S2O8. Moreover, if TEMPO was taken in an equivalent amount or two-fold excess over the oxidant, the entire oxidant was spent by the side reaction of oxidation of TEMPO, which is easily oxidized to about 0 V ref. Fc+/Fc or +0.42 V ref. Ag/AgCl.21,22 However, in 2017, Li's group23 described a novel Mn(II)-promoted tandem phosphorylation/cyclization reaction with phosphine oxides and suggested that unlike all the previously discussed reactions, the formation of a P-cycle was carried out by an ionic, not radical path. As a rule, the proposed cycles are tentative and cannot explain the use of excess of silver salt for example.15,16
In recent years, a series of successful electrocatalytic reactions with C–C, C–P bond formation catalyzed by metal complexes in reductive or oxidative conditions have been performed.24–33 In the present study, the synthesis of benzo[b]phosphole oxides from diphenylphosphine oxide and acetylenes catalyzed by Ag salt (0.1 equiv.) at room temperature under electrochemical conditions has been demonstrated. In this case, an excess of oxidant or Ag salt is not required due to electrochemical regeneration of the catalyst's active form near the electrode surface.
Results and discussion
Electrocatalytic oxidative synthesis
AgOAc is frequently used as an oxidant or a catalyst for successful Ag-mediated CH/PH functionalization of diphenylphosphine oxide by acetylenes.15–17,34,35It has been found that joint electrochemical oxidation of diphenylphosphine oxide and acetylene in the presence of 0.1 equiv. AgOAc at room temperature leads to the formation of benzo[b]phosphole oxide 3 with mild to high yields (Table 1).
| Compound | R1 | R2 | Spectral yield, % | Isolated yield, % |
|---|---|---|---|---|
| 3a | Ph | Ph | 100 | 90 |
| 3b | p-NO2–Ph | p-NO2–Ph | 90 | 85 |
| 3c | Pr | Pr | 85 | 80 |
| 3d | Ph | Et | 50 | 40 |
| 3e | Ph | Pr | 40 | 35 |
| 3f | Ph | Bu | 40 | 35 |
| 3g |

|
55 | 35 | |
31P NMR monitoring of the electrochemical oxidation of the reaction mixture revealed the need for 2 F electricity per 1 mole of diphenylphosphine oxide 1 for conversion of 1 into the target cyclization product 3 with yields up to 100% (Table 1 and Fig. 1). The electrolysis potential was 0.15–0.35 V (vs. Fc+/Fc). Thus, the diphenylphosphine oxide 1 (commercially available, δ = 22.07)36 is fully converted into benzo[b]phosphole oxide after passing 2 F electricity per 1 mole of phosphine oxide with δ = 37.0–40.0.16 The result of 31P NMR spectra monitoring is demonstrated in Fig. 1. The yield of the target benzo[b]phosphole oxides is 85–100% in the case of symmetric acetylenes and 40–50% for asymmetric acetylenes. In the case of 3d–f, only one cyclization product is formed.
 | ||
| Fig. 1 31P NMR monitoring of electrochemical oxidation of diphenylphosphine oxide and diphenylacetylene mixture before and after 1 and 2 F electricity was passed. | ||
Cyclic voltammetry
For the detailed investigation of the reaction pathway, the methods of cyclic voltammetry (CV) and electron-spin resonance spectroscopy (ESR) were used. At first, the redox properties of AgOAc and Ph2P(O)Ag – a key intermediate of the catalytic cycle (formed from AgOAc and diphenylphosphine oxide), acetylenes, and Ph2P(O)H – were studied by cyclic voltammetry. Acetylenes and Ph2P(O)H are not redox active.The CVs of AgOAc contain one reduction and two oxidation peaks at −0.48 V and −0.16 and 1.43 V (vs. Fc+/Fc) for Ag(I/0) and Ag(0/I/II), respectively, in CH3CN (Fig. 2). There is little data on the oxidation potentials for Ag0/Ag+/Ag2+ couples in organic media in literature. High standard potential of silver(II) in acidic water (E0 = 1.98 V vs. normal hydrogen electrode, NHE)37 is found. The potential for the Ag+/Ag0 couple (strongly dependent on solvent and counter-ions) is 0.04 in acetonitrile.38 AgBF4 reveals peaks at −0.28, 0.016, and 1.40 V (vs. Fc+/Fc) for Ag(I/0) and Ag(0/I/II) under similar conditions. Thus, the excess of the Bu4NBF4 supporting electrolyte does not substitute OAc− in the silver salt AgOAc and is suitable for investigation.
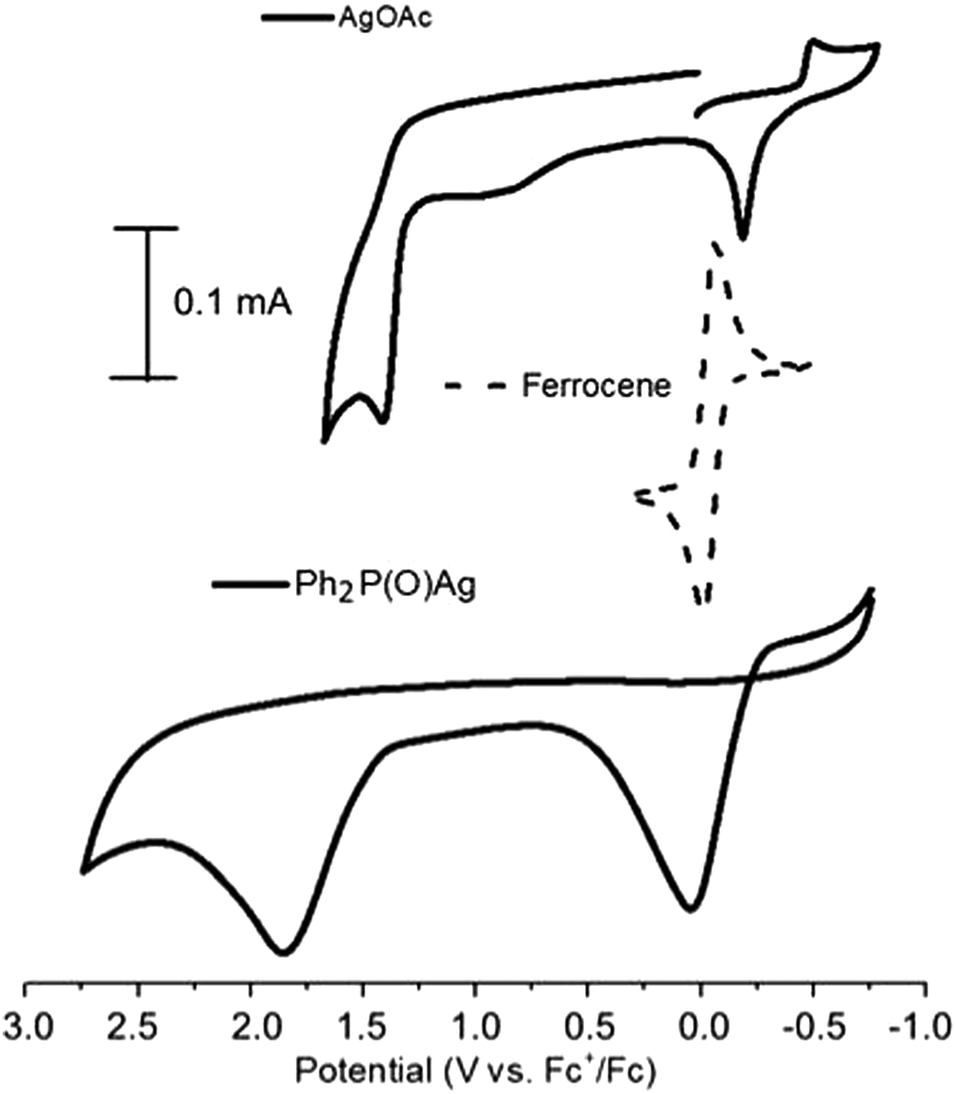 | ||
| Fig. 2 CVs of AgOAc and Ph2(O)PAg in CH3CN. Conditions: 0.1 V s−1 scan rate, 0.1 M Bu4NBF4, WE:GC, RE:Pt. | ||
We proposed that one key step in the investigated catalytic cycle should be AgP(O)Ph2 oxidation.16,35 Some contradictions should be noted in the studies of Ag-catalyzed C–H phosphonations. Most of the studies postulate the instant formation of a phosphinyl radical in the reaction of Ag(I) with Ph2P(O)H.39–41
However, at room temperature, when protected from visible light, this compound is stable and does not change its properties when stored even for several months. Sometimes, the participation of AgP(O)Ph2 was speculated, but it was not proved in studies reported in literature.16,35 We obtained Ph2(O)PAg through a previously reported method47–49 and characterized it spectrally. The investigation of the redox properties of the key Ph2(O)PAg intermediate by cyclic voltammetry has not been carried out to date. The CVs of Ph2P(O)Ag contain only two oxidation peaks at 0.05 and 1.85 V (Fig. 2). The first oxidation peak of Ph2P(O)Ag is close to the potential of electrolysis, which involves electrochemical phosphonation/cyclization. Thus, it was confirmed that the preparative electrochemical oxidation of diphenylphosphine oxide with acetylenes proceeds at the potential of the first oxidation peak of Ph2P(O)Ag.
ESR study and mechanistic considerations
Thus, the oxidation of Ph2(O)PAg salt at 0.05 V (vs. Fc+/Fc) is the key step in the electrochemical reaction. To establish the character of the catalytic cycle and the intermediate P(O)–Ag bond scission (ionic or radical type), we carried out a number of ESR experiments under anaerobic conditions. Joint oxidation of Ph2P(O)Ag and spin trap PBN (PBN = N-tert-butyl-α-phenylnitrone) mixture at 0.05 V (vs. Fc+/Fc) in an ESR-spectroelectrochemical cell is characterized by appearance and increase in the intensity of the ESR signal from the PBN-bound radical species of the Ph2P(O) adduct (Fig. 3 and Scheme 2).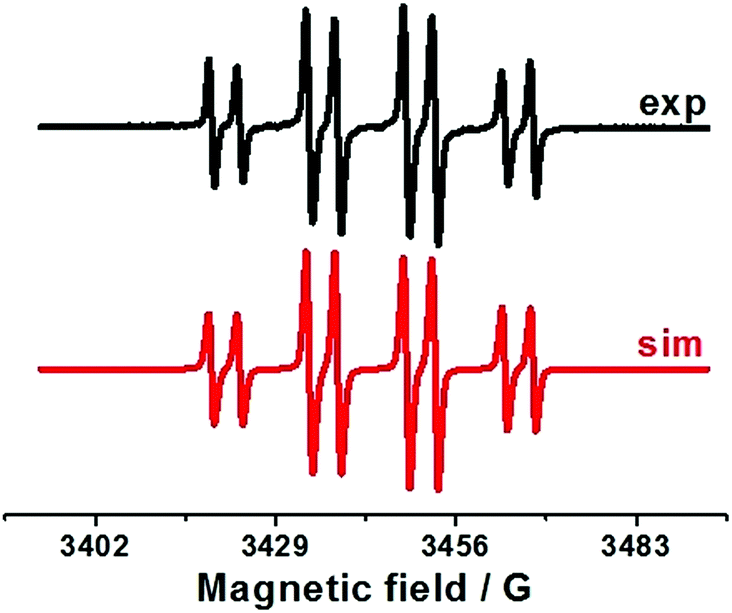 | ||
| Fig. 3 ESR spectra obtained during anodic joint electrolysis of Ph2P(O)Ag and spin trap PBN solution (left) at 0.05 V (vs. Fc+/Fc) in CH3CN, obtained at 293 K with simulations. | ||
 | ||
| Scheme 2 Trapping of phosphorus-centered radical by PBN. | ||
The ESR spectrum of the PBN adduct includes the splitting on the nuclei of nitrogen and hydrogen (in the α-position) and a significant splitting on the nucleus with nuclear spin I = 1/2, which is the nucleus of phosphorus at the β-position. The magnetic resonance parameters were obtained as a result of the simulation: g = 2.0059, aN = 14.5 G, aH = 4.3 G, and aP = 14.9 G. The observed g values at about g = 2.006 indicate the formation of PBN-bound radical species, and other magnetic resonance parameters are characteristic of phosphorus-containing PBN spin adducts.50,51 These parameters are close to those reported for the phosphinyl Ph2P(O)˙ radicals, but are slightly different from those since they were obtained elsewhere under other conditions.42–46
For example, in literature,44 the presence of the PBN/phosphinyl radical adduct, Ph2P(O)–PBN was revealed, but the presence of the benzoyl radical was also detected, and the spectra obtained were complicated. In literature,45 the spectrum of the phenyl–PBN radical adduct, formed after phenyl – radical generation, as a precursor to the phosphinyl radical has been reported. Ph2I+OTf (form Ph radical),45 camphorquinone (CQ)/diphenyl (2,4,6-trimethylbenzoyl)-phosphine oxide (TPO)/amine photoinitiating systems (form benzoyl radical),44 benzophenone, 2-isopropylthioxanthone, camphorquinone, 2,4,6-(4-methoxyphenyl)thiopyrylium tetrafluoroborate, and Eosin-Y42 were used as oxidants and photosensibilisators. Thus, on the way to phosphinyl radical, different types of radicals (phenyl, benzoyl, etc.) have been observed. This fact complicates the estimation of true PBN/phosphinyl radical adduct parameters. The data closest to our parameters were described by Buettner.52 Thus, electrooxidative conditions (without additional oxidants) for Ph2P(O)Ag oxidation are the most correct for the detection of the phosphinyl radical.
Thus, we can conclude that Ph2P(O)˙ reacts readily with the spin trap, and the spin adduct is quite stable.
The result suggested that this reaction followed a radical pathway. It should be noted that in addition to this spectrum (Fig. 3), a very wide line with g = 2.34 and ΔH peak–peak = 1200 G (the black line in Fig. 4) was observed. This signal can be apparently assigned to the metallic Ag(0),42 the particles of which precipitate at the bottom of the cell.
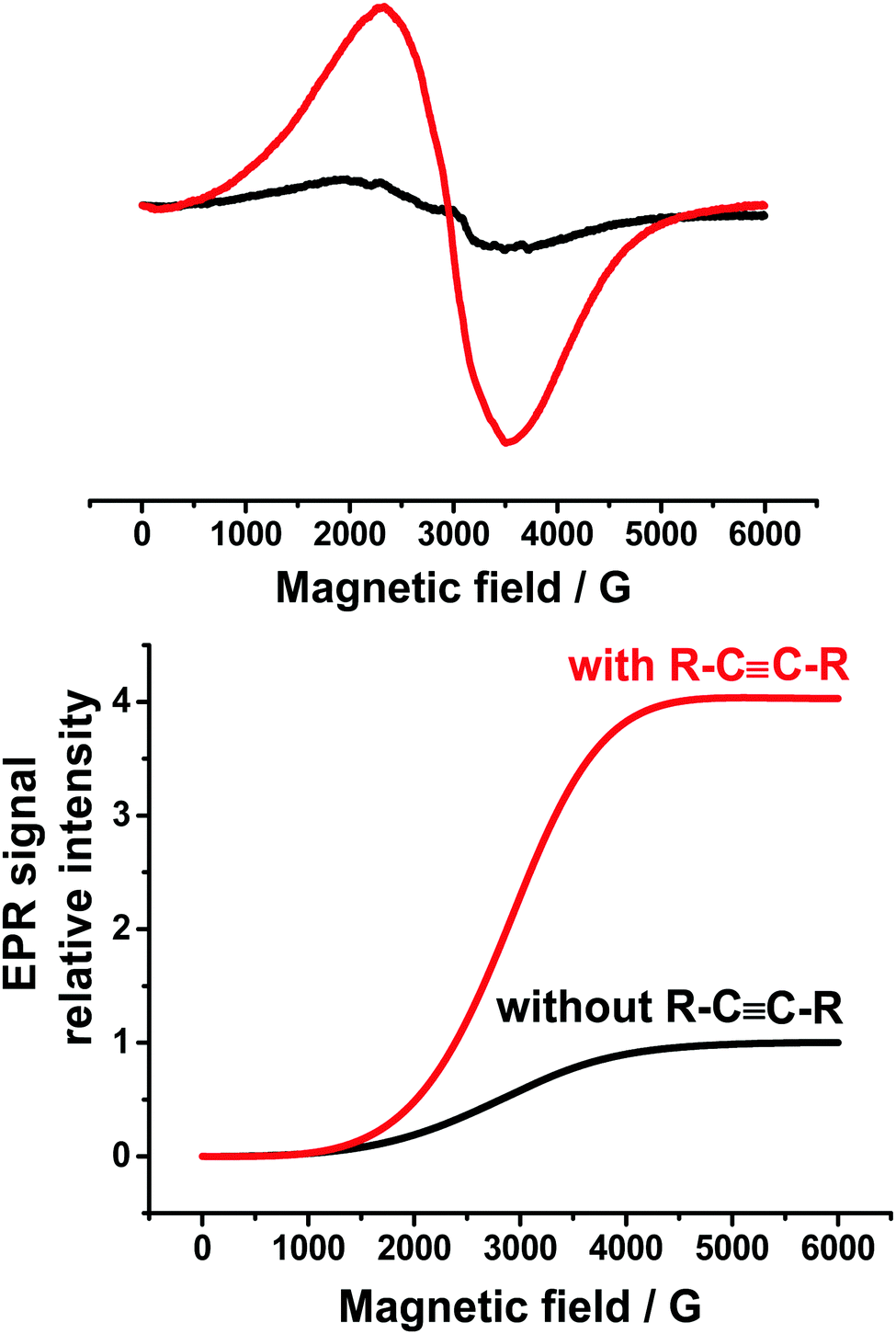 | ||
| Fig. 4 Comparison of Ag(0) signals in the presence (red) or absence (black) of diphenylacetylene (upper), and the intensities of these signals (lower). | ||
Interestingly, the addition of diphenylacetylene does not qualitatively change the type of the ESR spectra obtained during electrolysis of this mixture at the potential of 0.15 V. However, the intensity of the spectrum of Ph2P(O)˙–PBN decreases, but the intensity of the Ag(0) signal increases (Fig. 4) approximately four times. This circumstance well confirms the possible mechanism of the catalytic cycle including the observed intermediates Ph2P(O)Ag, Ph2P(O)˙, and Ag(0).
A mixture of Ph2P(O)H and PBN or Ph2P(O)Ag and PBN in the absence of applied potential at room temperature does not exhibit noticeable ESR spectra.
Thus, catalytic cycle in Scheme 3 was suggested based on the ESR and CV experiments. The proposed catalytic pathway includes the formation of Ph2P(O)Ag in the first step; its oxidation leads to Ph2P(O)˙ radical and Ag+ regeneration. Then, the radical addition to alkyne affords an alkenyl radical 4. Next, the intramolecular addition of the alkenyl radical 4 to the aryl ring at the ortho position of the phosphorus atom, followed by the oxidation with Ag+ and the elimination of a proton, affords the phosphorus heterocycle 3. The Ag0-intermediate is formed in the presence of acetylene in the transient step, as detected by ESR. Considering these details, it is possible to explain the decrease of ˙P(O)Ph2 ESR signal intensity in the presence of alkyne (concurrent radical trapping with this substrate) and subsequent increase of ESR signal of silver aggregates (due to oxidation of resulting cyclization product). Contrary to the methods reported in the literature, for example, the Duan protocol,16 Ag(I) completely regenerates during catalytic cycle since it is easily oxidized at anode rather than precipitated.
 | ||
| Scheme 3 Proposed catalytic cycle. | ||
Experimental
Solvents were purified, dried, and distilled before use. The supporting electrolyte Bu4NBF4 used for electrochemical studies was purchased from Sigma-Aldrich and dried overnight at 100 °C under vacuum prior to use. Anhydrous acetonitrile (>99.8%) used for electrochemistry was purchased from Sigma-Aldrich.Cyclic voltammetry measurements were performed using the E2P potentiostat of BASi Epsilon (USA) composed of a measuring block, Dell Optiplex 320 computer with installed Epsilon ES-USB-V200 program, and C3 electrochemical cell. A stationary glassy-carbon electrode (3.0 mm diameter) was used as the working electrode. Platinum wire of 0.5 mm diameter was used as an auxiliary electrode. Ag/AgCl was used as the reference electrode. Ferrocenium was used as an internal standard. The potential of Fc+/Fc is 0.45 V vs. Ag/AgCl. Measurements were performed under argon.
All NMR experiments were performed using the Bruker AVANCE-400 spectrometer. Frequencies, 400.13 MHz, in 1H NMR, 161.90 MHz in 31P NMR were referenced to the residual signal of the solvent. 31P chemical shifts were referenced to the signals of 85% H3PO4 (0.00 ppm).
The ESR spectra were obtained using a Brucker Elexsys E500 X-range spectrometer with an ESR electrolysis cell operating in the three-electrode scheme placed into the resonator. Platinum spiral was used as the working electrode, platinum filament was used as the auxiliary electrode, and Ag/AgCl was used as the reference electrode. Prior to each experiment, helium was passed for 5 min through the solution in the cell. When passing was complete, the cell was filled with helium. The spectra were obtained in the potentiostatic mode. The spectra were simulated by WinSim.
The preparative electrolysis was performed using a direct current source B5-49 in a three-electrode cell with 40 ml volume with a separation of the anode and cathode compartments. The potential value of the working electrode was obtained using a direct-current V7-27 voltmeter in relation to the Ag/AgCl (0.01 M, NaCl) reference electrode that had two sections separated with Vycor, the second of which contained a saturated solution of the background salt in CH3CN. The surface area of the working platinum (Pt) U-shaped electrode was measured to be 48.00 cm2. A ceramic plate with the pore size of 900 nm was used as a membrane. During the preparative synthesis, the electrolyte was continuously stirred using a magnetic stirrer with a continuous inflow of inert gas that was run through a purification system to remove any traces of oxygen and other gaseous impurities. PyHBF4 was used as the supporting electrolyte.
The electrochemical cell contained 0.4 mmol of diphenylphosphine oxide (CAS 4559-70-0), 0.4 mmol of appropriate acetylene, and 0.04 mmol of AgOAc in 20 ml of CH3CN with 0.1 M of PyHBF4. The solution was agitated using a magnetic stirrer. Then, 2 F of electricity per one mole of diphenylphosphine oxide (22 mA for 0.4 mmol) was passed through the solution. The electrolysis potential was 0.15–0.35 V vs. Fc+/Fc. The electrolysis time is just over an hour. All syntheses were carried out at ambient temperature. After the electrolysis was over, the reaction mixture was filtered and concentrated under vacuum. Then, the dry residue was dissolved in CHCl3 and PyHBF4 (poorly soluble in CHCl3) was filtered off. The reaction mixture in CHCl3 was concentrated under vacuum again, and the residue was purified by silica gel chromatography with hexane/ethyl acetate = 1/1 to afford the product as a white to yellow color solid. The spectra of the cyclization products are consistent with the literature data.16,53 PyHBF4 can be further purified by recrystallization from alcohol and used again.
3a: 1H NMR (400 MHz, CDCl3) δ 7.90–7.80 (m, 2H), 7.75–7.70 (m, 5H), 7.55–7.60 (m, 1H), 7.55–7.50 (m, 5H), 7.40–7.35 (m, 6H). 31P NMR (161.9 MHz, CDCl3) δ 38.0 (s).
3b: 1H NMR (400 MHz, CDCl3) δ 7.80 (m, 3H), 7.65 (m, 2H), 7.50 (m, 3H), 7.35 (m, 7H), 7.00 (m, 2H). 31P NMR (161.9 MHz, CDCl3) δ 38.0 (s).
3c: 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 3H), 7.45 (m, 2H), 7.35 (m, 4H), 3.15 (m, 2H), 2.30 (m, 1H), 2.05 (m, 2H), 1.97 (m, 2H), 1.60 (m, 1H), 1.30 (t, JHH = 7.3, 3H), 0.94 (t, JHH = 7.3, 3H). 31P NMR (161.9 MHz, CDCl3) δ 39.2 (s).
3d: 1H NMR (400 MHz, CDCl3): 7.80–7.70 (m, 4H), 7.50–7.45 (m, 4H), 7.40–7.35 (m, 6H), 3.25–3.20 (m, 2H), 1.35 (t, JHH = 7.3, 3H). 31P NMR (161.9 MHz, CDCl3) δ 39.7(s).
3e: 1H NMR (400 MHz, CDCl3): 7.80 (m, 4H), 7.62 (m, 2H), 7.44 (m, 8H), 3.17 (m, 2H), 2.15 (m, 1H), 2.01 (m, 1H), 1.30 (t, JHH = 7.3, 3H). 31P NMR (161.9 MHz, CDCl3) δ 39.5(s).
3f: 1H NMR (400 MHz, CDCl3) δ 7.84–7.57 (m, 6H), 7.57–7.47 (m, 4H), 7.43–7.40 (m, 4H), 3.16 (t, JHH = 7.0, 2H), 1.50–1.35 (m, 2H), 1.30 (t, JHH = 7.3, 3H), 0.97–0.85 (m, 2H). 31P NMR (161.9 MHz, CDCl3) δ 38.7 (s).
3g: 1H NMR (400 MHz, CDCl3) δ 7.73–7.68 (m, 6H), 7.51–7.50 (m, 4H), 7.15–7.12 (m, 6H), 7.07–7.02 (m, 6H). 31P NMR (161.9 MHz, CDCl3) δ 37.1 (s), 37.5 (s).
AgP(O)Ph2 was obtained by mixing and stirring of 1 equiv. of AgOAc and 1 equiv. of diphenylphosphine oxide at room temperature overnight in CH3CN. 1H NMR (400 MHz, CDCl3) δ 7.74–7.69 (m, 4H), 7.46–7.42 (t, 2H), 7.36–7.32 (m, 4H). 31P NMR (161.9 MHz, CDCl3) δ 32.9(s).
Conclusions
Thus, the electrochemical oxidation of diphenylphosphine oxide with acetylene in equal amounts was performed without chemical oxidants and additives at room temperature with 0.1 equivalent of Ag salt as a catalyst. The yield of the target benzo[b]phosphole oxide was up to 100%. The redox properties of the key intermediate were investigated. The oxidation of Ph2P(O)Ag with the formation of phosphinyl radical is a key catalytic step for electrochemical reaction. The total regeneration of Ag(I) during electrolysis leads to the use of catalytic amounts of Ag salt.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was supported by the Russian Science Foundation (grant No. 14-23-00016).References
- M. Stolar and T. Baumgartner, Chem. – Asian J., 2014, 9, 1212 CrossRef CAS PubMed
.
- V. Gilard, R. Martino, M. Malet-Martino, U. Niemeyer and J. Pohl, J. Med. Chem., 1999, 42, 2542 CrossRef CAS PubMed
- M. M. Mader and P. A. Bartlett, Chem. Rev., 1997, 97, 1281 CrossRef CAS PubMed
- J. W. Darrow and D. G. Drueckhammer, J. Org. Chem., 1994, 59, 2976 CrossRef CAS
- A. Kumar, P. V. Sharmar, K. Gurram and N. Rane, Bioorg. Med. Chem. Lett., 2006, 16, 2484 CrossRef CAS PubMed
- Y. Matano, A. Saito, T. Fukushima, Y. Tokudome, F. Suzuki, D. Sakamaki, H. Kaji, A. Ito, K. Tanaka and H. Imahori, Angew. Chem., Int. Ed., 2011, 50, 8016 CrossRef CAS PubMed
- A. Fukazawa, E. Yamaguchi, E. Ito, H. Yamada, J. Wang, S. Irle and S. Yamaguchi, Organometallics, 2011, 30, 3870 CrossRef CAS
- Y. Ren and T. Baumgartner, J. Am. Chem. Soc., 2011, 133, 1328 CrossRef CAS PubMed
- H. Tsuji, K. Sato, Y. Sato and E. Nakamura, Chem. – Asian J., 2010, 5, 1294 CAS
- H. Tsuji, K. Sato, Y. Sato and E. Nakamura, J. Mater. Chem., 2009, 19, 3364 RSC
- A. Fukazawa, Y. Ichihashi, Y. Kosaka and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1729 CrossRef CAS PubMed
- Y. Matano and H. Imahori, Org. Biomol. Chem., 2009, 7, 1258 CAS
- J. Crassous and R. Réau, Dalton Trans., 2008, 6865 RSC
- Y. Ren and T. Baumgartner, Dalton Trans., 2012, 41, 7792 RSC
- Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975 CrossRef CAS PubMed
- Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754 CrossRef CAS PubMed
- W. Ma and L. Ackermann, Synthesis, 2014, 2297 CAS
- P. Zhang, Y. Gao, L. Zhang, Z. Li, Y. Liu, G. Tang and Y. Zhao, Adv. Synth. Catal., 2016, 358, 138 CrossRef CAS
- D. Ma, W. Chen, G. Hu, Y. Zhang, Y. Gao, Y. Yin and Y. Zhao, Green Chem., 2016, 18, 3522 RSC
- V. Quint, F. Morlet-Savary, J.-F. Lohier, J. Lalevée, A.-C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436 CrossRef CAS PubMed
- V. Jeena and R. S. Robinson, Chem. Commun., 2012, 48, 299 RSC
- S. D. Rychnosky, R. Vaidyanathan, T. Beauchamp, R. Lin and P. J. Farmer, J. Org. Chem., 1999, 64, 6745 CrossRef
- W.-S. Guo, Q. Dou, J. Hou, L.-R. Wen and M. Li, J. Org. Chem., 2017, 82, 7015 CrossRef CAS PubMed
- Y. H. Budnikova, Russ. Chem. Rev., 2002, 71, 111 CrossRef CAS
- Y. H. Budnikova, T. V. Gryaznova, V. V. Grinenko, Y. B. Dudkina and M. N. Khrizanforov, Pure Appl. Chem., 2017, 89, 311 CAS
- Y. H. Budnikova, D. G. Yakhvarov and O. G. Sinyashin, J. Organomet. Chem., 2005, 690, 2416 CrossRef CAS
- M. Khrizanforov, S. Strekalova, V. Khrizanforova, V. Grinenko, K. Kholin, M. Kadirov, T. Burganov, A. Gubaidullin, T. Gryaznova, O. Sinyashin, L. Xu, D. A. Vicic and Y. Budnikova, Dalton Trans., 2015, 44, 19674 RSC
- Yu. H. Budnikova and O. G. Sinyashin, Russ. Chem. Rev., 2015, 84, 917 CrossRef CAS
- T. V. Grayaznova, Y. B. Dudkina, D. R. Islamov, O. N. Kataeva, O. G. Sinyashin, D. A. Vicic and Yu. H. Budnikova, J. Organomet. Chem., 2015, 785, 68 CrossRef CAS
- T. Gryaznova, Y. Dudkina, M. Khrizanforov, O. Sinyashin, O. Kataeva and Y. Budnikova, J. Solid State Electrochem., 2015, 19, 2665 CrossRef CAS
- M. N. Khrizanforov, S. O. Strekalova, K. V. Kholin, V. V. Khrizanforova, M. K. Kadirov, T. V. Gryaznova and Y. H. Budnikova, Catal. Today, 2017, 279, 133 CrossRef CAS
- V. V. Khrizanforova, M. N. Khrizanforov, T. V. Gryaznova and Y. H. Budnikova, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 1602 CrossRef CAS
- M. N. Khrizanforov, S. O. Strekalova, K. V. Kholin, V. V. Khrizanforova, V. V. Grinenko, T. V. Gryaznova and Y. H. Budnikova, RSC Adv., 2016, 6, 42701 RSC
- H. J. Zhang, W. Li, Z. Wu, W. Ruan and T. B. Wen, Chem. Commun., 2015, 51, 3450 RSC
- Y.-M. Li, M. Sun, H.-L. Wang, Q.-P. Tian and S.-D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972 CrossRef CAS PubMed
- M. S. Hill, M. F. Mahon and T. P. Robinson, Chem. Commun., 2010, 46, 2498 RSC
- G. Wulfsberg and J. S. Coe, Coord. Chem. Rev., 1984, 54, 131 CrossRef
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed
- B. Zhang, C. G. Daniliuc and A. Studer, Org. Lett., 2014, 16, 250 CrossRef CAS PubMed
- J. Zheng, Y. Zhang, D. Wang and S. Cui, Org. Lett., 2016, 18, 1768 CrossRef CAS PubMed
- Y. Gao, G. Tang and Y. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 589 CrossRef CAS
- J. Lalevée, F. Morlet-Savary, M. Ali Tehfe, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 5032 CrossRef
-
J. P. Fouassier, Photoinitiators for Polymer Synthesis: Scope, Reactivity, and Efficiency, John Wiley & Sons, 2012, p. 476 Search PubMed
- F. Morlet-Savary, J. E. Klee, F. Pfefferkorn, J.-P. Fouassier and J. Lalevée, Macromol. Chem. Phys., 2015, 216, 2161 CrossRef CAS
- L. Noël-Duchesneau, E. Lagadic, F. Morlet-Savary, J.-F. Lohier, I. Chataigner, M. Breugst, J. Lalevée, A.-C. Gaumont and S. Lakhdar, Org. Lett., 2016, 18, 5900 CrossRef PubMed
- M. K. Kadirov, Y. G. Budnikova, K. V. Kholin, M. I. Valitov, S. A. Krasnov, T. V. Gryaznova and O. G. Sinyashin, Russ. Chem. Bull., 2010, 59, 466 CrossRef CAS
- F. Effenberger and H. Kottmann, Tetrahedron, 1985, 41, 4171 CrossRef CAS
- R. E. Montgomery, J. Inorg. Nucl. Chem., 1966, 28, 1750 CrossRef
- B. B. Hunt and B. C. Saunders, J. Chem. Soc., 1957, 2413 RSC
- J. A. Baban and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1986, 1607 RSC
- P. D. Costagliola, F. Benedetto, M. Benvenuti, G. P. Bernardini, C. Cipriani, P. F. Lattanzi and M. Romanelli, Am. Mineral., 2003, 88, 1345 CrossRef CAS
- G. R. Buettner, Free Radicals Biol. Med., 1987, 3, 259 CrossRef CAS PubMed
- H. Tsuji, K. Sato, L. Ilies, Y. Itoh, Y. Sato and E. Nakamura, Org. Lett., 2008, 10, 2263 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |