
Effects of appended hydroxyl groups and ligand chain length on copper coordination and oxidation activity†
Attawit
Jehdaramarn
ab,
Soraya
Pornsuwan
a,
Phongnarin
Chumsaeng
ac,
Khamphee
Phomphrai
c and
Preeyanuch
Sangtrirutnugul
 *ab
*ab
aCenter of Excellence for Innovation in Chemistry (PERCH-CIC), Department of Chemistry, Faculty of Science, Mahidol University, 272 Rama VI Rd., Bangkok, 10400, Thailand. E-mail: preeyanuch.san@mahidol.ac.th
bCenter for Catalysis, Department of Chemistry, Faculty of Science, Mahidol University, 272 Rama VI Rd., Bangkok, 10400, Thailand
cDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Wangchan, Rayong 21210, Thailand
First published on 28th November 2017
Abstract
Treatment of a series of (imino)pyridine ligands bearing appended hydroxyl groups 2-((pyridin-2-ylmethylene)amino)phenol (Hpyph), 2-((pyridin-2-ylmethylene)amino)ethanol (Hpyet), and 3-((pyridin-2-ylmethylene)amino)propanol (Hpypr) with one equiv. of CuCl2·2H2O afforded the corresponding Cu(II) complexes in low to moderate yields. The crystal structure of (μ-Cl)2[CuCl(κ2-N,N-Hpyet)]2 reveals a symmetric dinuclear structure with the bidentate N,N-coordination mode of (imino)pyridine with no Cu–OH interaction. On the other hand, the dinuclear Cu(II) complex of the related propyl ligand Hpypr possesses a significantly different crystal structure involving nucleophilic addition of the hydroxyl group to the aldehyde group of 2-pyridinecarboxaldehyde. The Cu complex/Cu0/TEMPO/Na2CO3 (TEMPO = 2,2,6,6-tetramethylpiperidinyl-1-oxyl) catalyst system generally exhibited good activity for aerobic oxidation of benzyl alcohol to benzaldehyde in H2O at room temperature. The dinuclear Cu(II) complex (μ-Cl)2[CuCl(κ2-N,N-Hpyet)]2 was demonstrated as an effective catalyst toward aerobic oxidation of various benzyl alcohol derivatives, cinnamyl alcohol, and 2-thiophenemethanol.
Introduction
Copper-catalyzed oxidation of alcohols to their corresponding aldehydes using oxygen or air as an oxidant is generally highly efficient and can be performed under environmentally friendly conditions.1 Sammelhack and co-workers first reported the CuCl/TEMPO/DMF (TEMPO = 2,2,6,6-tetramethylpiperidinyl-1-oxyl, DMF = N,N-dimethylformamide) catalyst system, which was effective for oxidation of primary alcohols to aldehydes under atmospheric O2.2 Later works by Sheldon,3 Koshinen,4 and Stahl5 involved copper catalyst systems supported by the 2,2′-bipyridine ligand in the presence of an additional base including KOtBu, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and N-methylimidazole (NMI) with O2 or ambient air as an oxidant in CH3CN. Other related N-based ligands have also been investigated as catalyst supports by our group and others.6–9 For environmental reasons, there has recently been growing interest in copper-catalyzed aerobic alcohol oxidation in water. Zhang9 and Rheingold10 reported water-soluble copper catalysts supported by N-based ligands, which were active toward aerobic oxidation of benzyl alcohols in water. Lipshutz11 also demonstrated that the designer surfactant TPGS-750-M was effective at facilitating Cu-catalyzed aerobic alcohol oxidation in an aqueous solution (Table 1).

Notably, active copper species for aerobic oxidation of alcohols are a key component in the galactose oxidase (GOase) enzyme.12 The distorted square pyramidal copper(II) active site in GOase consists of two nitrogen donors from two histidine imidazoles and, in the axial position, a phenol group of tyrosine, which facilitates oxidative transformation.13,14 Further evidence for the role of the hydroxyl group in oxidation was reported by Puddephatt and co-workers, in which appended phenol and hydroxyethyl groups in the ligand framework were shown to promote reactions between a dimethylplatinum(II) complex and O2.15,16 Despite these findings, no related studies have been carried out on molecular copper catalyst systems. Based on the established oxidation activities of copper catalysts and potential involvement of appended hydroxyl groups in metal-catalyzed oxidation, we prepared multidentate ligands featuring N,N,O donors from pyridine, Schiff base, and hydroxyl groups with three different ligand backbones. The corresponding Cu(II) complexes were used as catalysts for aerobic alcohol oxidation in an alkaline aqueous solution and their catalytic activities were compared. In particular, this work aims to evaluate the effects of appended hydroxyl groups associated with ligand rigidity and chain length on the coordination environments at the Cu(II) center and their subsequent catalytic activities toward oxidation reactions.
Experimental
Materials
2-Pyridinecarboxaldehyde, 2-aminophenol, 2-aminoethanol, and 3-aminopropanol (Aldrich) were of reagent grade and used as received. The commercial grade ethyl acetate, dichloromethane, and hexane were distilled prior to use. All other chemicals and solvents used in this work were of reagent or analytical grade and were used as received.Physical measurements
Fourier transform nuclear magnetic resonance spectra were recorded using a Bruker's Ascend 400 high-resolution magnetic resonance spectrometer. Chemical shifts were reported in δ units (parts per million) using residual solvent peaks as references. Mass spectrometric analysis was carried out using a Bruker micrOTOF spectrometer in the ESI mode, whereas elemental analyses were performed on a Perkin Elmer 2400 CHN. Fourier transform infrared (FTIR) spectra were collected on a Bruker Alpha instrument (Bruker Optics GmbH, Ettlingen, Germany) (400–4000 cm−1). UV-visible spectra of the aqueous solutions of Cu(II) complexes 1–3 were obtained using a JASCO model V-530 and recorded in the 200–800 nm range. Powder X-ray diffraction (PXRD) was performed using a Bruker AXS model D8 Discover with a monochromatic Cu Kα (40 kV, 40 mA) source with a step size of 0.020° and a step time of 0.2 s step−1. Electron spin resonance (ESR) spectra were obtained at room temperature using a Bruker Elexys-500 X-band spectrometer. The solid powder samples were packed in 1.5 mm OD capillary tubes. All measurements were performed at a frequency of ∼9.8 GHz and a microwave power of 2 mW. The center field was set at 3200 G with a sweep width of 1500 G, and the modulation amplitude was 4 G. Single crystal X-ray analyses of complexes 2 and 3 were carried out using a VISTEC. X-ray crystallographic data were collected at low temperatures on a Bruker D8 Venture using a Photon II detector and IμS 3.0 Microfocus source, Mo Kα radiation (λ = 0.71073 Å). Data integration was performed using SAINT and intensity data were corrected based on the intensities of symmetry-related reflections measured at different angular settings (SADABS). The space groups were determined using XPREP, whereas the structure was solved by the direct method using intrinsic phasing (XT program) and refined by full-matrix least squares against F2 using the XL program. Product yields and percent substrate conversions obtained from catalytic experiments were analyzed by GLC on a 6890N Agilent Technologies gas chromatograph equipped with a 5973N Agilent Technologies quadrupole mass detector.Synthesis of Schiff base ligands
To 40 mL of an EtOH solution of 2-pyridinecarboxaldehyde (950 μL, 10 mmol), aminoalcohol (10 mmol) was added. The reaction mixture was refluxed for a certain amount of time, after which all volatiles were removed under vacuum.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH), 8.69 (m, 1H, ArH), 8.38 (d, J = 8 Hz, 1H, ArH), 7.94 (t, J = 8 Hz, 1H, ArH), 7.50 (m, 1H, ArH), 7.29 (m, 1H, ArH), 6.92 (d, J = 8 Hz, 1H, ArH), 6.85 (m, 1H, ArH). 13C{1H} NMR (100 MHz, DMSO-d6): δ 159.4, 154.5, 151.4, 149.5, 136.8, 128.3, 125.4, 121.5, 119.7, 119.6, 116.3, 114.4 (aromatic Cs). FT-IR (cm−1): 3378 (br, O–H), 1628 (CN), 1151 (C–OH).
NH), 7.96 (m, 1H, pyH), 7.87 (m, 1H, pyH), 7.46 (m, 1H, pyH), 4.65 (br s, 1H, OH), 3.68 (br s, 4H, CH2CH2). 13C{1H} NMR (100 MHz, DMSO-d6): δ 162.7, 154.2, 149.3, 136.8, 125.0, 120.4 (aromatic Cs), 63.1 60.5 (CH2CH2). FT-IR (cm−1): 3278 (br, O–H), 1649 (CN), 1059 (C–OH).
NH), 8.69 (m, 1H, ArH), 8.38 (d, J = 8 Hz, 1H, ArH), 7.94 (t, J = 8 Hz, 1H, ArH), 7.50 (m, 1H, ArH), 7.29 (m, 1H, ArH), 6.92 (d, J = 8 Hz, 1H, ArH), 6.85 (m, 1H, ArH). 13C{1H} NMR (100 MHz, DMSO-d6): δ 159.4, 154.5, 151.4, 149.5, 136.8, 128.3, 125.4, 121.5, 119.7, 119.6, 116.3, 114.4 (aromatic Cs). FT-IR (cm−1): 3378 (br, O–H), 1628 (CN), 1151 (C–OH).
NH), 7.96 (m, 1H, pyH), 7.87 (m, 1H, pyH), 7.46 (m, 1H, pyH), 4.65 (br s, 1H, OH), 3.68 (br s, 4H, CH2CH2). 13C{1H} NMR (100 MHz, DMSO-d6): δ 162.7, 154.2, 149.3, 136.8, 125.0, 120.4 (aromatic Cs), 63.1 60.5 (CH2CH2). FT-IR (cm−1): 3278 (br, O–H), 1649 (CN), 1059 (C–OH).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.
NH), 7.94 (d, J = 8 Hz, 1H, ArH), 7.86 (dt, J = 8, 2 Hz, 1H, ArH), 7.45 (m, 1H, ArH), 4.50 (t, J = 5 Hz, 1H, OH), 3.68 (dt, J = 8, 1 Hz, 2H, NCH2), 3.48 (m, 2H, CH2OH), 1.78 (quin, J = 6 Hz, 2H, CH2). 13C{1H} NMR (100 MHz, DMSO-d6): δ 161.8, 154.2, 149.3, 136.8, 125.0, 120.4 (aromatic Cs), 58.5, 57.2, 33.6 (CH2CH2CH2).
NH), 7.79 (dt, J = 8, 2 Hz, 1H, ArH), 7.32 (m, 1H, ArH), 5.06 (s, 1H, pyCH), 1.31–4.09 (m, 6H, CH2). 13C{1H} NMR (100 MHz, DMSO-d6): δ 148.5, 136.7, 123.4, 121.2 (aromatic Cs), 89.6 (pyCH), 67.3, 43.6, 26.5 (CH2CH2CH2). FT-IR (cm−1): 3285 (br, O–H), 1649 (str, CN), 1058 (str, C–OH).
:1 ratio.
NH), 7.94 (d, J = 8 Hz, 1H, ArH), 7.86 (dt, J = 8, 2 Hz, 1H, ArH), 7.45 (m, 1H, ArH), 4.50 (t, J = 5 Hz, 1H, OH), 3.68 (dt, J = 8, 1 Hz, 2H, NCH2), 3.48 (m, 2H, CH2OH), 1.78 (quin, J = 6 Hz, 2H, CH2). 13C{1H} NMR (100 MHz, DMSO-d6): δ 161.8, 154.2, 149.3, 136.8, 125.0, 120.4 (aromatic Cs), 58.5, 57.2, 33.6 (CH2CH2CH2).
NH), 7.79 (dt, J = 8, 2 Hz, 1H, ArH), 7.32 (m, 1H, ArH), 5.06 (s, 1H, pyCH), 1.31–4.09 (m, 6H, CH2). 13C{1H} NMR (100 MHz, DMSO-d6): δ 148.5, 136.7, 123.4, 121.2 (aromatic Cs), 89.6 (pyCH), 67.3, 43.6, 26.5 (CH2CH2CH2). FT-IR (cm−1): 3285 (br, O–H), 1649 (str, CN), 1058 (str, C–OH).
Synthesis of Cu(II) complexes (1–3)
A equimolar mixture of CuCl2·2H2O and ligands 1–3 was stirred in 40 mL of CH2Cl2 at room temperature for 24 h. All volatiles were then removed in vacuo and the remaining solids were crystallized from suitable solvents.N).
N), 1058 (C–OH).
N), 1042 (C–O–C).
General procedure for alcohol oxidation
To 3 mL of an aqueous solution of the Cu(II) complex (0.050 mmol Cu) and Cu0 sheets (a total surface area = 1.0 cm2), 1.0 mmol of alcohol was added. To ensure the same concentration of the Cu active species, 0.050 mmol of Cu was calculated based on 0.025 mmol of the dinuclear copper complexes 1–3. 2 mL of an aqueous solution of TEMPO (0.050 mmol) and Na2CO3 (0.10 mmol) was added and the reaction mixture was stirred in air at room temperature for a given time. Then, the aqueous solution was extracted with 3 × 10 mL of CH2Cl2. The combined organic layers were dried using anhydrous Na2SO4 and filtered, after which the solvent was removed. The percent conversions were quantified using the GC-MS method, in which EtOAc was used as a solvent in the presence of anisole (0.10 mmol) as the internal standard.Results and discussion
Synthesis of ligands and Cu(II) complexes
The (imino)pyridine ligands Hpyph, Hpyet, and Hpypr were prepared according to the previous literature,17,18 with slight modifications via condensation of 2-pyridinecarboxaldehyde and its respective aminoalcohols in EtOH under refluxed conditions (Scheme 1). The phenylene-containing product Hpyph appeared as a yellow solid, which was purified and isolated in 60% yield by crystallization in diethyl ether. Meanwhile, ligands with an alkyl framework Hpyet and Hpypr were obtained as yellow oils. Based on their 1H NMR spectra in DMSO-d6, the ethylene linker Hpyet exists only in an open chain form, whereas the propyl derivative Hpypr is in equilibrium between the open chain (a) and cyclic (b) forms. The 1H NMR integration reveals that the crude product of Hpypr has the open chain form as a major species (∼8:1 ratio of (a):(b) in DMSO-d6, Fig. S5, ESI†). In contrast, Puddephatt et al., whose work was published during the course of our study, reported the cyclic form of Hpypr (b) as a dominant product (∼80%) in CDCl3 solution.16
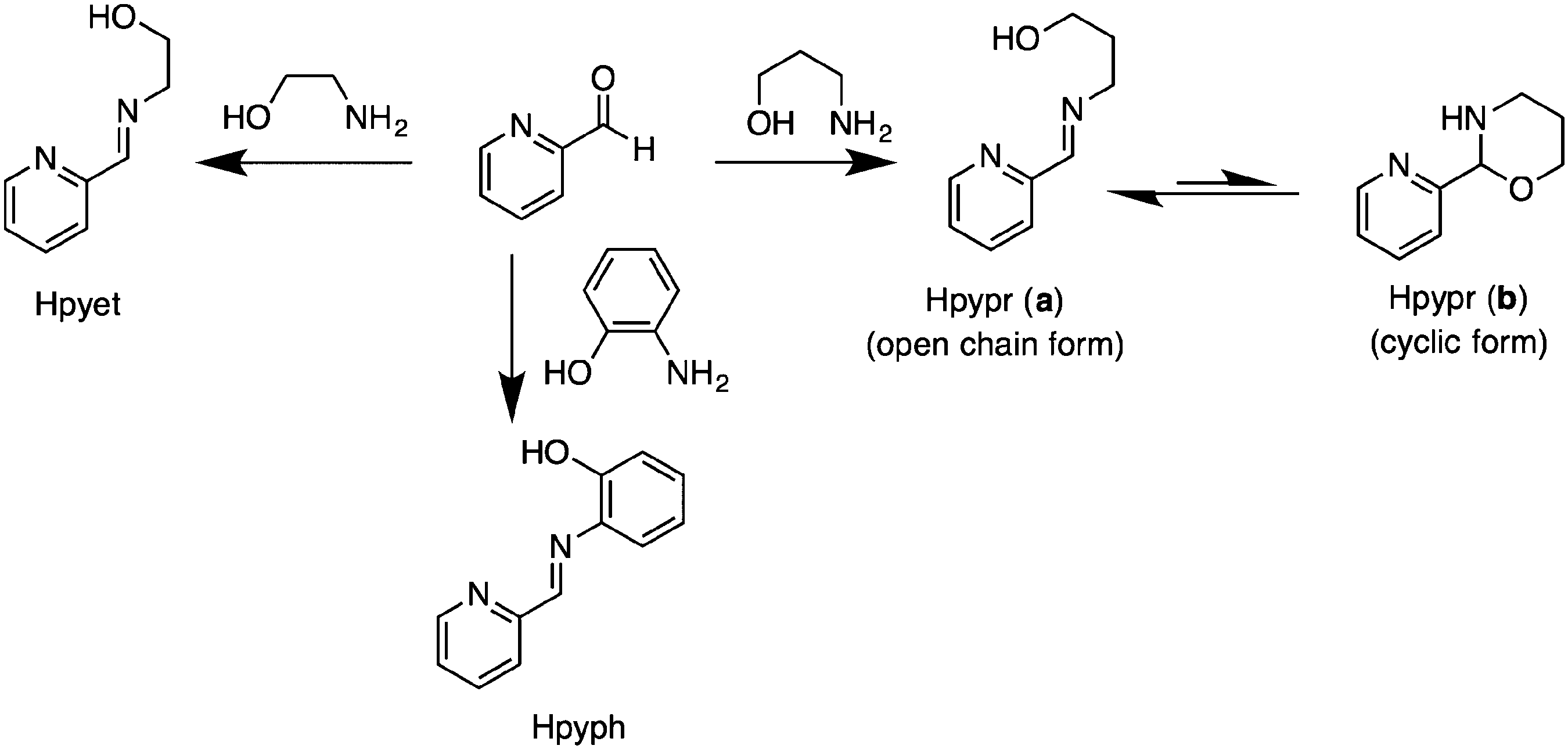 | ||
| Scheme 1 Ligand synthesis. | ||
Treatments of CuCl2·2H2O with an equimolar amount of Hpyph in CH2Cl2 at room temperature for 24 h resulted in a brown solid, while, under the same conditions, reactions with Hpyet or Hpypr gave green crystals (Scheme 2). The phenylene-containing Cu(II) complex 1 was isolated via slow evaporation of the CH3OH solution at room temperature, which afforded a brown crystalline solid in 46% yield. Attempts to obtain a solid state structure of 1 have so far been unsuccessful. On the other hand, layering the DMF solutions of Cu(II) complexes 2 and 3 with Et2O (for 2) or EtOAc (for 3) at room temperature gave green crystalline solids suitable for X-ray analysis in 35% and 30% yields, respectively. The FT-IR spectra of 1 and 2 contain a broad absorption band at ∼3430 cm−1, whereas no absorption band corresponding to O–H stretching was observed in that of 3 (Fig. S8, S10, and S12, ESI†). Furthermore, the powder XRD analyses of 2 and 3, obtained from both the crude samples and the isolated crystalline solids, reveal the same XRD patterns, confirming that the obtained crystals represent the major products of Cu complexation (Fig. S14 and S15, ESI†). It should be noted that Pal and co-workers have previously reported the synthesis and crystal structure of Cu(pyph)Cl, generated by a similar reaction of Hpyph with CuCl2·2H2O and KOH in refluxing CH3OH.17 For our experiment, even in the absence of a base, it is possible that the acidic phenolic proton may be deprotonated. To gain insights into the structure of 1, the powder XRD spectrum of the isolated complex 1 and that simulated from single crystal data of Cu(pyph)Cl were compared, showing different XRD patterns (Fig. S13, ESI†). On the basis of powder XRD results and the presence of an OH absorption band in the FT-IR spectrum, a dimeric structure similar to that of 2 is proposed for the phenylene derivative 1 (Scheme 2 and Fig. 1).
 | ||
| Scheme 2 Synthesis of Cu(II) complexes 1–3. | ||
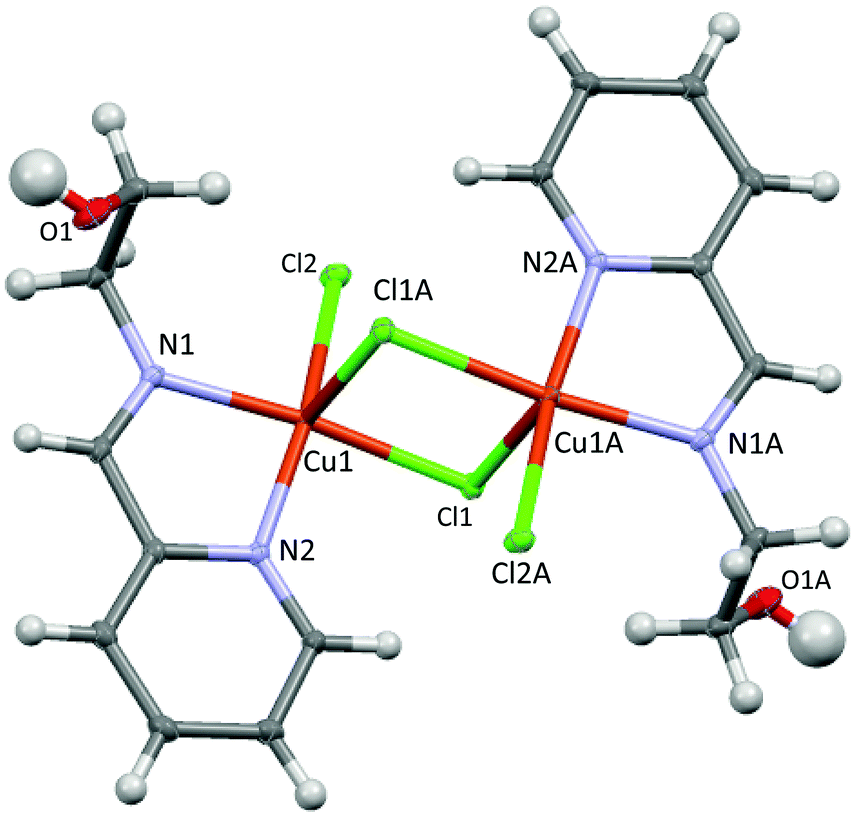 | ||
| Fig. 1 The ORTEP representation of 2 with thermal ellipsoids at the 50% probability level. Selected bond parameters: Cu1–N1 = 2.032(1), Cu1–N2 = 2.030(1), Cu1–Cl1 = 2.2833(6), Cu1–Cl2 = 2.2679(6), Cu1–Cl1A = 2.791(1) Å; N1–Cu1–N2 = 80.26(3), Cl1–Cu1–Cl2 = 92.06(1), N1–Cu1–Cl1 = 173.07(3), N2–Cu1–Cl2 = 169.55(3), Cl1–Cu1–Cl1A = 88.33(1)°. | ||
Structure description of complexes 2 and 3
X-ray data of 2 reveal a chloride-bridged asymmetric dinuclear Cu(II) complex with a square pyramidal geometry at the copper center (τ = 0.06, where τ = 0 for a square pyramid and τ = 1 for a trigonal bipyramid),19 as shown in Fig. 1. The Cu(1) and Cu(1A) ions are related by inversion and each copper center is chelated by pyridine and imine N donors, both of which are at basal sites. A terminal Cl and a bridging Cl ion complete basal positions, whereas another bridging Cl is located in the axial position. The Npy(2)–Cu(1)–Nim(1) bite angle is small at 80.26(4)°, whereas the Cu(1)–Npy(2) (2.030(1) Å) and Cu(1)–Nim(1) (2.032(1) Å) bond distances are closely similar. The terminal Cu(1)–Cl(2) and bridging Cu(1)–Cl(1) bond distances at basal sites are 2.2833(6) and 2.2679(6) Å, respectively, which are considerably shorter than that of the bridging, axial Cu(1)–Cl(1A) bond (2.791(1) Å). Furthermore, through bridging Cl ligands, the intramolecular Cu–Cu separation is found to be 3.647(1) Å, comparable to other related dinuclear complexes with the Cu(μ-Cl)2Cu core.20,21 It is worth mentioning that Puddephatt and co-workers have recently reported results obtained from DFT calculations of the related complexes Pt(L)Me2 (L = Hpyet and Hpypr).16 The calculations suggest that the stable conformations involve a hydroxyl substituent directing toward the Pt(II) center and forming a weak hydrogen bond. In contrast, the related crystal structure of Cu(II) complex 2 exhibits no apparent Cu–OH interactions.Unlike 2, the solid state structure of 3 reveals an unsymmetric dinuclear Cu(II) complex, featuring five coordinate Cu1 and four coordinate Cu2 (Fig. 2). Both Cu1 and Cu2 ions are bridged by the oxygen atom O2 of the alkoxy group and a chloride ion. The bridging Cu1–O2–Cu2 bond angle is 110.96(9)°. The Cu1 ion has a distorted square pyramidal geometry based on the τ value19 of 0.26 and is supported by a tridentate N,N,O ligand, a terminal Cl, and a bridging Cl ion. On the other hand, the Cu2 ion possesses a distorted square planar geometry and is surrounded by the bidentate N,O ligand with a O2–Cu2–N3 bite angle of 81.73(9)°, a terminal Cl, and a bridging Cl. The sum of six independent angles at Cu2 is ∼703°, significantly deviated from the expected 720° for the square planar structure. Interestingly, the Cu–Cu separation in 3 is 3.199(2) Å, which is significantly shorter than that in 2. In addition, despite the presence of chiral centers such as Cu1 and C10 atoms in the molecule, only one diastereomer crystallized from a mixture of DMF:EtOAc solvent.
 | ||
| Fig. 2 The ORTEP representation of 3 with thermal ellipsoids at the 50% probability level. Selected bond parameters: Cu1–N1 = 2.007(3), Cu1–N2 = 2.004(3), Cu1–O2 = 1.962(2), Cu2–O2 = 1.920(2), Cu2–N3 = 2.004(3), Cu1–Cl1 = 2.242(1), Cu1–Cl2 = 2.591(2), Cu2–Cl2 = 2.275(1), Cu2–Cl3 = 2.209(1) Å; N1–Cu1–N2 = 80.8(1), Cl1–Cu1–O2 = 94.67(6), Cl2–Cu1–O2 = 77.08(6), O2–Cu1–N2 = 91.04(9), Cl2–Cu2–Cl3 = 96.36(3), O2–Cu2–N3 = 81.73(9), Cl2–Cu2–O2 = 86.22(6), N3–Cu2–Cl3 = 95.57(7), Cu1–O2–Cu2 = 110.96(9), Cu1–Cl2–Cu2 = 81.92(3)°. | ||
For the (imino)pyridine ligands, the coordination behaviors of the appended hydroxyl group are rather unexpected. The related NNO and ONO ligand systems featuring an appended alcohol functional group have been crystallographically characterized or proposed as tridentate ligands with M–OH interactions.16,18,22,23 In contrast, our results have revealed no Cu–OH interaction in the solid state structure, for the ethylene-based ligand. Furthermore, for a slightly longer propylene linker, the intermolecular nucleophilic addition of the OH group was evident according to the crystal structure. In particular, the formation of the dinuclear Cu(II) complex 6 is likely a result of Cu-catalyzed imine hydrolysis to generate Cu-coordinated 2-pyridinecarboxaldehyde followed by nucleophilic addition of the appended propyl alcohol at the aldehyde group. Several works have previously documented copper-promoted hydrolysis of the Schiff base ligands to generate the constituent aldehyde and amine precursors.24,25 Hammershøi and Cuevas also independently demonstrated that the coordinated imine ligands were susceptible to nucleophilic attacks.26–28 However, to the best of our knowledge, Cu-catalyzed imine hydrolysis followed by nucleophilic addition at aldehyde to afford an 8-membered dinuclear metallacycle is unprecedented.
The effect of the linker chain length of pyridine-diimine-based ligands on the complex's stability has been previously investigated by Ghosh and co-workers. It was found that while the Cu(II) complex of the ethylene-linked pyridine-diimine ligand was stable, a related propylene-linked ligand underwent hydrolysis and rearrangement in the presence of Cu(II) or H+ ions.25 The lower stability of the propylene-based chelate ligand was attributed to more steric strain, as a result of two adjacent six-membered metallacyclic rings. However, in this work, since the hydroxyl groups of the (imino)pyridine ligands do not coordinate to the Cu(II) centers, we reason that the drastic differences in the ligand activity stem from the alkyl chain length. In particular, the longer alkyl chain of the propyl alcohol enables the hydroxyl group to reach farther away from the Cu coordination sphere for the subsequent nucleophilic addition.
Spectroscopic and ESR studies
UV-vis spectra of the Cu(II) complexes in DMSO contain weak absorption bands at 447, 709, and 714 cm−1 assignable to Cu(II) d–d transitions for 1, 2, and 3, respectively (Fig. S16–S18, ESI†). Meanwhile, intense absorption bands between 200 and 350 cm−1 were attributed to π–π* transition (intraligand) and ligand-to-metal charge transfer.29,30 The ESR powder spectra of 1–3 contain the two signals, g⊥ and g‖, as illustrated in Fig. 3. Table 2 shows the magnetic parameters for the g-values of each Cu complex. The elongated axial symmetry characterized by g‖ > g⊥ > 2.0023 is consistent with distorted square pyramid and square planar geometries at Cu(II) ions, as confirmed by the solid state structures of 2 and 3 (vide supra).31 The g-value parameters obtained from 1–3 are comparable to our previously reported ESR data for the mononuclear distorted square planar complex of the type (NN′)CuCl2 (g⊥ ∼ 2.07–2.08 and g‖ ∼ 2.25–2.28),7 suggesting weak exchange interactions between copper ions in the dinuclear Cu complexes. Another interesting feature of these ESR data is a significantly smaller separation between g⊥ and g‖ for 3 (Δg = 0.125 compared to 0.204 (for 1) and 0.188 (for 2); Table 2). The decrease in the Δg value is attributed to a stronger interaction at the d9 Cu electron, as a result of a bridging paramagnetic O donor compared to a bridging Cl atom (Fig. 1 and 2). Furthermore, based on the closely related g-value parameters of 1 and 2 together with their FT-IR and powder XRD data, we propose that the solid state structure of 1 possesses a similar coordination environment to that of 2 (vide supra).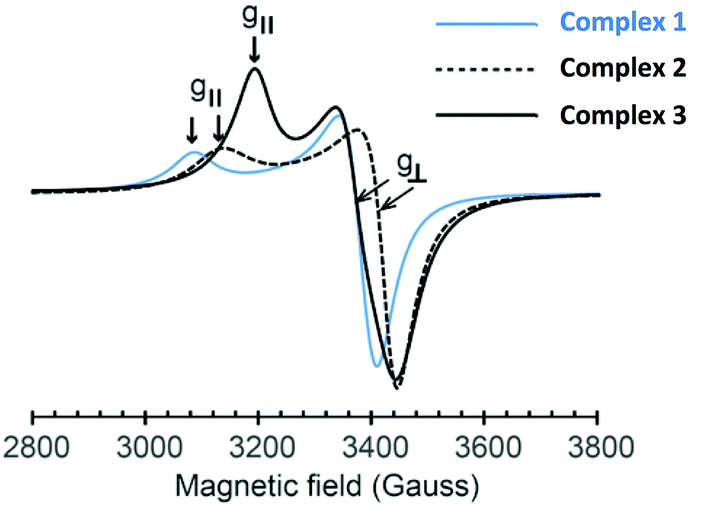 | ||
| Fig. 3 ESR powder spectra of the Cu complexes 1–3 at room temperature. The g-values of each complex are shown as g⊥ and g‖ indicated in arrow signs. | ||
Aerobic alcohol oxidation in water
The Cu(II) complexes 1–3 were investigated as catalysts for aerobic alcohol oxidation in an aqueous solution. First, the catalytic activities of 1–3 were compared using 1.0 mmol of benzyl alcohol as the model substrate, catalyzed by 5 mol% Cu(II) species, 5 mol% TEMPO, Cu0 sheets with a total area of 1 cm2, and 10 mol% Na2CO3 in air at room temperature with the H2O solvent (Table 3). In comparison, the activities of 2 and 3 are comparable, giving benzaldehyde as a sole product in excellent conversions after 16 h, and higher than that of 1 (entries 1–3). Interestingly, different coordination environments at the Cu centers in the dinuclear Cu(II) complexes 2 and 3 did not have a significant impact on oxidation activities. The reduced catalytic activity of 1 was explained by a decrease in the electron density at the copper center as a result of the more electron-withdrawing phenylene groups.32 Next, the effect of base was investigated using catalyst 2. Under similar conditions, 10 mol% of Na2CO3 at room temperature furnished a near quantitative conversion of benzyl alcohol to benzaldehyde, whereas other bases including Cs2CO3, K3PO4, and N-methylimidazole (NMI) resulted in lower conversions (entries 4–6). Decreased conversions were also obtained with reduced catalyst loadings (from 5 mol% to 1 mol% Cu), the use of CuCl2 with no ligand, and the in situ generated Cu(II) complex (entries 7–9). Similarly, lower conversions were observed with the catalyst systems in the absence of either Cu0 sheets (75%, entry 10) or Cu(II) catalyst (18%, entry 11). These results suggest that the most effective catalyst system requires both Cu(II) species and Cu0 to undergo a comproportionation reaction33,34 and generate Cu(I) species, which are more active toward catalytic oxidation.35|
|
|||
|---|---|---|---|
| Entry | Cu catalyst | Base | Conversionb (%) |
| a Reaction conditions: benzyl alcohol (1.0 mmol), Cu catalyst (5 mol% based on Cu), TEMPO (0.050 mmol), base (0.10 mmol), four Cu0 sheets with a total area of 1 cm2 in H2O (5 mL) under aerobic conditions with 0.010 mmol of anisole as an internal standard, room temperature, 16 h. b Determined by GC-MS. c Reaction time = 24 h. d The catalyst loading = 1 mol% of 2. e Without Cu0 sheets. | |||
| 1c | 1 | Na2CO3 | 83 |
| 2 | 2 | Na2CO3 | 93 |
| 3 | 3 | Na2CO3 | 94 |
| 4 | 2 | Cs2CO3 | 84 |
| 5 | 2 | K3PO4 | 58 |
| 6c | 2 | NMI | 87 |
| 7d | 2 | Na2CO3 | 86 |
| 8 | CuCl2 | Na2CO3 | 37 |
| 9 | CuCl2/Hpyet | Na2CO3 | 62 |
| 10e | 2 | Na2CO3 | 75 |
| 11 | — | Na2CO3 | 18 |

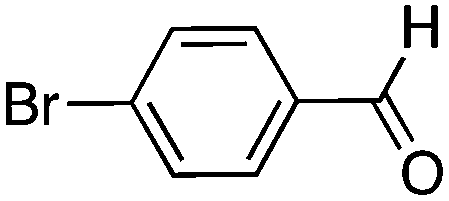
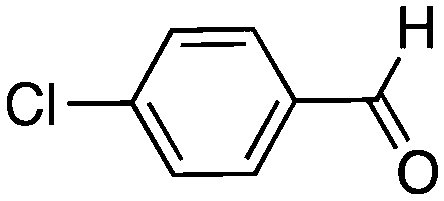
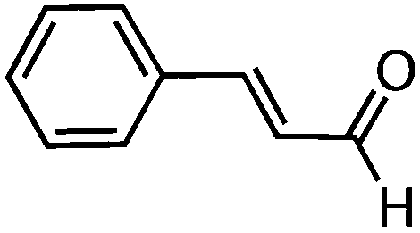
To investigate the substrate scope, catalyst 2 was selected for further studies. Aerobic oxidation of benzylic alcohol derivatives was evaluated as shown in Table 4. The electron-donating OMe and OH groups generally gave good to excellent conversions after 16 h at room temperature (entries 1–3). On the other hand, the electron-withdrawing NO2 group on benzyl alcohol afforded slower reactions as only moderate conversions were observed after 48 h (entries 4 and 5). Halogen-substituted benzyl alcohols gave mixed results: 4-iodobenzyl alcohol was completely converted to the oxidized product after 16 h, whereas 4-bromo- and 4-chlorobenzyl alcohols needed 48 h to reach high conversions (87% and 93%, respectively; entries 6–8). Cinnamyl alcohol was oxidized to the corresponding aldehyde in a quantitative conversion after 8 h (entry 9). The sulfur atom in 2-thiophenemethanol is well tolerated, giving a complete conversion after 24 h (entry 10). Attempts to oxidize aliphatic primary and secondary alcohols including cyclohexane, 1-hexanol, and 2-methyl-1-pentanol under similar catalytic conditions at room temperature and at 100 °C were unsuccessful.
|
|
|||
|---|---|---|---|
| Entry | Aldehyde | Time (h) | Conversionb (%) |
| a Reaction conditions: alcohol substrate (1.0 mmol), Cu complex 2 (0.025 mmol), TEMPO (0.050 mmol), Na2CO3 (0.10 mmol), four Cu0 sheets with a total area of 1 cm2 in H2O (5 mL) under aerobic conditions at room temperature with 0.010 mmol of anisole as an internal standard. b Based on GC analysis. Since the corresponding aldehyde is the only product observed, % conversion = the ratio of product × 100/(product + starting material). c Reaction time = 48 h. d Reaction time = 24 h. | |||
| 1 |
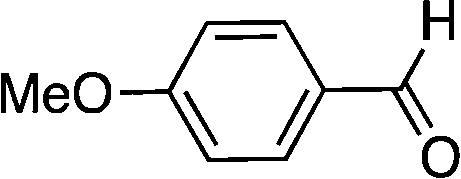
|
16 | >99 |
| 2 |
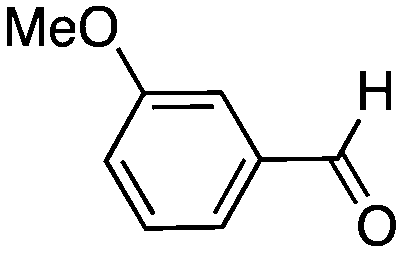
|
16 | 69 |
| 3 |

|
16 | >99 |
| 4 |
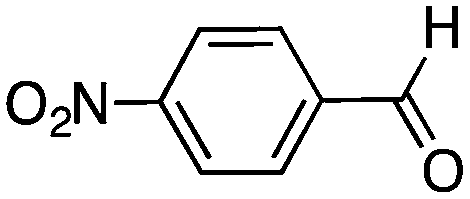
|
48 | 42 |
| 5 |
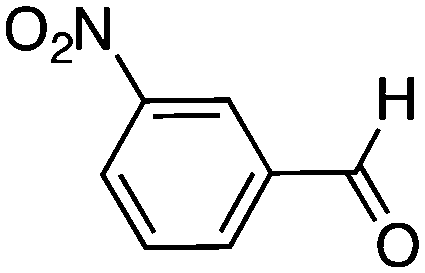
|
48 | 51 |
| 6 |
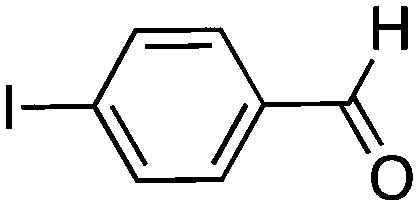
|
16 | >99 |
| 7 |
|
16 | 59 (87)c |
| 8 |
|
16 | 58 (93)c |
| 9 |
|
8 | >99 |
| 10 |
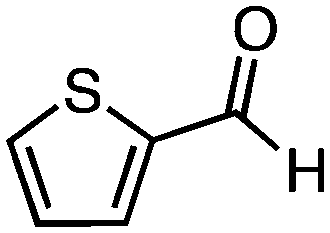
|
16 | 76 (>99)d |
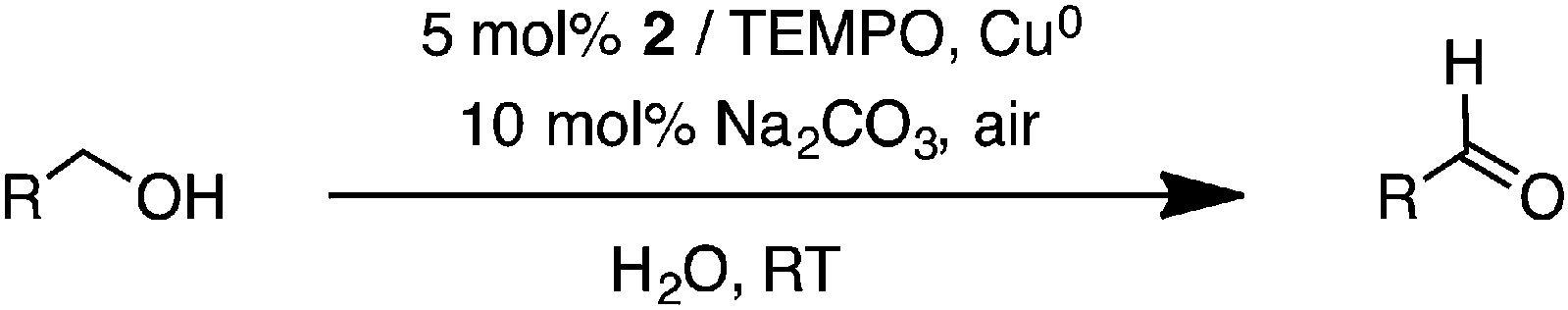
Conclusion
The appended hydroxyl substituents of the (imino)pyridine ligands Hpyph, Hpyet, and Hpypr behave differently in the reaction with CuCl2 under the same conditions. In the case of the ethylene-based ligand Hpyet, the solid structure of 2 reveals a bidentate N,N binding with no Cu–OH interactions. Surprisingly, a slightly longer propyl analogue yielded a considerably different crystal structure for the dinuclear Cu(II) complex 3, as a result of a nucleophilic addition of the appended propyl alcohol to the aldehyde group of 2-pyridinecarboxaldehyde. Despite the lack of X-ray data for the phenyl derivative 1, a symmetrical dimeric structure similar to that of 2 was proposed based on FT-IR, PXRD, and ESR data. Activity comparisons of the Cu(II) complex/Cu0/TEMPO/Na2CO3 catalyst system in water revealed that different coordination environments at the dinuclear copper centers in 2 and 3 do not have a significant impact on the catalytic oxidation of benzyl alcohol. On the other hand, the lower activity of 1 was contributed to the presence of more electron-withdrawing phenylene groups in the ligand framework. In general, this work provides insights into different chemical behaviors of the appended hydroxyl groups based on slightly different ligand chain lengths. Lessons learned from the corresponding Cu coordination and subsequent catalytic alcohol oxidation activities of these complexes in water should be applicable to other metal complexes of related hydroxyl-containing ligand systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Thailand Research Fund (Grant No. RSA6080082). The authors also gratefully acknowledge the Center for Innovation in Chemistry (PERCH-CIC), Faculty of Science, Mahidol University, and Mahidol University under the National Research Universities Initiative. A. J. also appreciates financial assistance from the Science Achievement Scholarship of Thailand (SAST).References
- O. Das and T. K. Paine, in Transition metal catalysis in aerobic alcohol oxidation, ed. F. Cardona and C. Parmeggiani, The Royal Society of Chemistry, 2015, ch. 2, pp. 40–69, 10.1039/9781782621652-00040.
- M. F. Sammelhack, C. R. Schmid, D. A. Cortes and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374–3376 CrossRef.
- P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414–2415 RSC.
- E. T. T. Kumpulainen and A. M. P. Koskinen, Chem. – Eur. J., 2009, 15, 10901–10911 CrossRef CAS PubMed.
- J. M. Hoover, J. E. Steves and S. S. Stahl, Nat. Protoc., 2012, 7, 1161–1166 CrossRef CAS PubMed.
- P. Thongkam, S. Jindabot, S. Prabpai, P. Kongsaeree, T. Wititsuwannakul, P. Surawatanawong and P. Sangtrirutnugul, RSC Adv., 2015, 5, 55847–55855 RSC.
- M. Kongkaew, K. Sitthisuwannakul, V. Nakarajouyphon, S. Pornsuwan, P. Kongsaeree and P. Sangtrirutnugul, Dalton Trans., 2016, 45, 16810–16819 RSC.
- P. J. Figiel, A. Sibaouih, J. U. Ahmad, M. Nieger, M. T. Räisänen, M. Leskelä and T. Repo, Adv. Synth. Catal., 2009, 351, 2625–2632 CrossRef CAS.
- G. Zhang, X. Han, Y. Luan, Y. Wang, X. Wen, L. Xu, C. Ding and J. Gao, RSC Adv., 2013, 3, 19255–19258 RSC.
- G. Zhang, Y. Z. Zhang, W.-F. Lo, J. Jiang, J. A. Golen and A. L. Rheingold, Polyhedron, 2016, 103, 227–234 CrossRef CAS.
- B. H. Lipshutz, M. Hageman, J. C. Fennewald, R. Linstadt, E. Slack and K. Voigtritter, Chem. Commun., 2014, 50, 11378–11381 RSC.
- J. A. D. Cooper, W. Smith, M. Bacila and H. Medina, J. Biol. Chem., 1959, 234, 445–448 CAS.
- J. W. Whittaker, Chem. Rev., 2003, 103, 2347–2363 CrossRef CAS PubMed.
- P. Gamez, I. A. Koval and J. Reedijk, Dalton Trans., 2004, 4079–4088 RSC.
- M. E. Moustafa, P. D. Boyle and R. J. Puddephatt, Chem. Commun., 2015, 51, 10334–10336 RSC.
- K. A. Thompson, C. Kadwell, P. D. Boyle and R. J. Puddephatt, J. Organomet. Chem., 2017, 829, 22–30 CrossRef CAS.
- D. Sunirban, S. A. Maloor, S. Pal and S. Pal, Cryst. Growth Des., 2006, 6, 2013–2018 Search PubMed.
- S. Striegler and M. Dittel, Inorg. Chem., 2005, 44, 2728–2733 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. V. Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. Rodríguez, A. Llobet, M. Corbella, A. E. Martell and J. Reibenspies, Inorg. Chem., 1999, 38, 2328–2334 CrossRef.
- C. P. Pradeep and S. K. Das, Polyhedron, 2009, 28, 630–636 CrossRef CAS.
- G. S. Nyamato, S. O. Ojwach and M. P. Akerman, Dalton Trans., 2016, 45, 3407–3416 RSC.
- C. P. Pradeep and S. K. Das, Coord. Chem. Rev., 2013, 257, 1699 CrossRef CAS.
- X. R. Bu, C. R. Jackson, D. V. Derveer, X. Z. You, Q. J. Meng and R. X. Wang, Polyhedron, 1997, 16, 2991–3001 CrossRef CAS.
- M. S. Ray, R. Bhattacharya, S. Chaudhuri, L. Righi, G. Bocelli, G. Mukhopadhyay and A. Ghosh, Polyhedron, 2003, 22, 617–624 CrossRef CAS.
- A. M. Arnaiz, A. Carbayo, J. V. Cuevas, V. Diez, G. García-Herbosa, R. González, A. Martínez and A. Muñoz, Eur. J. Inorg. Chem., 2007, 4637–4644 CrossRef CAS.
- L. Bendahl, A. Hammershøi, D. K. Jensen, S. Larsen, A. Riisager, A. M. Sargeson and H. O. Sørensen, J. Chem. Soc., Dalton Trans., 2002, 3054–3064 RSC.
- T. Birkle, A. Carbayo, J. V. Cuevas, G. García-Herbosa and A. Muñoz, Eur. J. Inorg. Chem., 2012, 2259–2266 CrossRef CAS.
- N. A. Bailey, D. E. Fenton, R. Moody, C. O. Rodriguez de Barbarin and I. N. Sciambarella, J. Chem. Soc., Dalton Trans., 1987, 2519–2529 RSC.
- K. R. J. Thomas, P. Tharmaraj and V. Chandrasekhar, Polyhedron, 1995, 14, 977–982 CrossRef CAS.
- E. Garribba and G. Micera, J. Chem. Educ., 2006, 83, 1229–1232 CrossRef CAS.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromolecules, 2012, 45, 7388–7396 CrossRef CAS.
- L.-M. Zhang and T. C. W. Mak, J. Am. Chem. Soc., 2016, 138, 2909–2912 CrossRef CAS PubMed.
- B. L. Ryland, S. D. McCann, T. C. Brunold and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 12166–12173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for all new compounds reported in this work, and crystallographic data contained in a CIF file of the Cu(II) complexes. CCDC 1564878 (2) and 1564879 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj03113k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |