
Mobilization of iron from ferritin: new steps and details
A.
La
a,
T.
Nguyen
a,
K.
Tran
a,
E.
Sauble
a,
D.
Tu
a,
A.
Gonzalez
a,
T. Z.
Kidane
a,
C.
Soriano
a,
J.
Morgan
a,
M.
Doan
a,
K.
Tran
a,
C.-Y.
Wang
 b,
M. D.
Knutson
b and
M. C.
Linder
*a
b,
M. D.
Knutson
b and
M. C.
Linder
*a
aDepartment of Chemistry and Biochemistry, California State University, Fullerton, CA 92834-6866, USA. E-mail: mlinder@fullerton.edu
bDepartment of Food Science and Human Nutrition, University of Florida, Gainsville, FL 32611, USA
First published on 4th December 2017
Abstract
Much evidence indicates that iron stored in ferritin is mobilized through protein degradation in lysosomes, but concerns about this process have lingered, and the mechanistic details of its aspects are lacking. In the studies presented here, 59Fe-labeled ferritin was induced by preloading hepatic (HepG2) cells with radiolabeled Fe. Placing these cells in a medium containing desferrioxamine resulted in the loss of ferritin-59Fe, but adding high concentrations of reducing agents or modulating the internal GSH concentration failed to alter the rates of ferritin-59Fe release. Confocal microscopy showed that Fe deprivation increased the movement of ferritin into lysosomes and hyperaccumulation was observed when lysosomal proteolysis was inhibited. It also resulted in the rapid movement of DMT1 to lysosomes, which was inhibited by bafilomycin. Ferrihydrite crystals isolated from purified rat liver/spleen ferritin were solubilized at pH 5 and 7 by GSH, ascorbate, citrate and lysosomal fluids obtained from livers and J774a.1 macrophages. The inhibition of DMT1/Nramp2 and siRNA knockdown of Nramp1 each reduced the transfer of 59Fe from lysosomes to the cytosol; and hepatocyte-specific knockout of DMT1 in mice prevented the release of Fe from the liver responding to EPO treatment, but did not inhibit lysosomal ferritin degradation. We conclude that ferritin-Fe mobilization does not occur through changes in cellular concentrations of reducing/chelating agents but by the coordinated movement of ferritin and DMT1 to lysosomes, where the ferrihydrite crystals exposed by ferritin degradation dissolve in the lysosomal fluid, and the reduced iron is transported back to the cytosol via DMT1 in hepatocytes, and by both DMT1 and Nramp1 in macrophages, prior to release into the blood or storage in ferritin.
Significance to metallomicsHow stored iron – inside the hollow protein ferritin – is made available when needed is not fully understood. Here we show that treating cells with chelating and reducing agents externally or by manipulating them internally does not induce the release of iron from ferritin. Instead, the movement of ferritin from the cytosol into lysosomes and the breakdown of its protein “shell” are required, and intact ferritin accumulates when lysosomal proteases are inhibited. The ferrihydrite crystals exposed by the degradation are readily dissolved by lysosomal “juices” and by glutathione and ascorbate found in lysosomes. DMT1, which accompanies ferritin trafficking to lysosomes, and Nramp1 then transport the dissolved and reduced iron back to the cytosol in ionic form, and hence it can be used internally or be sent to other cells and organs via the blood. These new findings not only corroborate the earlier ones but also provide a much fuller picture of the process by which stored iron in mammalian cells is mobilized. |
Introduction
From the aspect of nutritional biochemistry and metabolism, iron is unique among minerals in the way that it is handled by mammalian organisms.1 Since its solubility in aqueous solution at neutral pH is very low, uptake from the diet is very poor, and is particularly low when compared to the total amount in the organism. In human adults, absorption is in the range of 1–2 mg per day (10–15% of intake), which is a small fraction of the 2000–4000 mg total in a human adult. In contrast to what occurs with other nutrients, most of the mammalian iron is in the blood, making it vulnerable to significant losses that may be accidental or natural (menstrual bleeding, blood donation, etc.). Moreover, there may not be an organized route for iron excretion. Indeed, the body is geared towards retaining the iron it has absorbed, and the excess is stored in a large hollow protein, ferritin. For women, in addition to menstrual iron losses, large amounts (∼400 mg) are transferred to the fetus during pregnancy, and smaller amounts go daily into the milk during lactation. Thus, women tend towards being iron deficient, particularly in their fertile years. For men, the lack of bleeding or iron excretion may lead to excessive iron accumulation over the lifespan, which can result in tissue/cell damage. Thus men tend towards iron overload, particularly as they age. In contrast to other minerals, the concentrations and total amounts of iron in cells and organs are also not constant; they vary enormously from one individual to the next, depending on these and other factors. Most of the variations in concentration are due to the quantities of iron stored in ferritin, which range from zero to large amounts. These facts contrast with the situation for most other metals (like copper and zinc) where the percentages of dietary content absorbed are much higher, the cell and organ concentrations are quite constant, and the rates of excretion adapt to maintain homeostasis.2–4Iron is important not just for the transport of oxygen by red blood cells and muscle myoglobin but also as a cofactor for electron transport proteins as well as a variety of enzymes ranging from aconitases and catalases to ribonucleotide reductases. A large quantity of iron (about 20 mg in human adults) cycles in and out of the red blood cell hemoglobin daily, which particularly involves the spleen, liver and bone marrow.1 As a result, cells in these organs tend to be the richest in iron and ferritin. The amounts of iron to be stored determine the amounts of ferritin protein to be synthesized or degraded, and hence the levels of the ferritin protein reflect the levels of iron stored in ferritin. This balance occurs through the regulation of ferritin synthesis by iron at the translational level – through a well-known mechanism involving iron response elements (IREs) and IRE-binding proteins (IRPs),5 as well as through the regulation of ferritin stability by iron.6 The latter is less well understood but appears to involve the inhibition by iron of ferritin binding7 to the cargo receptor NCOA4, which mediates its entry into lysosomes,8,9 as well as the promotion by iron of NCOA4 degradation.10 The levels of iron in the cytosolic “labile iron pool” (which fluxes in and out of functional and storage compartments) cause changes in the binding of IRPs of the IRE in the 5′UTR of ferritin mRNA, binding (occurring with low iron concentration) resulting in the inhibition of translation/ferritin biosynthesis, and the reverse occurs when extra iron enters in and removes the IRP, leading to increased production.5 Ferritin is a large, 24-subunit, hollow, very efficient iron storage molecule. It has been estimated that up to 4500 iron atoms can be accommodated within a single molecule,11 where they deposit as ferrihydrite crystals that can be visualized using electron microscopy. At any given moment, individual ferritin molecules will have different amounts and patterns of ferrihydrite crystals in their interior. Therefore, iron entering into the cells can initially be accommodated within partially filled and already existing ferritin molecules, while at the same time increasing the formation of new ferritin molecules through the removal of IRPs from ferritin mRNA. Storage in ferritin also provides a means of detoxifying iron ions that might otherwise stimulate the production of reactive oxygen species (ROS) through Fenton chemistry, resulting in cell damage,1,11,12 although other effects of excess iron may contribute.13 Iron enters ferritin through eight 3-fold channels, the deposition of ferrihydrite resulting from the oxidation of Fe(II) by direct interaction with O2 or through the catalytic ferroxidase activity of H ferritin subunits, one of the two kinds of subunits (H and L) that make up the overall structure.14 Excess iron is thus driven to deposit innocuously within ferritin.
How iron stored in ferritin is then released (as needed) has been the subject of many different kinds of studies carried out by a large number of investigators, including ourselves. The knowledge of these processes has been detailed in a recent review.1 However in brief, there is robust evidence that the main route used by cells to extract iron from ferritin requires its protein “shell” to be degraded in lysosomes. This means that ferritin moves from the cytosol into lysosomes for this to occur, a process that involves the specific autophagic cargo receptor (NCOA4) already mentioned.7–10 The resulting complex interacts with ATG proteins (LC3, ATG5 and ATG7) associated with the membranes of the autophagosomes,15,16 which has recently been explored and related to a new form of non-apoptotic cell death (ferroptosis)17–19 and is also of interest in cancer.20,21 Within the lysosomes it is clear that the digestion of the protein part of ferritin is needed for the iron inside to be released and return to the cytosol, as demonstrated by us and others in many different cell types, using specific inhibitors of lysosomal proteases, as well as chloroquine (which increases the lysosomal pH), and by other means.1,22,23 Finding that this is the main mechanism for releasing iron from ferritin was initially unexpected and even seemed counterintuitive for some investigators – which is not surprising. In test tubes, the iron in ferritin is easily removed by dialysis in solutions containing reducing and chelating agents that can penetrate the ferritin protein shell (to produce apoferritin).24 However, this is not what has been observed by studies in vivo, and there is no published evidence that the in vitro process does occur in real life in mammalian cells.1 Moreover in retrospect, it would seem disadvantageous for cells to release iron from ferritin in response to the influx (or production) of high concentrations of reducing or chelating agents (such as ascorbate or citrate), since this is unlikely to be a specific or well-controlled process, and might lead to un-needed recycling of ferritin iron that could promote ROS formation. Some of the studies reported here further address this issue, showing that the influx of ascorbate, glutathione (GSH) and other reducing agents does not result in the efflux of iron from ferritin intracellularly.
In previous studies of ferritin turnover6 and release of iron from ferritin,25 we have demonstrated that both ferritin iron and ferritin protein turned over rapidly and with parallel kinetics6 when cultured cells loaded with iron were depleted of it by exposure to the strong chelator DFO (desferrioxamine) or incubating in a low-iron medium.6,25 The half-lives for both the ferritin iron and ferritin protein were virtually identical in a given cell type (in the range of 10–24 hours), and the half-lives were markedly increased when cells were exposed to inhibitors of lysosomal proteases or chloroquine, but not when exposed to inhibitors of the proteasome.25 This occurred in cells modeling different kinds of iron utilization, from enterocytes (Caco2 cell monolayers) to hepatocytes (rat hepatoma cells), and reticulocytes (K562 cells treated with tributyrin to induce hemoglobin production). The studies reported here provide additional evidence of ferritin movement to lysosomes and address two further questions, namely how the iron in ferrihydrite crystals from ferritin is solubilized, and how it is then transported back into the cytosol, to be used by the cell for endogenous iron-dependent proteins/enzymes, or released to the blood to support iron needs in other parts of the organism.
Materials and methods
Materials
Leupeptin, chloroquine, iodixanol (Opti-prep), and affinity purified goat anti-horse spleen ferritin were obtained from Fisher Scientific (Pittsburgh, PA). Rabbit anti-horse spleen ferritin, affinity purified rabbit anti-horse spleen ferritin, alpha lipoic acid, thioglycolic acid, glutathione, bathophenanthroline disulfonate, and brilliant blue stain were obtained from Sigma (St. Louis, MO). Rabbit anti-Nramp1 and anti-Nramp2 were obtained from Alpha Diagnostics International (San Antonio, TX). Bafilomycin A1 was obtained from LC Laboratories (Woburn, MA). The mouse anti-human Lamp2 (H4B4) antibody was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA). Paraformaldehyde was obtained from Polysciences Inc. (Warrington, PA). Lysosomal tracker 1 was a kind gift from Drs David E. Lewis and Scott C. Hartsel (Department of Chemistry, University of Wisconsin-Eau Claire). Diethyl maleate, 2-cyclohexen-1-one, and flavin mononucleotides were obtained from VWR International (West Chester, PA). Goat anti-rabbit alkaline phosphatase conjugated antibody, prestained high range SDS-PAGE standards, 5-bromo-4-chloro-3-indolyl phosphate (BCIP), nitroblue tetrazolium (NBT) and PVDF membranes were obtained from BioRad (Hercules, CA). LysoTracker Red DND-99, rabbit anti-rat Lamp2 antibody, Alexa 555 donkey anti-rabbit antibody, Alexa 555 donkey anti-mouse antibody, Alexa 647 chicken anti-goat antibody, and Alexa 647 donkey anti-rabbit antibody were obtained from Invitrogen (Carlsbad, CA). OptiSeal polyallomer centrifuge tubes of 13 × 48 mm and 16 × 60 mm dimensions were obtained from Beckman Coulter (Fullerton, CA). The DMT1 inhibitor (6f)26 was a gift from Xenon Pharmaceuticals Inc., Burnaby, BC, Canada, via Jay A. Cadieux and Paul Goldberg. Sprague-Dawley rats were obtained from Simonsen Laboratories (Gilroy, CA).Cells, cell culturing, and treatments
All cells were obtained from the American Type Culture Collection (Manassas, VA). HepG2 and J774a.1 cells were grown in minimal essential medium with 10% fetal bovine serum (FBS), 1 mM pyruvate, and 1% non-essential amino acids. Rat hepatoma H4-II-E-C3 cells were grown in Dulbecco's modified Eagle's medium, with 15% horse serum and 5% fetal bovine serum, all purchased from Sigma-Aldrich (St. Louis, MO).Animals and treatments
All animal studies were performed according to approved protocols from the Institutional Animal Care and Use (IACUC) committee. Adult male mice (1295ve) were obtained from Taconic (Hudson NY). Alternatively, this strain of mouse with and without the hepatic conditional knockout of DMT1 was bred and raised in-house, as previously described.27 Experiments were conducted when mice were between 10 and 12 weeks of age. Groups of mice were either made to bleed 10% of their blood (about 150 μL) from the submandibular vein to induce iron depletion, or injected twice daily i.p. with 10 U of erythropoietin (Procrit; Amgen, Inc.) to stimulate erythropoiesis. Controls were untreated or injected with isotonic saline. During treatments, all mice were fed an iron deficient diet (Dyets, Inc.; AIN 76A). Blood was collected from the liver via the vena cava after whole body heparinization, to remove blood iron from the tissue and measure hematocrit; the liver was frozen for total and ferritin iron analyses. In some cases, fresh livers from adult Fisher rats were used to isolate lysosomes.Measurements of ferritin iron release and ferritin protein
Ferritin iron release and loss of ferritin protein were measured as previously described.6 Briefly, cultured cells and cellular ferritin were loaded with 59Fe overnight by incubation with 59Fe-ferric ammonium citrate (180 μM) in a serum-containing medium. Washed cells were then incubated in fresh medium containing 100 μM desferrioxamine mesylate (DFO; Sigma), with or without other additives for various time periods. These additives included ascorbate, glutathione, thioglycolate, reduced FMN, alpha-lipoic acid, 2-cyclohexen-1-one, and diethylmaleate. In some experiments DFO was absent. The cells were then lysed with lysis buffer (150 mM NaCl, 20 mM Tris, 200 μM PMSF, and 1% Triton X-100, pH 7.5). As in the previous studies, heat supernatants were concentrated 15 fold (Centricon-30; Millipore, Bedford, MA), and 6 or 12 μL portions were used in rocket immunoelectrophoresis (IEP). Autoradiography was performed on IEP gels, and rocket images were recorded using phosphorimaging of the 59Fe. Changes in the total density of the rockets (due to 59Fe) over time indicated the relative amount of ferritin iron remaining at a given time point. Rocket areas calculated from autoradiographs were used to follow changes in ferritin protein concentration. The results were corrected for differences in cell number based on protein concentrations of lysates, determined by the Bradford or BCA assay, using the protocols and reagents from Bio-Rad (Richmond, CA) and bovine serum albumin as the standard.Isolation of ferritin iron cores and measurements of iron solubilisation
Ferritin was purified from the livers and kidneys of Sprague Dawley rats pre-injected i.p. with 50 mg of iron-dextran to induce ferritin accumulation, as previously described.28 The ferrihydrite crystals inside the ferritin (“iron cores”) were isolated by dissolving the ferritin subunits in 2% SDS at 95 °C for 15 min, following which the cores (red) were sedimented and washed twice with large volumes of 20 mM K phosphate, at pH 7, and then nanopure water. Suspensions were tested for iron concentration using bathophenanthroline after reduction with ascorbate29 (as previously described6), and portions (50 or 100 μL) were incubated with buffers containing reducing/chelating agents or 50–100 μL of lysosomal extracts (final vol. 1500 μL), at room temperature with shaking, in 100 mM acetate or MOPS buffers, at pH 5.0 and 7.0, respectively, for various periods of time. After microfuge sedimentation of the remaining cores, the dissolved iron was measured in the top 10% of the supernatant (which was clear) to ensure that we did not include small particles inadvertently released from the pellet during handling.Preparation of lysosomal extracts
Lysosomes were purified from the murine macrophage cell line (J774a.1) pre-treated with LysoTracker Red and MitoTracker Green after cell lysis by nitrogen bomb cavitation, and incubation with 1 mM CaCl2 to increase the density of mitochondria. Lysosomes in 1300 × g supernatants were separated by sedimentation in self-forming iodixanol gradients (Opti-Prep; Sigma-Aldrich), as previously described.25 Lysosomes were purified from 20% homogenates of adult mouse livers and kidneys in 0.25 M sucrose after incubation with 1 mM CaCl2 and Lysotracker Red, sedimentation of unbroken cells and debris at 340 and 1300 × g, and the separation of lysosomes in the supernatant in the same kind of iodixanol gradient. Fractions high in Lysotracker fluorescence and hexosaminidase activity (and low in MitoTracker fluorescence) were pooled, and their contents extracted by sonication or cycles of freezing and thawing.Tissue and cell iron quantitation
Iron quantification was done with bathophenanthroline disulfonate in 45% (w/v) sodium acetate, plus solid ascorbic acid.29 Portions of tissue homogenates (10% w-vol) in nanopure water or cell lysates were wet-ashed in 70% nitric acid (trace element grade; Fisher) with H2O2, and the dissolved residues were analyzed for Fe.Confocal microscopy
HepG2 cells grown on coverslips, and treated/not treated with various agents, were fixed in 4% paraformaldehyde for 10 min. Cells were permeabilized with 0.02% saponin-PBS for 20 min and blocked with 1% bovine serum albumin (BSA) in PBS for 10 min. Autofluorescence was quenched with 75 mM ammonium chloride and 20 mM glycine in PBS for 30 min, at pH 8. Cells were incubated with primary antibodies in 1% BSA-PBS overnight at 4 °C, and (after washings) with secondary antibodies (Alexa 555 and Alexa 647) for 2 h at room temperature. Finally, the cells were mounted in Bio-Rad anti-fade reagent. Autofluorescence levels were evaluated by viewing cells without antibodies, and antibody specificity was evaluated by incubation with secondary antibody alone. Confocal imaging was performed using a Leica TCS-SP2 confocal microscope. Images were prepared using Adobe Photoshop CS2 (Adobe; San Jose, CA) and exported to an image analysis program (ImageJ; NIH, Bethesda, MD) to quantify the area of colocalization.RNA analysis and RNAi treatments
Rat hepatoma cells (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were plated in 60 mm Petri dishes that had been pre-treated with poly-D-lysine. Cells were transfected with specific and scrambled (control) siRNAs from Dharmacon (Thermo Fisher Scientific) in LipofectamineTM RNAiMAX (Invitrogen; Carlsbad, CA) added to normal DMEM with horse and fetal bovine serum (see Cell culture section). siRNA for DMT1 was designed to target all forms of the DMT1 message. mRNA levels were quantitated by Real Time qPCR from cDNA prepared from total RNA extracted from cells with RNA-Bee RNA isolation reagent (Tel-Test; Friendswood, TX), using a BioRad iCycler. Primers and probes for rat and murine DMT1/Nramp2 were designed and provided by Applied Biosystems (Carlsbad, CA).
000) were plated in 60 mm Petri dishes that had been pre-treated with poly-D-lysine. Cells were transfected with specific and scrambled (control) siRNAs from Dharmacon (Thermo Fisher Scientific) in LipofectamineTM RNAiMAX (Invitrogen; Carlsbad, CA) added to normal DMEM with horse and fetal bovine serum (see Cell culture section). siRNA for DMT1 was designed to target all forms of the DMT1 message. mRNA levels were quantitated by Real Time qPCR from cDNA prepared from total RNA extracted from cells with RNA-Bee RNA isolation reagent (Tel-Test; Friendswood, TX), using a BioRad iCycler. Primers and probes for rat and murine DMT1/Nramp2 were designed and provided by Applied Biosystems (Carlsbad, CA).
Immunoblotting
Cells were lysed and sonicated in standard SDS-PAGE sample buffer but with the addition of protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA) and separated (without boiling) on 7.5% SDS polyacrylamide gels, electroblotted onto Immobilon-P transfer membranes (Millipore; Billerica, MA), blocked with 1% dried milk, and incubated with mouse anti-rat DMT1 antibody (Novus Biologicals; Littleton, CO) using goat anti-mouse secondary antibody conjugated with horseradish peroxidase (BioRad), developing the blots with Pierce® ECL Western Blotting Substrate (Thermo Fisher Scientific) visualized using a Kodak Image Station 4000MM (Eastman Kodak; Rochester, NY).Statistical analysis
Data are reported as means ± SD (N). Statistical significance was determined by the Student's t-test; p values <0.05 were considered significant.Results
Attempts to release iron from ferritin by modulating concentrations of reducing/chelating agents
Although iron can clearly be removed from purified ferritin in vitro by exposure to agents that reduce and chelate iron, such as thioglycolate,23 the effects of such agents on living cells have in general not been investigated, except in the case of ascorbate,1 which in fact increased rather than decreased the levels of ferritin iron. Consequently, we tested the effects of a variety of reducing agents, some of which also are strong chelators of iron, for their ability to reduce the amounts of iron in ferritin. The production of 59Fe-labeled ferritin in cultured hepatic (HepG2) cells was induced by exposure to 59Fe-labeled ferric ammonium citrate, and the levels of both the ferritin protein and ferritin iron were measured at various times thereafter by 59Fe autoradiography of ferritin protein rockets obtained by immunoelectrophoresis (see Methods). An example of the changes in size/area of the rockets (a measure of ferritin protein concentration) and density (a measure of ferritin 59Fe content) in response to iron deprivation is shown in Fig. 1A. As seen in previous studies,6,25 there was a decrease in both area and density over time indicative of ferritin iron release and turnover of the ferritin protein. Fig. 1B shows the results of three studies, each in duplicate, in which cells were exposed to reducing and chelating agents during 48 h of iron deprivation induced by placing the 59Fe-labeled cells in a fresh medium containing DFO. In the absence of these agents, the levels of ferritin protein (grey bar) and ferritin iron (dark bar) dropped about 50% from 2 to 48 h, but the ratio of iron to protein (light bar) remained the same as for the 2 h control. Exposure to large doses of various reducing agents over the same period, specifically thioglycolate, reduced flavin mononucleotides or GSH, gave virtually identical results. Thus, high concentrations of reducing/chelating agents did not decrease the iron content of the ferritin, although this would occur in vitro, nor did it change the apparent rate of ferritin degradation. GSH is a likely candidate for ferritin iron removal because it is normally found in cells (and their lysosomes) at high concentrations (2–5 mM).1 However, since we could not be sure that adding GSH to cells would actually increase intracellular GSH (particularly over the long term), we also tested the effects of other agents that increase (alpha-lipoic acid)30,31 and decrease intracellular GSH (2-cyclohexen-1-one and diethyl maleate).32,33 The latter agents decrease GSH levels in the cytosol by conjugating with GSH directly or through GSH transferases, resulting in enhanced water solubility of alkenes that leads to their excretion – as the GSH complex. Lipoic acid made no difference, while 2-cyclohexene-1-one enhanced the loss of not only ferritin iron but also the ferritin protein (the ratio remaining the same as that of the control). In the case of diethylmaleate, there was decreased iron in ferritin, the opposite of what one would expect if high GSH stimulated ferritin iron release. These results indicate that, at least over 2 days, high concentrations of agents that can reduce/chelate iron and remove it from ferritin in vitro do not release iron from ferritin in the context of whole cells.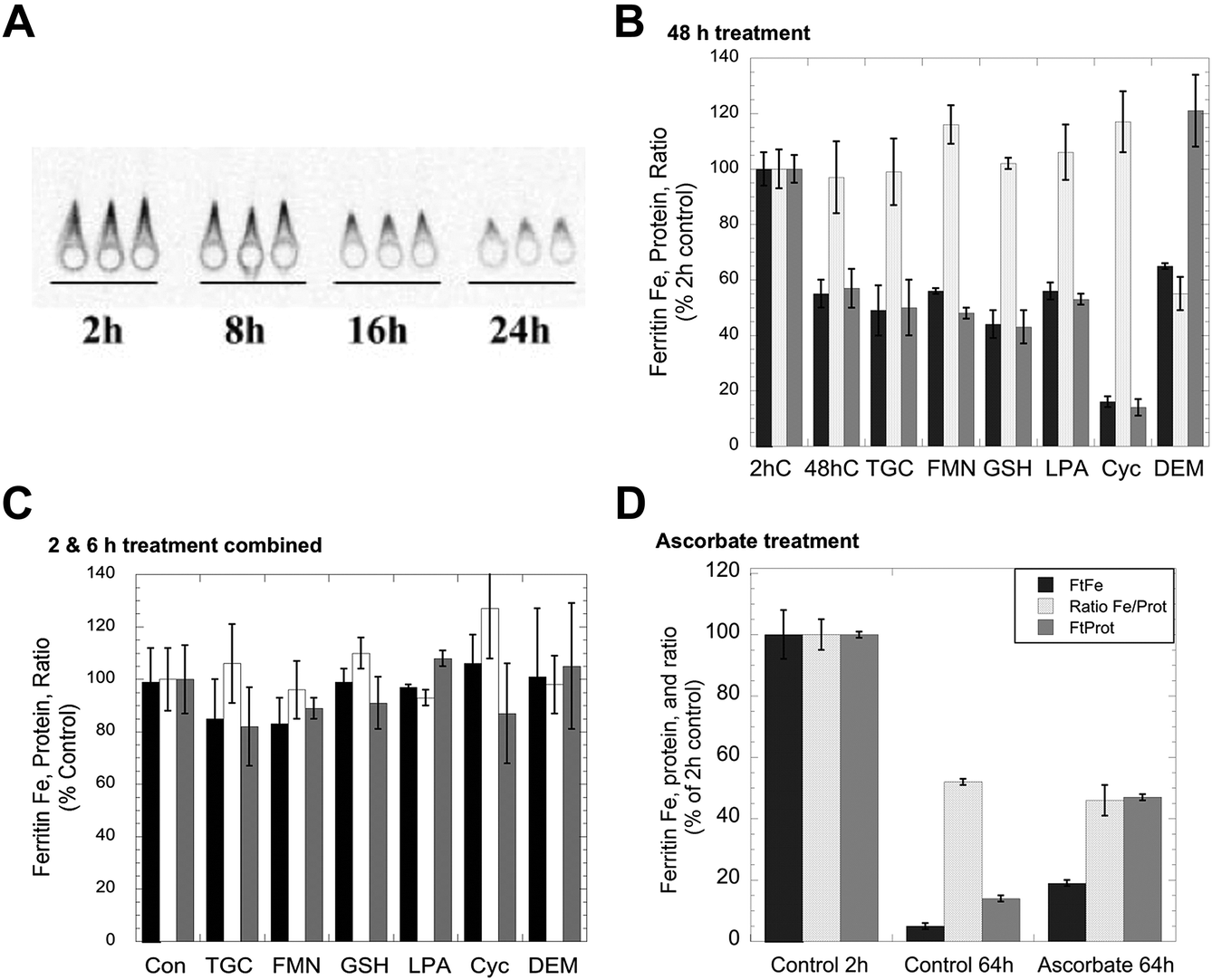 | ||
| Fig. 1 Effects of exposing whole cells to reducing and chelating agents on the release of iron from ferritin. HepG2 cells preloaded with 59Fe-labeled FAC to induce ferritin accumulation were washed and transferred to a culture medium containing 100 μM DFO to induce iron deprivation, in the absence and presence of various agents that reduce and/or chelate iron, for various periods of time, and the resulting cells were then assayed for ferritin protein and ferritin iron-radioactivity using rocket immunoelectrophoresis (IEP). (A) An example of an autoradiograph of 59Fe-labeled ferritin rockets for triplicate cell batches exposed to DFO (without other additions) for various periods of time. The areas of such rockets were used to determine the ferritin protein concentration per mg cell protein, and the densitometry of the rockets determined the relative 59Fe content. (B) The levels of ferritin iron (dark bars) and ferritin protein (grey bars) and the ratios of 59Fe/protein (light bars) after exposure of cells to various reducing/chelating agents for 48 h. Data are percent of 2 h values for a given experiment, and the results of two separate experiments were combined (means ± SD, N = 6). Treatment agents were thioglycolate (TGC; 1 mM, which reduces and chelates Fe); reduced flavin mononucleotide (FMN; 5 mM); glutathione (GSH; 10 mM); alpha-lipoic acid (LPA; 0.5 mM), which increase intracellular GSH concentrations;29,30 2-cyclohexen-1-one (Cyc; 1 mM), which lowers GSH concentrations rapidly31 and dimethylmaleate (DEM; 1 mM), which also lowers GSH concentrations.35 (C) Combined data for changes in the ferritin iron, protein and ferritin Fe/protein ratios for cells exposed or not exposed to the same agents for 2 or 6 h, and relative to controls (not exposed to reducing/chelating agents) within the same experiment (means ± SD, N = 9). (D) Data for the effects of incubation with DFO ± 1 mM ascorbate on ferritin iron (dark bars), ferritin protein (grey bars), and the ratios of 59Fe/protein (light bars) over 64 h. Values are means ± SD (N = 6) for data from two different experiments given as percent of controls (data obtained 2 h after the start of treatment). | ||
The effects of much shorter treatments (0.5, 2 and 6 h) were also examined and led to the same conclusions. Although the loss of ferritin protein and iron was much less (as seen for the combined data for 2 and 6 h; Fig. 1C), the ratios of ferritin iron to ferritin protein were virtually unchanged by any of the treatments, including 2-cyclohexene-1-one or diethyl maleate. The effects of ascorbate were also examined in this cell line (Fig. 1D). As reported previously,34–36 we found that high levels of ascorbate decreased the degradation of the ferritin protein and the release of its iron, i.e. the opposite of what would be expected if ascorbate promoted the release of iron from ferritin by acting upon it directly, in cells. After 64 h of iron deprivation (without ascorbate), the levels of ferritin iron and protein (as well as the Fe/protein ratio) fell markedly, but three times more ferritin iron and protein remained when ascorbate was present, and the Fe/protein ratio at 64 h was the same as that of the control.
The movement of ferritin to lysosomes during iron deprivation, as determined by confocal microscopy
Confocal microscopy presents a way of following protein trafficking in cells, and so we used it on HepG2 cells to follow the migration of ferritin to lysosomes in response to iron deprivation, using LAMP2 as a lysosomal marker. Data from confocal images were used to quantitate the changes in ferritin translocation to lysosomes by counting the area and number of merged particles (representing ferritin in lysosomes) under various conditions. Fig. 2A shows a sample (colored) image of the co-localization of ferritin and LAMP2. In Fig. 2B, sample black and white images from an experiment show just the particles for ferritin alone (top), the lysosomal marker alone (middle), and the merged (ferritin-LAMP2) particles alone, which can be quantitated (see C). Fig. 2B also provides visual evidence that iron deprivation (with DFO) in the presence of a lysosomal protease inhibitor, leupeptin, results in the accumulation of ferritin in lysosomes: the numbers and sizes of particles in the right bottom corner of the figure, representing ferritin–lysosome merges, were greater when the inhibitor was present. Such images from 12 confocal pictures taken from three separate cultures were used to calculate the mean area, particle numbers and particle sizes of the ferritin–lysosome merges, and these results are shown in Fig. 2C. Treatment with chloroquine (Cl; which raises lysosomal pH) or leupeptin (Leu; which inhibits cathepsin b) resulted in 2–3× more ferritin colocalizing with LAMP2 than in the controls (C). The number of merged particles doubled, but the total area occupied by the particles increased even more, particularly in the case of leupeptin treatment. These data provide additional evidence of ferritin entering lysosomes and of its degradation by proteases within the organelle.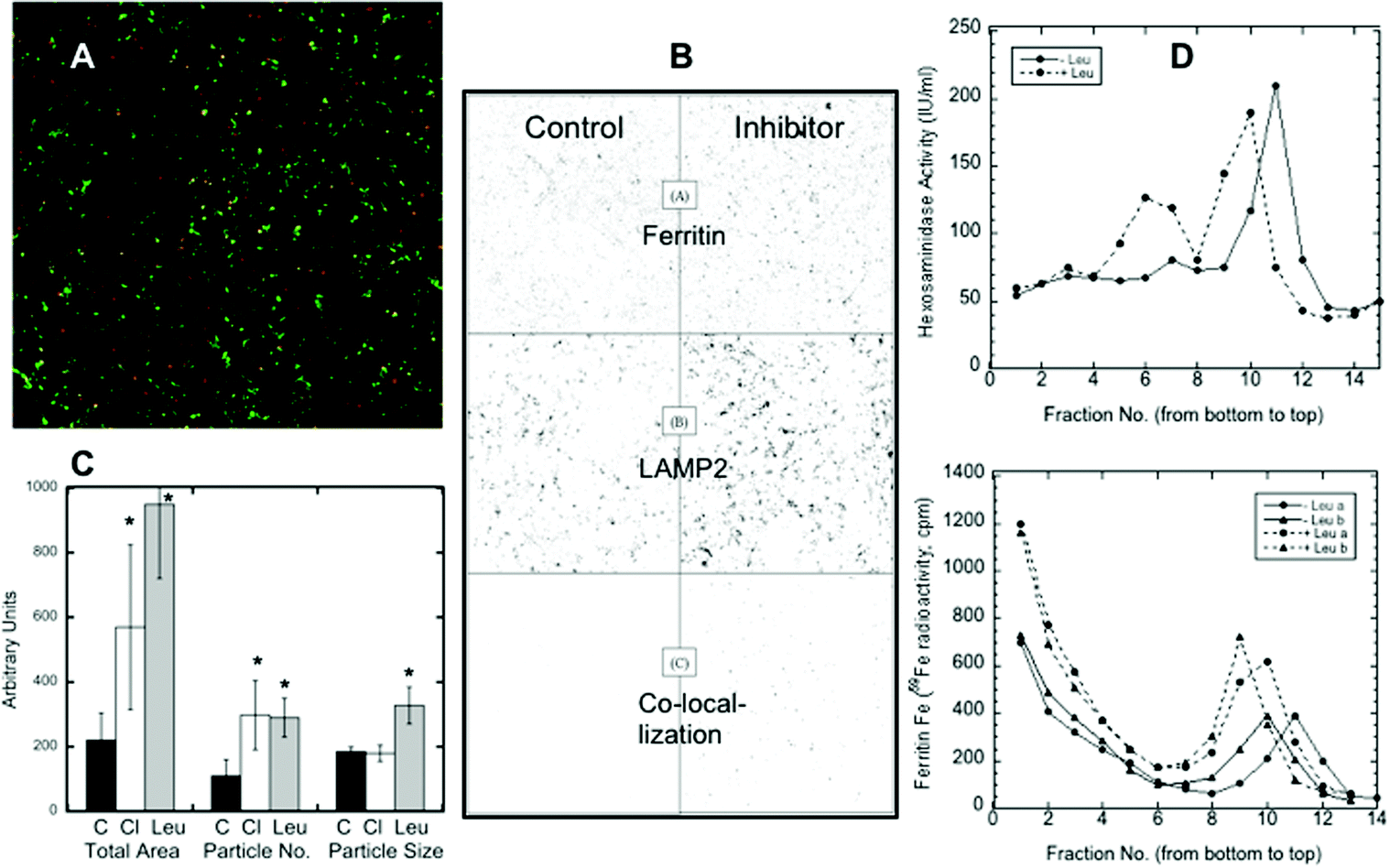 | ||
| Fig. 2 The accumulation of ferritin in lysosomes in response to iron deprivation, and the effects of lysosomal protease inhibitors, using confocal microscopy of cultured HepG2 cells. (A) A representative confocal micrograph of hepatoma cells preloaded with iron (to form ferritin), then transferred to a new culture medium containing DFO in the presence of the lysosomal protease inhibitor, leupeptin, for 48 h. The merged image shows ferritin (red), the lysosomal marker (LAMP2) (green), and the ferritin-lysosome merged particles (yellow). (B) A black and white image of the representative confocal micrograph results for an experiment showing ferritin (top), LAMP2 (lysosomes) (middle), and the merges (colocalizations) of ferritin and LAMP2 (bottom) for cells not exposed (control) or exposed (inhibitor) to the lysosomal protease inhibitor, leupeptin, for 48 h. (C) The effects of leupeptin or chloroquine on the co-localization of ferritin with the lysosomal (LAMP2) marker after iron deprivation, as determined by the total area, number of particles, and size of particles quantitated based on 12 scans of black-white colocalization images, from three separate experiments. Values are means ± SD (N = 12). Except in the case of the particle size with chloroquine, all differences between control and drug treated samples were statistically significant (p < 0.01–0.001). (D) The changes in the co-sedimentation of lysosomes (detected by fluorescence assays of beta-hexosaminidase) and ferritin (detected with 59Fe) sedimented in iodixanol gradients, during iron deprivation (with DFO) in the absence and presence of the lysosomal inhibitor, leupeptin. Data from separate experiments (a and b), with (dashed lines) and without leupeptin (solid lines), were superimposed. Fractions were collected and numbered starting from the bottom of the gradient. | ||
Further evidence of ferritin accumulation in lysosomes upon protease inhibition was obtained by comparing the sedimentation of the 59Fe-labeled ferritin and lysosomes in iodixanol gradients. (Fraction 1 was at the bottom of the gradient, and fractions 14–15 were at the top.) Fig. 2D shows (top graph) the sedimentation of lysosomes detected by measuring beta-hexosaminidase activity (fluorescence), which was increased (from about fraction 11 to fraction 9) when cells were treated with the lysosomal protease inhibitor leupeptin, implying larger size and/or density. The same was the case for ferritin in the lysosomes (Fig. 2D, bottom graph, detected as 59Fe in fractions 9–11). The bottom graph also shows that some ferritin (not with lysosomes) sedimented much more rapidly; the non-lysosomal ferritin (identified with 59Fe) was found near the bottom of the gradient (fractions 1–4). [Ferritin has a variable iron content, which explains the spectrum of densities. Purified ferritin alone, applied to the same kind of gradient (data not shown), sedimented to the bottom fractions like the non-lysosomal ferritin.] The bottom graph in Fig. 2D also shows that there was more ferritin in the lysosomes (radioactivity was higher) upon leupeptin treatment (dashed lines). Also, since leupeptin increased the degree of sedimentation of the lysosomes and their ferritin content, it seems likely that the density of ferritin contributed to this outcome.
How the ferrihydrite crystals in ferritin may be solubilized in lysosomes
When ferritin in lysosomes is degraded by proteases, the ferrihydrite crystals inside would be exposed to the lysosomal fluid. So, the question arises as to how the iron in the crystals would dissolve in the lysosomal fluid in preparation for its transport back into the cytosol. Two possible scenarios come to mind, one being that lysosomes contain enzymes (such as reductases) that interact directly with the ferrihydrite crystals – causing the release of soluble Fe(II) that is kept in solution using small peptides or other chelating metabolites, the other being non-enzymatic reduction and chelation by reagents in the lysosomal fluid. We favored the latter concept, reasoning that since lysosomes contain high concentrations of GSH and ascorbate,37,38 these molecules alone would solubilize the ferrihydrite. We tested this idea by first preparing substantial quantities of these crystals, and then incubating suspensions of them with GSH, ascorbate, or other agents, sedimenting the remaining ferrihydrite crystals and assaying the upper 10% of the supernatant for iron content (to ensure no inclusion of small particles). Ferritin from iron-treated rat livers and spleens was purified using standard procedures involving 70 °C heat treatment, pH 4.8 treatment, ammonium sulfate precipitation, and size exclusion chromatography (SEC) in large pore gels, as previously described.11,28 The resulting ferritin was heated to 90 °C in 2% SDS to dissolve away the protein portion, which was washed away by extensive treatment with nanopure water, the ferrihydrite crystals sedimenting easily to the bottom. Portions of the washed crystal suspension containing ∼3000 ng of iron were then incubated and shaken with various solutions for 15–120 min, and the percentage of solubilized iron was determined.Fig. 3A–C show the time course of the release of iron from the ferrihydrite by 1 and 10 mM ascorbate, GSH and citrate at lysosomal pH (pH 5). With ascorbate (Fig. 3A), a substantial portion of the iron was already solubilized after 15 min of incubation but then increased further in a fairly linear fashion over the next 105 min. There was a slightly higher rate of solubilisation with higher ascorbate concentration. The effect of solubilisation in both concentrations of glutathione (Fig. 3B) was almost immediate and very strong, but then seemed to reverse, the concentrations of the solubilized iron falling back to lower levels. Interestingly, the non-reducing iron chelator, citrate (Fig. 3C), also solubilized some of the iron, the effect also being more pronounced initially and falling back. The reversal of ferritin iron solubilization seen with GSH and citrate is most likely the result of exposure to oxygen during aeration caused by the shaking of the incubated samples and is less likely to occur in vivo, within the lysosomes, where there is less oxygen.
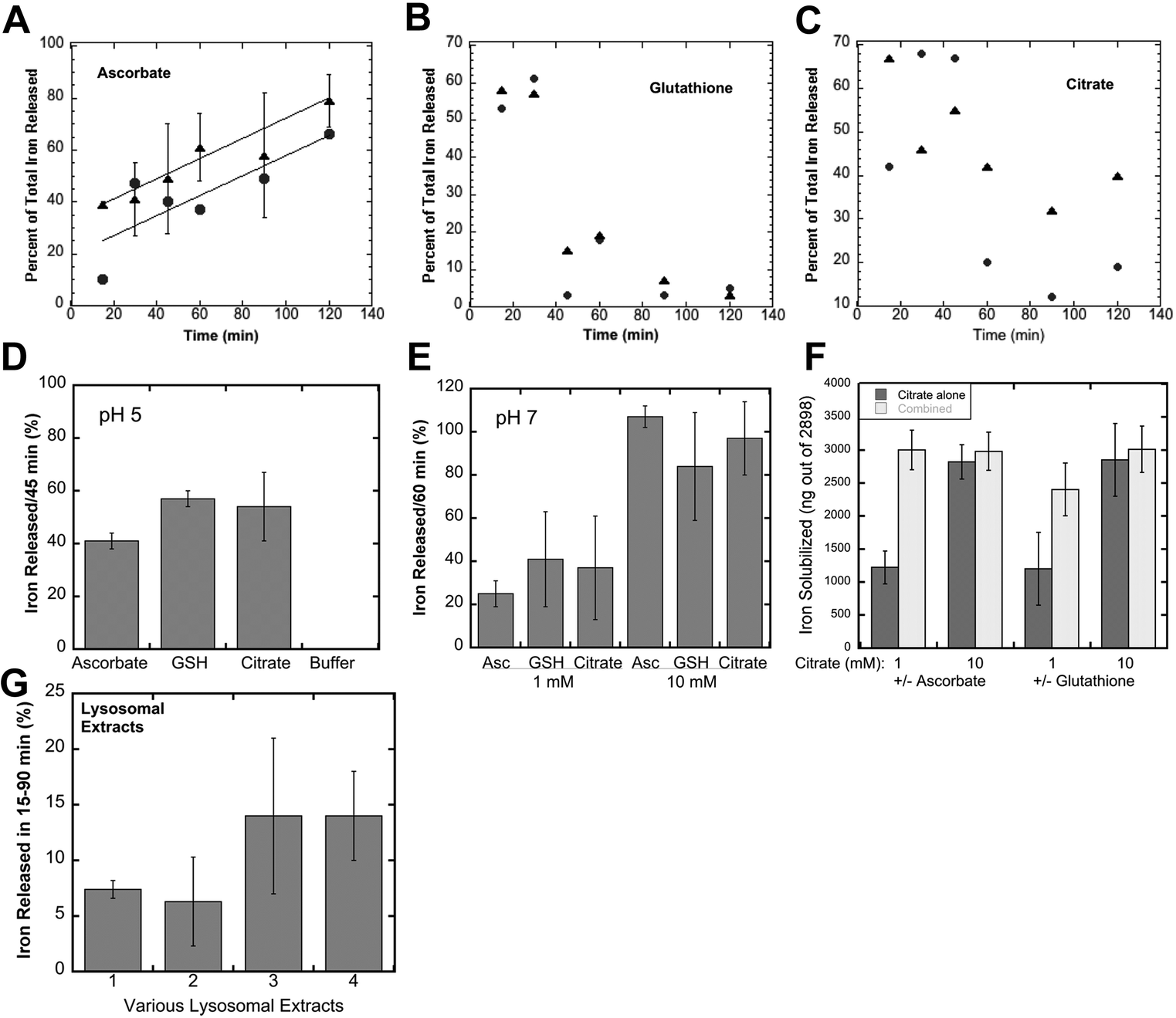 | ||
| Fig. 3 The effects of glutathione, ascorbate, citrate and lysosomal juices on the solubilization of the iron in ferritin iron crystallites (ferrihydrite). Suspensions of the iron crystallites (about 3000 ng Fe) from rat liver/spleen ferritin, obtained by dissolving the ferritin protein in 2% SDS and extensive washing with nanopure water, were incubated in either 100 mM acetate buffer, at pH 5, or 100 mM MOPS buffer, at pH 7, and 1 or 10 mM concentrations of the reducing and/or chelating agents for specific periods of time, and the amount or percentage of the Fe solubilized was measured. (A–C) The time course of iron solubilisation during incubation with 1 mM (black dots) or 10 mM (black triangles) ascorbate (A), glutathione (B) and citrate (C) at pH 5. Data for 1 mM incubations were individual determinations from single experiments, as were those for 10 mM glutathione; data for 10 mM ascorbate were combined from two experiments (N = 5) and given as mean ± SD; data for citrate are averages from duplicate determinations in a single experiment. The best lines through the data for 1 mM (lower line) and 10 mM ascorbate (upper line) in (A) had r2 values of 0.66 and 0.98, respectively. (D–F) Combined data (multiple experiments) for solubilisation of ferritin iron crystallites over 45–60 min, under various conditions (means ± SD; N = 7–15): (D) 10 mM ascorbate, glutathione (GSH), citrate or buffer alone for 45 min at pH 5; (E) 1 or 10 mM of the same reagents, for 60 min at pH 7; (F) combinations of 1 and 10 mM citrate minus/plus 1 or 10 mM ascorbate or GSH for 60 min at pH 7. (G) The effects of lysosomal extracts/fluids isolated from homogenized mouse liver + spleen (1 and 2), or from cultured macrophages (J774.a1) (3 and 4) on the solubilisation of suspensions of the same kind of ferritin iron crystallites over 15–90 min. The pH of the extracts was not adjusted, but was about 5.5. Data are given as the percentage of the ferrihydrite iron solubilised, mean ± SD (N = 4). | ||
Further studies were then carried out using shorter incubation times, to obtain a better sense of the magnitude of the effects, and to examine some other variables. Fig. 3D shows data obtained for the incubation of 10 mM reducing and chelating agents for 45 min at pH 5, or in buffer alone (far right on the graph). The latter failed to release any iron from the ferrihydrite crystals, but the addition of ascorbate or GSH released a large percentage. Citrate seemed to be just as good as GSH and ascorbate in solubilizing the iron from ferritin ferrihydrite crystals. Studies at pH 7 (incubated for 60 min) gave similar results (Fig. 3E). Ascorbate, GSH or citrate at 1 mM concentrations solubilized 25–40% of the ferrihydrite iron, and solubilized almost all of it at 10 mM concentrations in the same time period. Combinations of citrate with ascorbate or GSH were even more effective (Fig. 3F). Thus, substances found in high concentrations in lysosomes (GSH and ascorbate), as well as citrate (which may or may not be present in lysosomes), were able to directly and rapidly bring the iron in ferrihydrite crystals from ferritin into solution, in the absence of its protein “shell”. In this kind of a strongly reducing environment, the resulting iron would be in the Fe(II) state, ready for transport back to the cytosol through DMT1 or other iron transporters. Thus, intervention by reductases or other enzymes would not be required. Furthermore, many in-depth studies would be needed to fully explore and decipher the possible mechanisms by which, alone or together, these reducing and chelating agents are acting to solubilize and reduce the iron, although it is clear that reduction by GSH and solubilisation by citrate are rapid and almost equally effective, and that the ascorbate effect is more sustained.
To verify that lysosomes themselves can dissolve the iron in ferritin-derived ferrihydrite crystals, we isolated them from the macrophage cell line (J774a.1) as well as from mouse livers and kidneys using differential centrifugation and gradient sedimentation, lysed the lysosomal fractions, and tested the resulting fluids for ferrihydrite solubilization. As shown in Fig. 3G, the lysosomal fluids were also able to directly dissolve iron crystals derived from ferritin. The rates of ferritin iron release were linear and slower (data not shown) than those in our studies with reducing and chelating agents, and lysosomes from cell cultured macrophages were more active than those isolated from cells in the liver and spleen, most likely because these tissues also have many other cell types, and lysosomes are most abundant in macrophages.
Identification of transporters involved in returning ferritin-derived iron from the lysosomes to the cytosol
Divalent metal transporter 1 (DMT1; also known as Nramp2) is probably the best known transmembrane iron transporter in mammalian cells and the main one involved in the uptake of dietary iron by enterocytes, after its reduction by duodenal cytochrome b (dCyt b) and/or other reductases present on the cell surface.39,40 DMT1 also mediates iron release from endosomes containing the transferrin–transferrin receptor complex, during the endocytic cycle that delivers transferrin-Fe(III) to cells from the blood (the main way iron is provided to internal cells), after reduction within endocytic vesicles mainly by Steap 3,39 and with the return of apotransferrin to the blood. The association of some DMT1 with lysosomes has been reported.41 The DMT1 homolog Nramp1 is also capable of transporting iron.42Evidence that DMT1 was involved in mobilizing iron stored in ferritin via movement into lysosomes was sought using confocal microscopy and the same approach for the quantitation of co-localization already described for ferritin (Fig. 2). The colocalization of DMT1 with the lysosomal marker (LAMP2) was followed in HepG2 cells preloaded with iron to form ferritin and then deprived of iron with DFO. As shown in Fig. 4A, there was a rapid increase in the area of DMT1–lysosome colocalization in response to iron deprivation. Colocalization increased 10-fold over 2.5 h, a dramatic and rapid change, indicating marked trafficking of this transporter in connection with the movement of ferritin into lysosomes.
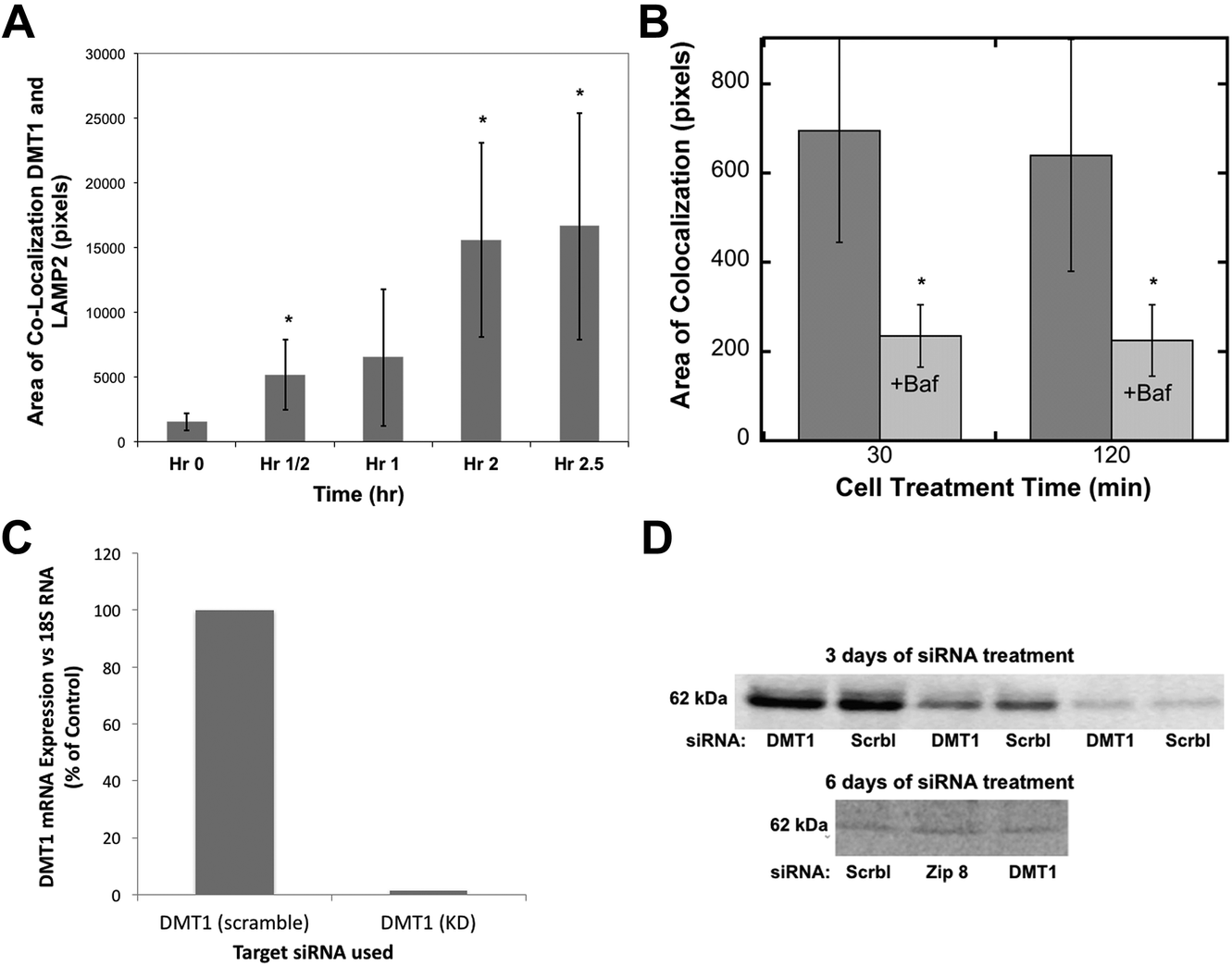 | ||
| Fig. 4 The movement of DMT1 to lysosomes during iron deprivation, effects of bafilomycin, and stability of the DMT1 protein after siRNA treatment. (A and B) Quantitation of the co-localization (total area) of DMT1 with the lysosomal marker LAMP2 in confocal images was as described for ferritin in Fig. 2, (A) over time in hours (h) following placement of HepG2 cells in a medium containing DFO to deplete the cells of Fe, or (B) at 30 and 120 min, without and with the added proton pump inhibitor, bafilomycin A. Data are means ± SD (N = 12). (C and D) The effect of siRNA on the levels of DMT1 mRNA and protein in rat hepatoma cells, (C) mRNA 3 days after treatment with specific (DMT1) or scrambled (Scrbl) siRNA, determined by qPCR (average values for duplicate determinations), and (D) DMT1 protein levels determined by western blotting after 3 and 6 days of treatment (days 1 and 3) with scrambled (Scrbl), Zip 8, or DMT1-specific siRNAs. The 3 day blot is representative of two experiments and shows the same samples applied to SDS-PAGE at different dilutions. The 6 day blot is representative of two experiments in which treatment with siRNA against Zip 8 had been included (and served as another control). | ||
Since the degradation of ferritin is dependent on lysosomal enzymes with low pH optima, we used the lysosomal protein pump inhibitor, bafilomycin, to determine whether it affected the migration of DMT1 to the lysosomes. Fig. 4B shows that the decreased proton pump activity greatly reduced the rates of co-localization, suggesting complex signaling to coordinate this process.
To obtain evidence that the movement of DMT1 to lysosomes is associated with returning ferritin iron to the cytosol, we attempted to knock down DMT1 expression with RNAi. Although the expression of DMT1 mRNA was almost completely abolished over three days of treatment (Fig. 4C), this did not result in a marked decrease in the levels of the DMT1 protein, even with multiple treatments (Fig. 4D), suggesting that it is fairly stable and turns over slowly.
As an alternative, we used a specific DMT1 inhibitor (6f), developed by Xenon Pharmaceuticals, Inc.26 to test the involvement of DMT1 in lysosome-to-cytosol iron return. Here, the lysosomal loading of iron was accomplished by the incubation of murine-derived macrophage-like cells (J774a.1) with iron-dextran, a polymer that is endocytosed to lysosomes. The iron-loaded cells were then treated with the Xenon inhibitor (Xen) or the DMSO vehicle in which it was dissolved, to monitor differences in the rates of lysosomal iron release (hepcidin was not added). Total cell iron and ferritin concentrations were then measured after 24 h. Ferritin concentrations would reflect the transfer of lysosomal iron into the cytosol – which induces ferritin synthesis and accumulation. Thus, the ferritin levels would be a measure of the amount of iron transferred from the lysosomes. As shown in Fig. 5A, treatment with the DMT1 inhibitor reduced the amount of ferritin produced by 40% compared to what occurred in the vehicle-treated control cells. In these experiments we also used siRNA treatment to reduce the activity of the alternative transporter, Nramp1. Treatment with the DMT1 inhibitor (Xen) plus control (scrambled) siRNA had the same effect as treating with Xen alone. However, adding siRNA specific to Nramp1 on top of Xen significantly further reduced the amount of iron entering the cytosol. Total cellular iron levels (Fig. 5B) showed the opposite trend: Xen and Nramp1 siRNA increased the retention of iron in the cells in an additive way, the least being retained in controls and the most when the DMT1 inhibitor and Nramp1 siRNA were used together. These results implied that both DMT1 and Nramp1 are involved in shuttling iron from lysosomes into the cytosol, at least in macrophages, and that a good portion of the iron released to the cytosol was leaving the cells, the rest accumulating in ferritin. The effects of the DMT1 inhibitor or Nramp1 siRNA alone on the formation of ferritin in response to iron released from lysosomes were also tested numerous times (Fig. 5C). The average drop in ferritin production due to Xen was highly significant; Nramp1 siRNA treatment also significantly inhibited the entry of iron into the cytosol from the lysosomes.
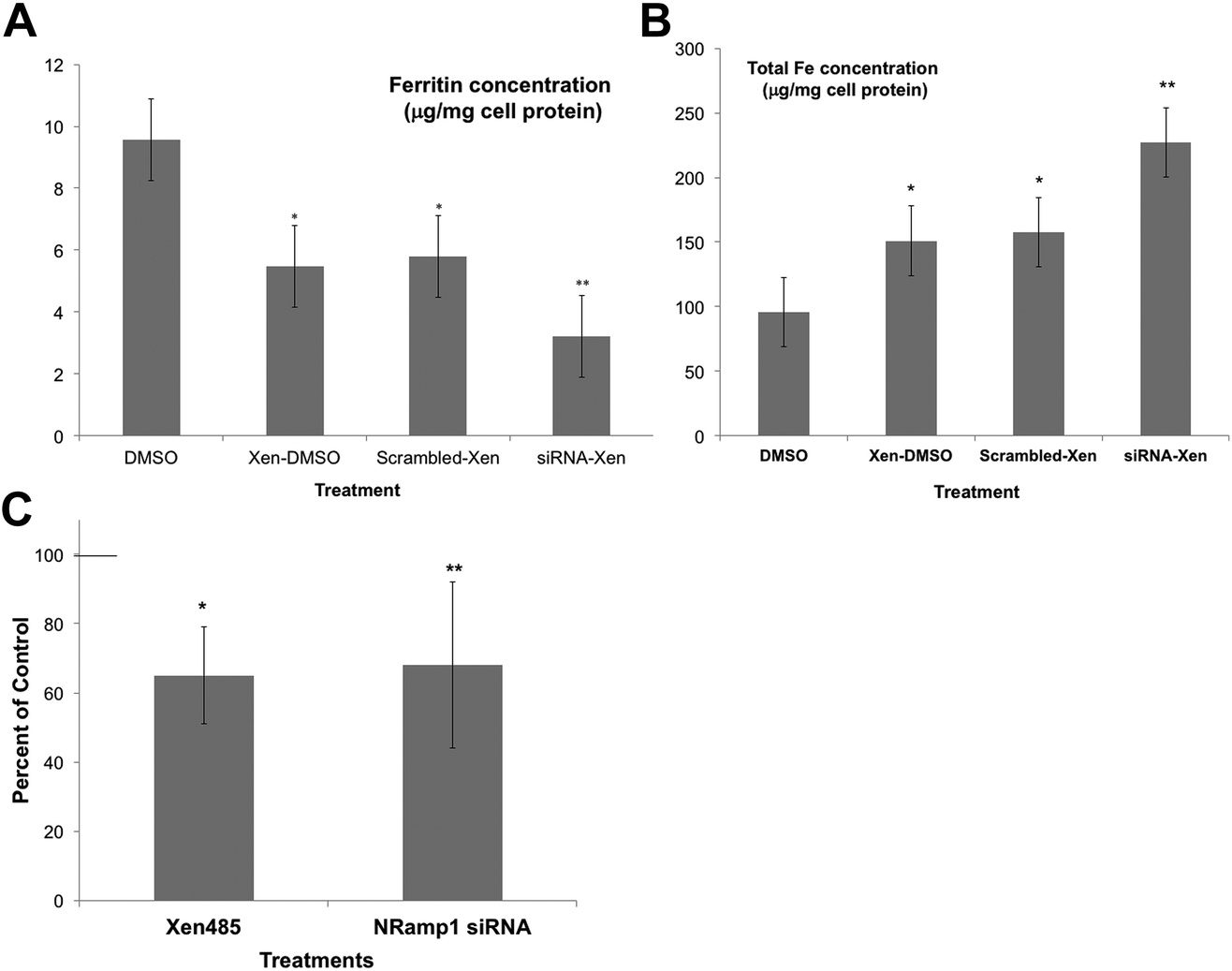 | ||
| Fig. 5 The effects of inhibiting DMT1 and knocking down the expression of Nramp1 on the retention of Fe by lysosomes, using cultured macrophages (J774.1). Macrophage lysosomes were preloaded with iron by exposing the cells to iron dextran, then transferred to new culture medium for 24 h that did/did not contain the DMT1 inhibitor (Xen) [6f],26 and/or scrambled or Nramp1-specific siRNA. (DMSO was the vehicle for Xen.) Cytosolic ferritin concentrations (μg mg−1 cell protein) (A) and total cellular iron (μg mg−1 cell protein) (B), determined in cell extracts for controls (DMSO only), and those cultured with only the drug (Xen-DMSO), the drug and scrambled siRNA (scrambled Xen), and with both the drug and Nramp1 siRNA (siRNA-Xen). Values are means ± SD (N = 4) for two separate experiments. *p < 0.01 for difference from control; **p < 0.02–0.001 for difference from “scrambled-Xen”. (C) The effects of the DMT1 inhibitor alone (Xen) and Nramp1-siRNA alone on the accumulation of cytosolic ferritin in response to the transfer of lysosomal iron, expressed as percent of controls: DMSO vehicle for the drug and scrambled siRNA for the siRNA. Values are means ± SD (N = 8) for 5 experiments. *p < 0.01 for difference from control; **p < 0.02 for difference from control. | ||
The importance of DMT1 for the transfer of ferritin-derived iron from lysosomes to the cytosol was also studied in adult male mice in which the gene for DMT1 was knocked out only in hepatocytes. Iron release from ferritin stores was stimulated either by bleeding of about 10% of their blood from the submandibular vein or by daily subcutaneous injections of erythropoietin (EPO). After bleeding and during EPO treatment, rats were fed a low iron diet to ensure that they could not replenish most of their iron losses from the diet. Care was taken to use WT and conditional DMT1 KO (hDMT1 KO) mice of the same age and sex (male) and with the same dietary history for a given study. Preliminary data indicated that bleeding reduced total iron in the liver over 3 days by about 30% in WT mice. However, in subsequent experiments with WT and hDMT1 KO mice, there were relatively small changes in either total iron or ferritin over the three day period, but the EPO treatment was more effective. We thus carried out several studies on male rats given 5 days of EPO treatment, and the results are presented below.
Five days of EPO treatment resulted in an average increase in the hematocrit in both WT and hDMT1 KO mice, from 47 ± 8 to 56 ± 6 (mean ± SD; N = 7). The total liver iron (Fig. 6A) concentrations fell significantly by about 30% in the case of the WT controls. The initial concentrations of total iron were lower in the hDMT1 KO mice. However, the important point was that EPO treatment resulted in no loss of total liver iron in the mice where DMT1 was knocked out in the hepatocytes. This supported our contention that knocking out DMT1 would diminish the ability of the liver to respond to iron deprivation. In contrast to total liver iron, hDMT1 knockout mice started with the same ferritin protein concentrations as the wild type (determined by rocket immunoelectrophoresis), and treatment with EPO resulted in the same marked drop in ferritin concentrations as in the WT (Fig. 6B). This indicated that ferritin had moved to lysosomes and been degraded. Thus, in the case of hDMT1 KO mice, the iron remained in the cells, not in ferritin but presumably in the lysosomes, implying again that DMT1 was essential for releasing the lysosomal iron derived from ferritin so it could leave the hepatocytes. Our finding that the hDMT1 KO mice had somewhat lower total iron concentrations in their livers than the WT controls differs from what has been reported earlier,27 but is mostly likely explained by the fact that in these last studies, the WT control mice were bred commercially, and although of the same age (and sex) as the KO mice, were only on the same diet as the “home-bred” hDMT1 KO mice for a few weeks before the experiments. Similar measurements of the responses of total iron and ferritin to EPO treatment or bleeding were made for the spleens of the WT and hDMT1 KO mice (data not shown). However, as anticipated,43,44 these treatments caused splenomegaly and induction of erythropoiesis in the spleens, which would prevent us from being able to differentiate between the mobilization of ferritin iron for bone marrow erythropoiesis versus retention and use for erythropoiesis within the spleen.
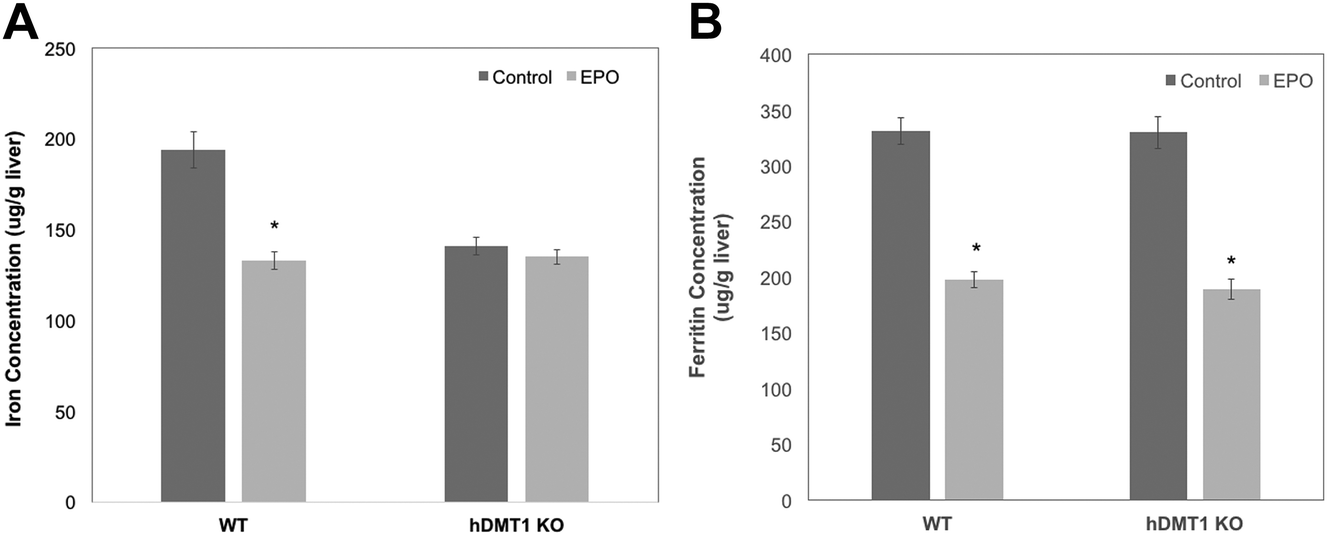 | ||
| Fig. 6 The effects of knocking out hepatocyte DMT1 on iron and ferritin in the livers of mice treated with erythropoietin (EPO) to induce the erythropoietic release of stored iron from this organ. Hepatocyte-specific DMT1-knockout mice (hDMT1) and matching wildtype (WT) mice were injected daily s.c. with 10 U EPO in 0.1 mL saline (light bars; EPO), or saline alone (dark bars; control) for 5 days. Their total liver Fe (A) and ferritin protein (B) concentrations (μg g−1 liver) were then measured and compared. Values are means ± SD (N = 8). *p < 0.001 for difference from control. | ||
Discussion
The purpose of the studies reported here was to add to our understanding of the mechanism by which iron stored in ferritin is released in response to cellular iron deprivation, and/or blood loss and other stimuli that enhance erythropoiesis in mammals. Ferritin is found primarily where it has been synthesized, namely in the cytosol. It is now well established that for the release of its iron, ferritin first undergoes autophagy into the lysosomes by binding to the cargo receptor, NCO4A.1,7–10 Previously, we and others had established that the iron in ferritin is only released after the exterior (protein) portion of the molecule is degraded by lysosomal proteases.1 Thus for example, not only does the loss of 59Fe-labeled iron in ferritin parallel the loss of the ferritin protein25 in cultured cells responding to iron deprivation, but the rate of loss of ferritin iron and protein is markedly reduced by treating with inhibitors of various lysosomal proteases, or chloroquine – which causes an increased lysosomal pH and thus reduced lysosomal enzyme activity.1 In this report, we provide confirmatory evidence of ferritin movement into lysosomes using confocal microscopy and provide for the first time quantitative evidence that ferritin accumulates in the lysosomes when its degradation is inhibited by lysosomal protease inhibitors. This indicates that there is no feedback inhibition of ferritin autophagy when the proteolytic activity of lysosomes is impaired and ferritin remains intact.The degradation of the ferritin protein is required for the release of the iron in its interior. Thus, the crystals of ferrihydrite inside will become exposed to the lysosomal fluid as degradation proceeds. The question then arises as to whether enzymatic activity is required to solubilize the ferrihydrite iron. Our data show that this is not the case. The lysosomal levels of at least two powerful reducing and chelating agents for iron (glutathione and ascorbate) are high,37,38 and our data clearly demonstrate that physiological concentrations can rapidly solubilize the iron in ferrihydrite, and that this also occurs in lysosomal fluids but at a slower rate. Although the exact concentrations in lysosomes are unknown, it is well established that iron release in lysosomes (from ferritin or after endocytosis of iron particles) lowers the cellular GSH concentrations substantially;45–47 cytosolic concentrations are in the range of 2–5 mM;37,38 lysosomes are a favorable milieu for Fenton reactions due to their content of reducing agents (GSH and ascorbate, as well as cysteine);48–50 and the formation of reactive oxygen species (ROS) in lysosomes is dependent not only on iron but also on reducing agents like GSH,37,47,48 and probably mainly on GSH.47 Indeed there is a great deal of interest in the role of lysosomal ROS formation (via iron and these reducing agents) in various diseases that result in cell death, which might even be exploited to kill cancer cells.51 Thus, although lysosomes may not have as high a redox potential as the cytosol,52 and their lower pH may somewhat reduce the rates of reduction, there is plenty of evidence that GSH is present in lysosomes53 and plays an important role in that organelle that can, in certain circumstances (such as when lysosomes are loaded with extra iron through endocytosis), lead to high ROS concentrations and increased cell death (termed “ferroptosis”, currently under investigation).47 We would suggest that normally, the formation of ROS does not occur, since after entry into the lysosomes, the rate of ferritin protein degradation, which is required for ferrihydrite crystals to be dissolved, plus the rate of returning solubilized iron to the cytosol by at least two transporters, prevents the accumulation of excess iron ions that might generate the acute production of ROS, i.e. as the ferritin protein shell is degraded, the ferrihydrite is rapidly solubilized and then rapidly returned to the cytosol, and this is a continuous process.
Upon solubilisation in lysosomes, the iron from ferrihydrite would then be in the Fe(II) state and thus capable of being transported out of the lysosomes back to the cytosol by known iron transporters that take up reduced iron, which we investigated. Divalent metal transporter 1 (DMT1, also known as Nramp2 and Slc11A2) is the main player in the uptake of dietary iron by intestinal epithelial cells and is also important for the transfer of Fe(II) from endosomes carrying the holo-transferrin–transferrin receptor complex after the receptor-mediated uptake of Fe(III) circulating on transferrin in the blood, after its reduction (in the endosomes) by the ferrireductase, Steap 3.39 The association of DMT1 with the lysosomes of HEp-2 cells had also previously been reported.41 Here we confirmed with confocal microscopy that DMT1 is associated with the lysosomes of hepatic cells and show for the first time that the amount of DMT1 associated with lysosomes increases markedly and rapidly in response to iron deprivation and the entry of ferritin into lysosomes. This implies a connection between the entry of storage iron into lysosomes and its transfer back into the cytosol as Fe(II) with the help of this transporter, from which the Fe(II) could be released into the blood (via ferroportin) to support bone marrow erythropoiesis. Interestingly, the migration of DMT1 to lysosomes was dependent on the lysosomal proton concentration, since inhibition of proton pumping with bafilomycin A inhibited the process. This also fits with the fact that the transport of metal ions by DMT1 is proton dependent.54
Further support for the involvement of DMT1 in returning iron from the lysosomes to the cytosol was obtained using livers from whole animals (mice) and cultured macrophages. Our studies with hepatocyte-specific DMT1 knockout mice and age and sex-matched controls indicated that DMT1 was indeed important for the mobilization of iron stored in the liver. In fact, hepatocyte-specific DMT1 knockout prevented the significant 30% loss of total liver iron that occurred in wild type mice responding to daily injections of erythropoietin (EPO) over 5 days. It did not however prevent the movement of ferritin into lysosomes and its degradation, implying that without DMT1 in the hepatocytes, iron was stuck in the lysosomes. This indicates not only that DMT1 is the primary transporter in hepatocytes responsible for the transfer of lysosomal iron to the cytosol and for exit from these cells, but also that the liver storage iron pool mobilized by excess EPO occurs primarily in hepatocytes and that these cells may also be holding most of the stored iron.
Using a specific DMT1 inhibitor, kindly provided by Xenon Pharmaceuticals (6f),26 we found that DMT1 was also important for the transfer of lysosomal iron to the cytosol in macrophages. Macrophages in the spleen and those in liver (Kupffer cells) are important in processing large quantities of red blood cell iron daily, which involves the autophagy of aged erythrocytes into lysosomes, the degradation of hemoglobin, and the return of the iron to the cytosol, where it is incorporated into ferritin for temporary storage and/or released into the blood for return to bone marrow erythropoiesis and/or transfer to hepatocytes. To examine the role of DMT1 in macrophages, we used the murine cell line J774.a1, which expresses both DMT1 and Nramp1.55 Lysosomes were loaded with iron by the exposure of cells to iron dextran (which is autophagized), and the levels of ferritin produced were measured to assess the amounts of iron released from the iron dextran in the lysosomes. [As described earlier, iron stimulates cytosolic ferritin synthesis through a translational mechanism involving iron response elements and iron response proteins (IREs and IRPs).5] Despite high iron levels in the lysosomes from the iron dextran, the DMT1 inhibitor consistently and significantly decreased the accumulation of ferritin in these cells, indicating that it diminished the transfer of lysosomal iron into the cytosol, and that DMT1 participated in this process. Since the related transporter, Nramp1, plays a role in macrophage iron metabolism and was detected in phagolysosomes,55,56 we also investigated its potential involvement in lysosomal iron release in the same macrophages. We found that the siRNA knockdown of Nramp1 mRNA also had a consistent negative effect on the transfer of lysosomal iron to the cytosol. Our findings that both Nramp 1 and 2 are differentially involved in this process are consistent with evidence from some previous reports, namely that Nramp1 is an Fe(II) transporter,57 the expression of which is mainly confined to macrophages (and neutrophils);55 it co-localizes with phagolysosomes;56 and the knockout of its expression results in the retention of iron in the spleen (rich in macrophages) rather than in the liver,58 particularly when the turnover of red blood cells was enhanced by phenylhydrazine. Collectively, these results indicate that in hepatocytes, DMT1 is the main transporter involved in the lysosome to cytosol transfer step in ferritin iron mobilization, whereas in macrophages both DMT1 (Nramp 2) and Nramp 1 participate, as also concluded by Soe-Lin et al.59
When we began these studies, there was some skepticism about the autophagy mechanism of ferritin iron mobilization,1 since in vitro, the iron in ferritin can be removed by direct treatment with small reducing and chelating agents that enter the interior compartment (where the ferrihydrite crystals are located) through channels in the protein structure, and the chelated dissolved Fe(II) can then diffuse out.1 In response to these concerns, we pre-treated cultured cells with 59Fe-ferric ammonium citrate to induce 59Fe-labeled ferritin production, then treated them with high concentrations of various reducing and chelating agents, or treated them with modulators of intracellular glutathione concentrations. The cells were monitored for levels of ferritin iron and ferritin protein over many hours, during which cells not treated with the reductants and chelators lost ferritin iron and protein in parallel, while growing in a medium deprived of iron. None of the treatments enhanced the loss of iron from ferritin, nor decreased the ferritin iron/protein ratio, as would be expected if these agents were removing iron from intact ferritin within the cells. Two agents that decrease the cystosolic levels of GSH (diethylmaleate and 2-cyclohexene-1-one) by conjugating with it and causing excretion32 did have some effects on ferritin that were contradictory, and could not be attributed directly to GSH. [Less GSH resulted in greater iron release (cyclohexene) or increased ferritin degradation (diethylmaleate).] So it is clear that these agents also have effects other than depleting cells of GSH. More importantly, their effects failed to support the idea that GSH directly removes iron from undegraded, cytosolic ferritin.
Our studies with reducing and chelating agents applied externally to cells also reconfirmed the results of others that ascorbate treatment actually decreases rather than increases the release of ferritin iron,1,34–36,60 and showed that the rate of turnover of ferritin (protein as well as iron) was slowed by ascorbate. This fits perfectly with earlier findings of Bridges61 that ascorbate inhibits the entry of ferritin into lysosomes, and suggests that ascorbate interferes with the mechanism of autophagy, or the signal which triggers this process for ferritin, in response to the need for the release of storage iron. These studies reconfirm the conclusions from multitudes of previous work,1 indicating that the iron in ferritin is not released in the cytosol by endogenous reducing and/or chelating agents that can carry out this process in vitro. Also, since the data from virtually all studies with mammalian cells,1 including those presented here, show that the degradation of ferritin depends upon first degrading the ferritin protein “shell”, it is highly unlikely that the kind of protein–protein interaction mechanism recently identified for bacterioferritin62 also occurs in mammals.
The inability of high concentrations of reducing and chelating agents to release ferritin iron in living cells is noteworthy and also makes physiological sense: taking large doses of vitamin C (or related iron reducers and chelators) might otherwise inadvertently promote the formation of reactive oxygen species by increasing the levels of Fe-ascorbate (etc.) in an uncontrolled manner, in contrast to what seems clearly to be the case, namely that iron release from ferritin only occurs through a complex-controlled response to iron deprivation (or need for more active iron) and is confined to a specific compartment to protect other parts of the cell. However, just how the process of ferritin autophagy (to bring ferritin into lysosomes) is triggered still remains to be discovered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the gift of the DMT1 inhibitor used in these studies from Xenon Pharmaceuticals, Inc., Burnaby, B.C., Canada, provided through Jay Cadieux and Paul Goldberg.References
- M. C. Linder, Mobilization of stored iron in mammals: a review, Nutrients, 2013, 5, 4022–4050 CrossRef PubMed.
- M. C. Linder, Biochemical Aspects of Human Nutrition, 2010, pp. 143–179, ISBN 978-81-7895-478-3 Search PubMed.
- M. C. Linder, Ceruloplasmin and other copper binding components of blood plasma and their functions: an update, Metallomics, 2016, 8, 887–905, 10.1039/c6mt00103c.
- S. Hojyo and T. Fukada, Zinc transporters and signaling in physiology and pathogenesis, Arch. Biochem. Biophys., 2016, 611, 43–50 CrossRef CAS PubMed.
- C. P. Anderson, M. Shen, R. S. Eisenstein and E. A. Leibold, Mammalian iron metabolism and its control by iron regulatory proteins, Biochim. Biophys. Acta, 2012, 1823, 1468–1483 CrossRef CAS PubMed.
- J. Truty, R. Malpe and M. C. Linder, Iron prevents ferritin turnover in hepatic cells, J. Biol. Chem., 2001, 276, 48775–48780 CrossRef CAS PubMed.
- M. Gryzik, A. Srivastava, G. Longhi, M. Bertuzzi, A. Gianonocelli, F. Carmona, M. Poli and P. Arosio, Expression and characterization of the ferritin binding domain of Nuclear Receptor Coactivator-4 (NCOA4), Biochim. Biophys. Acta, 2017, 30232–30235 Search PubMed , pii: S0304-4165(17).
- J. D. Mancias, X. Wang, S. P. Gygi, J. W. Harper and A. C. Kimmelman, Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy, Nature, 2014, 509, 105–109, DOI:10.1038/nature13148.
- W. E. Dowdle, B. Nyfeler, J. Nagel, R. A. Elling, S. Liu and E. Triantafellow, et al., Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo, Nat. Cell Biol., 2014, 16, 1069–1079 CrossRef CAS PubMed.
- J. D. Mancias, L. P. Vaites, S. Nissim, D. E. Biancur, A. J. Kim, X. Wang, Y. Liu, W. Goessling, A. C. Kimmelman and J. W. Harper, Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis, eLife, 2015, 4, e10308 Search PubMed.
- H. N. Munro and M. C. Linder, Ferritin: Structure, biosynthesis, and role in iron metabolism, Physiol. Rev., 1978, 58, 317–346 CAS.
- R. T. Scarl, C. M. Lawrence, H. M. Gordon and C. S. Nunemaker, STEAP4: its emerging role in metabolism and homeostasis of cellular iron and copper, J. Endocrinol., 2017, 234, R123–R134 CrossRef PubMed.
- P. Kraml, The role of iron in the pathogenesis of atherosclerosis, Physiol. Res., 2017, 66, S55–S67 CAS.
- M. Mehlenbacher, M. Poli, P. Arosio, P. Santambrogio, S. Levi, N. D. Chasteen and F. Bou-Abdallah, Iron oxidation and core formation in recombinant heteropolymeric human ferritins, Biochemistry, 2017, 56, 3900–3912 CrossRef CAS PubMed.
- W. Hou, Y. Xie, X. Song, X. Sun, M. T. Lotze, H. J. Zeh III and R. Kang, Autophagy promotes ferroptosis by degradation of ferritin, Autophagy, 2016, 12, 1425–1428 CrossRef CAS PubMed.
- J. D. Mancias and A. C. Kimmelman, Mechanisms of selective autophagy in normal physiology and cancer, J. Mol. Biol., 2016, 428, 1659–1680 CrossRef CAS PubMed.
- S. Doll and M. Conrad, Iron and ferroptosis: A still ill-defined liaison, IUBMB Life, 2017, 69, 423–434 CrossRef CAS PubMed.
- G. O. Latunde-Dada, Ferroptosis: Role of lipid peroxidation, iron, and ferritinophagy, Biochim. Biophys. Acta, 2017, 1861, 1893–1900 CrossRef CAS PubMed.
- M. Gao, P. Monian, Q. Pan, W. Zhang, J. Xiang and X. Jiang, Ferroptosis is an autophagic cell death process, Cell Res., 2016, 26, 1021–1032 CrossRef CAS PubMed.
- S. Torii, R. Shintoku, C. Kubota, M. Yaegashi, R. Torii, M. Sasaki, T. Suzuki, M. Mori, Y. Yoshimoto, T. Takeuchi and K. Yamada, An essential role for functional lysosomes in ferroptosis of cancer cells, Biochem. J., 2016, 473, 769–777 CrossRef CAS PubMed.
- R. F. Dielschneider, E. S. Henson and S. B. Gibson, Lysosomes as oxidative targets for cancer therapy, Oxid. Med. Cell. Longevity, 2017, 3749157, DOI:10.1155/2017/3749157.
- Y. Zhang, M. Mikhael, D. Xu, Y. Li, S. Soe-Lin, B. Ning, W. Li, G. Nie, Y. Zhao and P. Ponka, Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit, Antioxid. Redox Signaling, 2010, 13, 999–1009 CrossRef CAS PubMed.
- T. Asano, M. Komatsu, Y. Yamaguchi-Iwai, F. Ishikawa, N. Mizushima and K. Iwai, Distinct mechanisms of ferritin delivery to lysosomes in iron depleted and iron replete cell, Mol. Cell. Biol., 2011, 31, 2040–2052 CrossRef CAS PubMed.
- D. Gelvan, E. Fibach, E. G. Meyron-Holtz and A. M. Konijn, Ferritin uptake by human erythroid precursors is a regulated iron uptake pathway, Blood, 1996, 88, 3200–3207 CAS.
- T. Z. Kidane, E. Sauble and M. C. Linder, Release of iron from ferritin requires lysosomal activity, Am. J. Physiol.: Cell Physiol., 2006, 291, C445–C455 CrossRef CAS PubMed.
- J. A. Cadieux, Z. Zhang, M. Mattice, A. Brownlie-Cutts, J. Fu, L. G. Ratkay, R. Kwan, J. Thompson, J. Sanghara, J. Zhong and P. Goldberg, Synthesis and biological evaluation of substituted pyrazoles are blockers of divalent metal transporter 1 (DMT1), Bioorg. Med. Chem. Lett., 2012, 22, 90–95 CrossRef CAS PubMed.
- C.-Y. Wang and M. D. Knutson, Hepatocyte divalent metal ion transporter-1 is dispensable for hepatic iron accumulation and nontransferrin-bound iron uptake in mice, Hepatology, 2013, 58, 788–798 CrossRef CAS PubMed.
- M. C. Linder, H. R. Kakavandi, P. Miller and G. N. Nagel, Dissociation of ferritins, Arch. Biochem. Biophys., 1989, 269, 485–496 CrossRef CAS PubMed.
- W. James and B. Zak, Determination of serum copper and iron in a single sample, Am. J. Clin. Pathol., 1958, 29, 590–592 Search PubMed.
- L. A. Powell, K. M. Warpeha, W. Xu, B. Walker and E. R. Trimble, High glucose decreases intracellular glutathione concentrations and upregulates inducible nitric oxide synthase gene expression in intestinal epithelial cells, J. Mol. Endocrinol., 2014, 33, 797–803 CrossRef PubMed.
- J. H. Suh, S. V. Shenvi, B. M. Dixon, H. Liu, A. K. Jaiswal, R. M. Liu and T. M. Hagen, Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3381–3386 CrossRef CAS PubMed.
- C. W. Parker, C. M. Fischman and H. J. Wedner, Relationship of biosynthesis of slow reaction substance to intracellular glutathione concentrations, Proc. Natl. Acad. Sci. U. S. A., 1983, 77, 6870–6873 CrossRef.
- T. T. Peternelj, S. A. Marsh, N. A. Strobel, A. Matsumoto, D. Briskey, V. J. Dalbo, P. S. Tucker and J. S. Coombes, Glutathione depletion and acute exercise increase O-GlcNAc protein modification in rat skeletal muscle, Mol. Cell. Biochem., 2015, 400, 265–275 CrossRef CAS PubMed.
- K. E. Hoffman, K. Yanelli and K. R. Bridges, Ascorbic acid and iron metabolism: Alterations in lysosomal function, Am. J. Clin. Nutr., 1991, 54, 1188S–1192S CAS.
- K. Ollinger and K. Roberg, Nutrient deprivation of cultured rat hepatocytes increases the desferrioxamine-available iron pool and augments the sensitivity to hydrogen peroxide, J. Biol. Chem., 1997, 272, 23707–23711 CrossRef CAS PubMed.
- J. A. Larson, H. L. Howie and M. So, Neisseria meningitis accelerates ferritin degradation in host epithelial cells to yield an essential iron source, Mol. Microbiol., 2004, 53, 807–820 CrossRef CAS PubMed.
- T. Kurz, J. W. Eaton and U. T. Brunk, The role of lysosomes in iron metabolism and recycling, Int. J. Biochem. Cell Biol., 2011, 43, 1686–1697 CrossRef CAS PubMed.
- A. Terman, T. Kurz, M. Navratil, E. Arriaga and U. Brunk, Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging, Antioxid. Redox Signaling, 2010, 12, 503–535 CrossRef CAS PubMed.
- M. D. Knutson, Steap Proteins: Implications for iron and copper metabolism, Nutr. Rev., 2007, 65, 335–340 Search PubMed.
- S. Wyman, R. J. Simpson, A. T. McKie and P. A. Sharp, Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro, FEBS Lett., 2008, 582, 1901–1906 CrossRef CAS PubMed.
- T. Tabuchi, Y. Yoshimori, K. Yamaguchi, T. Toshida and F. Kishi, Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells, J. Biol. Chem., 2000, 275, 22220–22228 CrossRef PubMed.
- C. Curie, J. M. Alonso, M. Le Jean, J. R. Ecker and J. F. Briat, Involvement of NRAMP1 from Arabidopsis thaliana in iron transport, Biochem. J., 2000, 347, 749–755 CrossRef CAS PubMed.
- R. F. Paulson, L. Shi and D.-C. Wu, Stress erythropoiesis: new signals and new stress progenitor cells, Curr. Opin. Hematol., 2011, 18, 139–145 CrossRef PubMed.
- R. M. Pellegrino, F. Riondato, L. Ferbo, M. Boero, A. Palmieri, L. Osella, P. Pollicino, B. Miniscalco, G. Saglio and A. Roetto, Altered erythropoiesis in mouse models of Type 3 hemochromatosis, BioMed Res. Int., 2017, 2017, 2408941 CAS.
- M. Radu, M. C. Munteaunu and S. Petrache, Depletion of intracellular glutathione and increased lipid peroxidation mediate cytotoxicity of hematite nanoparticles in MRC-5 cells, Acta Biochim. Pol., 2010, 57, 355–360 CAS.
- M. C. Hohnholt and R. Dringen, Iron-dependent formation of reactive oxygen species and glutathione depletion after accumulation of magnetic iron oxide nanoparticles by oligodendroglial cells, J. Nanopart. Res., 2011, 13, 2305–2314 CrossRef.
- R. L. Bertrand, Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events, Med. Hypotheses, 2017, 101, 69–74 CrossRef CAS PubMed.
- T. Kurz, A. Terman, B. Gustafsson and U. T. Brunk, Lysosomes in iron metabolism, ageing and apoptosis, Histochem. Cell Biol., 2008, 129, 389–406 CrossRef CAS PubMed.
- S. K. Baird, T. Kurz and U. T. Brunk, Metallothionein protects against oxidative stress-induced lysosomal destabilization, Biochem. J., 2006, 394, 275–283 CrossRef CAS PubMed.
- R. L. Pisoni, T. L. Acker, K. M. Lisowski, R. M. Lemons and J. G. Thoene, A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: possible role in supporting lysosomal proteolysis, J. Cell Biol., 1990, 110, 327–335 CrossRef CAS PubMed.
- T. Kurz, B. Gustafsson and U. T. Brunk, Cell sensitivity to oxidative stress is influenced by ferritin autophagy, Free Radical Biol. Med., 2011, 50, 1647–1658 CrossRef CAS PubMed.
- Y.-M. Go and D. P. Jones, Redox compartmentalization in eukaryotic cells, Biochim. Biophys. Acta, 2008, 1780, 1273–1290 CrossRef CAS PubMed.
- M. Cao, H. Chen, D. Chen, Z. Xu, S. H. Liu, X. Chen and J. Yin, Naphthalimide-based fluorescent probe for selectively and specifically detecting glutathione in the lysosomes of living cells, Chem. Commun., 2016, 52, 721–724 RSC.
- A. Shawki, P. B. Knight, B. D. Maliken, E. J. Niespodzany and B. Mackenzie, H(+)-coupled divalent metal-ion transporter-1; functional properties, physiological roles and therapeutics, Curr. Top. Membr., 2012, 70, 169–214 CAS.
- M. D. Knutson, M. R. Vafa, D. J. Haile and M. Wessling-Resnick, Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages, Blood, 2003, 102, 4191–4197 CrossRef CAS PubMed.
- S. Gruenheid, E. Pinner, M. Desjardins and P. Gros, Natural resistance to infection with intracellular pathogens: The Nramp1 protein is recruited to the membrane of the phagosome, J. Exp. Med., 1997, 185, 717–730 CrossRef CAS PubMed.
- J. R. Forbes and P. Gros, Iron, manganese, and cobalt transport by Nramp1 (Slc11a1) and Nramp2 (Slc11a2) expressed at the plasma membrane, Blood, 2003, 102, 1884–1892 CrossRef CAS PubMed.
- S. Soe-Lin, S. S. Apte, B. Andriopoulos Jr, M. C. Andrews, M. Schranzhofer, T. Kahawita, D. Garcia-Santos and P. Ponka, Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5960–5965 CrossRef CAS PubMed.
- S. Soe-Lin, S. S. Apte, M. R. Mikhael, L. K. Kayembe, G. Nie and P. Ponka, both Nramp1 and DMT1 are necessary for efficient macrophage iron recycling, Exp. Hematol., 2010, 38, 609–617 CrossRef PubMed.
- K. R. Bridges and K. E. Hoffman, The effects of ascorbic acid on the intracellular metabolism of iron and ferritin, J. Biol. Chem., 1986, 261, 14273–14277 CAS.
- K. R. Bridges, Ascorbic acid inhibits lysosomal autophagy of ferritin, J. Biol. Chem., 1987, 262, 14773–14998 CAS.
- K. Eshelman, H. Yao, A. N. D. Punchi Hewage, J. J. Deay, J. R. Chandler and M. Rivera, Inhibiting the BfrB:Bfd interaction in Pseudomonas aeruginosa causes irreversible iron accumulation in bacterioferritin and iron deficiency in the bacterial cytosol, Metallomics, 2017, 9, 646–659 RSC.
| This journal is © The Royal Society of Chemistry 2018 |