
General oriented assembly of uniform carbon-confined metal oxide nanodots on graphene for stable and ultrafast lithium storage†
Jiashen
Meng
a,
Ziang
Liu
a,
Chaojiang
Niu
*a,
Linhan
Xu
b,
Xuanpeng
Wang
a,
Qi
Li
a,
Xiujuan
Wei
a,
Wei
Yang
a,
Lei
Huang
a and
Liqiang
Mai
 *ac
*ac
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China. E-mail: niuchaojiang11@whut.edu.cn; mlq518@whut.edu.cn
bDepartment of Physics and Collaborative Innovation Center for Optoelectronic Semiconductors and Efficient Devices, Xiamen University, Xiamen 361005, China
cDepartment of Chemistry, University of California, Berkeley, California 94720, USA
First published on 8th November 2017
Abstract
A facile and general method for the oriented assembly of uniform carbon-confined metal oxide nanodots on graphene was developed via a well-designed process including surfactant-induced assembly, mismatched coordination reaction and subsequent in situ carbonization. On the basis of experimental analyses and density functional theory calculations, the key mismatched coordination reaction mechanism is clearly revealed, resulting in the formation of small amorphous metal–ligand complexes. This versatile oriented assembly strategy is then generally applied to obtain various carbon-confined metal oxide (SnO2, Cr2O3, Fe3O4 and Al2O3) nanodots on graphene. Notably, the as-prepared C@SnO2@Gr electrode as an LIB anode material possesses a high reversible discharge capacity of 702 mA h g−1 and an excellent capacity retention of over 100% tested at 2 A g−1 after 1200 cycles.
Conceptual insightsRationally designed nanostructured metal oxides have been intensively studied as anode materials for new-generation lithium-ion batteries (LIBs) due to their abundant sources and high theoretical capacities. However, the controlled synthesis of uniform nanostructured metal oxides with fast ion and electron transport still remains a great challenge. Herein, this work firstly demonstrated a facile, efficient and general strategy for the oriented assembly of a series of uniform carbon-confined metal oxide nanodots on graphene via a rationally-designed process including surfactant-induced assembly, mismatched coordination reaction and subsequent in situ carbonization. Moreover, detailed experimental analyses and density functional theory calculations clearly revealed the formation mechanism. Importantly, this versatile oriented assembly strategy was then generally applied to obtain various carbon-confined metal oxide (SnO2, Cr2O3, Fe3O4 and Al2O3) nanodots on graphene. These as-prepared architectures possess high activity, high conductivity, short diffusion length and good strain accommodation. As a proof-of-concept application, the as-prepared C@SnO2@Gr electrode as an anode exhibits remarkable rate capability and cycle stability for LIBs. Therefore, our general synthetic strategy and the proposed mechanism open new avenues for the development of a wide range of functional nanostructured metal oxides for further potential applications which are not limited to energy storage. |
With the rapid rise of portable electronics, electric vehicles and hybrid electric vehicles, the demand for high-performance energy storage devices has become more and more urgent.1–3 Lithium-ion batteries (LIBs) are undoubtedly one of the best candidates.3 To meet the growing energy demands in a sustainable low-carbon economy, intensive efforts have been focused on developing safe LIBs with low cost, high energy/power density, and long cycling lifespan.4–8 LIBs are chemical devices that store energy, which involves reversible shuttling processes of lithium ions between host anode and cathode materials with concomitant redox reactions during the charge/discharge process.9 However, these redox reactions still suffer from sluggish kinetics because of the limited electron transfer and ion diffusion in both host materials.10 Compared to commercial supercapacitors, the unsatisfactory rate performances of current LIBs seriously limit their applications, especially in electric vehicles.11 Therefore, the development of advanced LIBs with supercapacitor-like rate performance remains a formidable challenge.
Metal oxides are remarkably attractive candidates as LIB anodes owing to their abundant sources and high theoretical capacities (>700 mA h g−1) compared to commercial graphite.12–14 However, the large volume variation and low electronic conductivity are two major and disastrous problems for their further application in LIBs.15 For instance, tin dioxide (SnO2), an n-type semiconductor (a bandgap of 3.6 eV), is regarded as a promising anode for LIBs, because a high theoretical capacity of 1494 mA h g−1 can be achieved by both conversion reaction and alloying reaction.16 However, the large volume change (up to about 300%) of SnO2 leads to serious pulverization and aggregation, resulting in poor cycling stability and rate performance. To address these scientific problems, intensive efforts have been dedicated to developing various methods to endow metal oxides with enhanced rate performance and excellent structural stability for advanced LIBs.17,18 An effective strategy is the combination of carbon-based materials and nanostructured metal oxides to construct well-defined composites, which can efficiently improve the electron conductivity and enable structural integrity.19–23 In particular, graphene, a two-dimensional atomic carbon layer has recently become one promising substrate for supporting nanostructured metal oxides.24 Zhao et al. reported SnO2 nanoparticles on graphene with enhanced lithium storage by in situ reduction of graphene oxide and oxidation of Sn2+.25 However, due to the nature of “bare” SnO2 nanoparticles on graphene, the electron transfer to active SnO2 and the structural stability in the whole electrodes are still limited. In this regard, the development of uniform carbon-confined, nanoscaled metal oxides on graphene would be a highly promising strategy to achieve high energy density and power density in LIBs. Intensive efforts have been dedicated to the synthesis of carbon-confined metal oxide nanoparticles on graphene in energy storage.26–28 Zhang et al. synthesized carbon-coated SnS/SnO2 nanoparticles on graphene with enhanced sodium storage via a rational multi-step design.28 However, developing a facile, efficient and general synthesis method for these metal oxide architectures is quite challenging.
Recently, metal–organic frameworks (MOFs) have been used as promising candidates for the synthesis of carbon-based materials in energy storage, because of their high surface area, tunable porosity and controllable structures by modulating well-defined coordination reactions between metal ions/clusters and organic ligands.29–31 However, due to their fast knetics and well-defined coordination reactions in the solution system, the resulting MOF crystals exhibit delicate shapes and relatively large sizes (>100 nm). It was obvious that after the confinement space pyrolysis, the nanoparticles always aggregated to form similar structures with MOFs, which may limit the ion diffusion and restrict the ability to accommodate large volume variation.32 Therefore, the key point of our idea is the formation of small amorphous metal–ligand complexes (<20 nm) by modulating coordination reactions between selected metal ions and organic ligands.
Herein, we develop a facile and general approach towards the oriented assembly of uniform carbon-confined metal oxide nanodots on graphene through a surfactant-induced assembly, mismatched coordination reaction and subsequent in situ carbonization process. The detailed experimental analyses and density functional theory (DFT) calculations revealed the mismatched coordination reaction mechanism. To realize the homogeneous growth, the surface of graphene oxide (GO) was modified by the amide carbonyl groups of poly(vinylpyrrolidone) (PVP) to enhance the Coulomb force with metal ions, thereby facilitating the formation of a uniform precursor nanodot layer on GO. This versatile strategy can be generally applied to obtain various carbon-confined metal oxides on graphene, including SnO2, Cr2O3, Fe3O4 and Al2O3. These as-prepared architectures possess high activity, high conductivity, short diffusion length and good strain accommodation. As a proof-of-concept application, the carbon-confined SnO2 nanodots on graphene exhibit outstanding rate capability and long-term cycle life.
The overall synthetic strategy for the oriented assembly of uniform carbon-confined metal oxide nanodots on graphene is schematically presented in Fig. 1. First, the surface of GO was modified by PVP to obtain sufficient and evenly distributed functional groups on the GO surface for further coordination reactions (step I). The rich amide carbonyl groups of PVP on GO can coordinate with metal ions driven by Coulomb force, resulting in the homogeneous oriented assembly of metal ions on GO.33 Second, during the solvothermal process (step II), the coordination reaction occurs between selected metal ions and organic ligands. However, due to the formation of mismatched bonding angles and/or distorted polyhedra, these selected metal ions and organic ligands cannot form a long-range order structure, resulting in the formation of amorphous metal–ligand complexes, which belong to coordination polymers. The resulting metal–ligand complexes are mainly attributed to the formation of coordination bonds and intermolecular forces (including van der Waals forces, π–π interactions, hydrogen bonding, and stabilization of π bonds by polarized bonds).34,35 These intermolecular forces tend to be weak, with a long equilibrium distance (bond length) compared to covalent bonds, which are benefical for the stability of metal–ligand complexes. To distinguish the difference from crystalline MOFs (another subclass of coordination polymers), these reaction processes are called mismatched coordination reactions. Meanwhile, the introduction of PVP-modified graphene oxides as soft templates can further realize the oriented assembly of dispersed small amorphous metal–ligand complexes. As a result, uniform amorphous metal–ligand complex nanodots on graphene oxide are formed as the target precursor by mismatched coordination reactions. Finally, after in situ carbonization in argon, the morphology-preserved carbon-confined metal oxide nanodots on graphene are easily obtained (step III). In brief, the overall oriented assembly process experiences the surfactant-induced assembly, mismatched coordination reaction and the in situ carbonization, which can be extended to obtain various carbon-confined metal oxide nanodots on graphene.
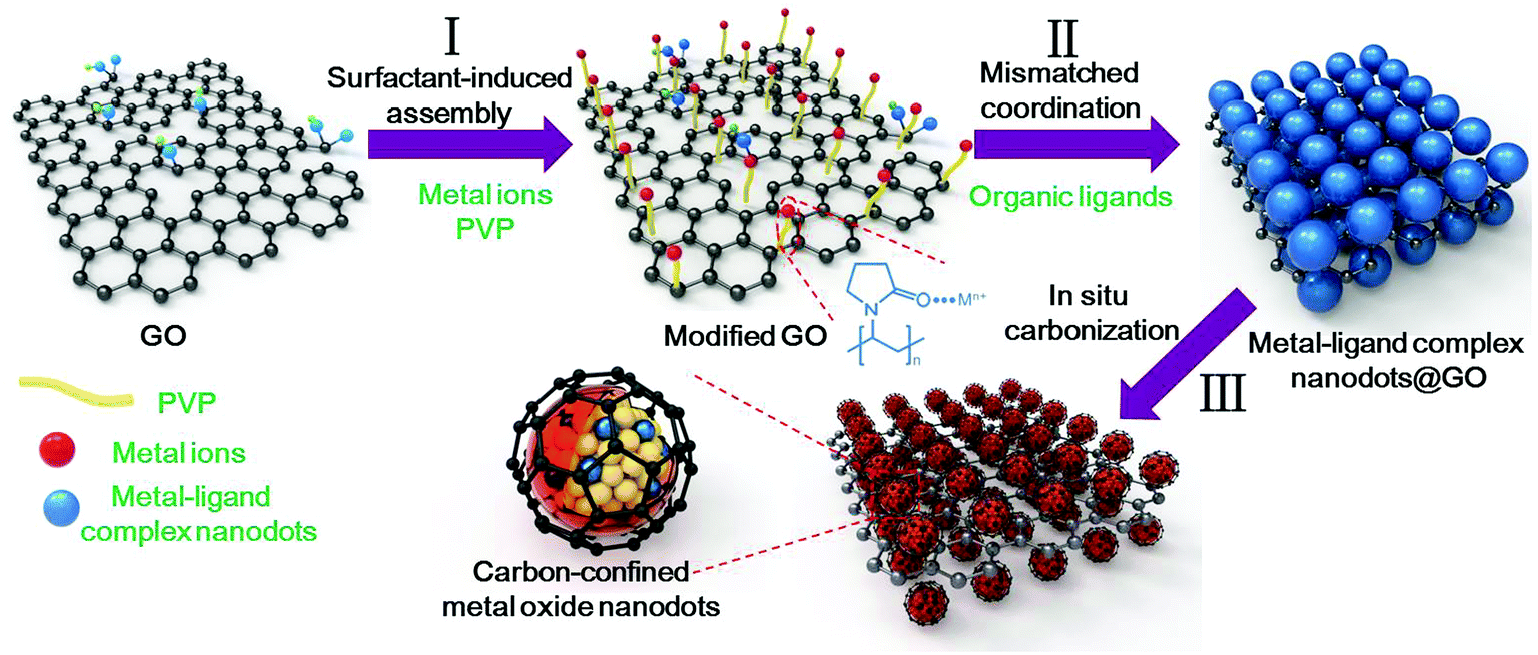 | ||
| Fig. 1 Schematics of the formation process of uniform carbon-confined metal oxide nanodots on graphene, including a surfactant-induced assembly, mismatched coordination reaction and in situ carbonization. | ||
To further reveal this method, the synthesis process of uniform carbon-confined SnO2 nanodots on graphene (denoted as C@SnO2@Gr) as an example was characterized and discussed in detail (Fig. 2). First, PVP-modified GO sheets were obtained by the self-assembly of PVP in the precursor solution. On the basis of Fourier-Transform Infrared (FTIR) spectra, the typical vibrations of PVP also appear in those of PVP-modified GO, indicating the existence of PVP on GO (Fig. S1, ESI†). Then, uniform Sn-based precursor nanodots on graphene oxide (denoted as Sn-precursor@GO) are prepared via the mismatched coordination reaction between Sn2+ ions and 1,4-dicarboxybenzene during the solvothermal process. Field-emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) images indicate that Sn-precursor nanodots (<10 nm) are uniformly dispersed on a graphene substrate without obvious agglomeration (Fig. 2a and b and Fig. S2, ESI†). The corresponding selected area electron diffraction (SAED) pattern and X-ray diffraction (XRD) pattern show no obvious diffraction rings and peaks, both confirming the amorphous nature and the loss of long-range order in the whole precursor (inset of Fig. 2b and d). The energy-dispersive X-ray (EDX) elemental mappings indicate that the Sn, O and C elements were distributed homogeneously over all of the selected area (Fig. 2c). On the basis of Fourier-Transform Infrared (FTIR) spectra, the typical vibrations of the 1,4-dicarboxybenzene also appear in those of the Sn-precursor@GO sample, further demonstrating the occurrence of a mismatched coordination reaction (Fig. S3a, ESI†). By contrast, without the introduction of organic ligands, large bare crystalline SnO nanoparticles were formed in the reaction system due to the hydrolysis of Sn2+ ions (Fig. S4, ESI†). After direct pyrolysis in argon, the Sn-precursor@GO sample was converted into the C@SnO2@Gr product via in situ carbonation. Due to the monodispersion of the precursor nanodots, the further aggregation of the product can be efficiently avoided during annealing. The broad diffraction peaks are well indexed to the pure SnO2 phase (JCPDS card no: 01-077-0447) (Fig. 2d). On the basis of Rietveld refinement analysis from the XRD result, the average crystalline size of the C@SnO2@Gr sample is ∼5.1 nm (Fig. S5, ESI†). FESEM and TEM images clearly show uniform and monodisperse nanodots on graphene (Fig. 2e, f and Fig. S6, ESI†). High-resolution TEM (HRTEM) images show that SnO2 nanodots (∼4 nm in diameter) are confined by thin carbon shells (Fig. 2g and Fig. S7, ESI†), which is in accordance with the XRD Rietveld refinement result. The thickness of the carbon coating in the C@SnO2@Gr sample is ∼1 nm. According to the statistical analysis, the diameter distribution of SnO2 nanodots is in a regular manner (inset of Fig. 2f). Raman spectra were carried out to characterize the nature of carbon in the C@SnO2@Gr sample (Fig. S3b, ESI†). A high IG/ID band intensity ratio (1.02) verifies the increased graphitization compared to the Sn-precursor@GO sample (0.95) and GO sample (0.81). The carbon content in the C@SnO2@Gr sample is 11.39 wt% by thermogravimetrical-differential scanning calorimeter (TG-DSC) analysis (Fig. S3d, ESI†). The nitrogen adsorption–desorption isotherm of C@SnO2@Gr shows a high Brunauer–Emmett–Teller (BET) specific surface area of 148 m2 g−1 (Fig. S3e and f, ESI†). The pore size distribution is mainly in the range between 2 nm and 7 nm, belonging to mesopores, which can be attributed to the random and loose stacking of nanodots.
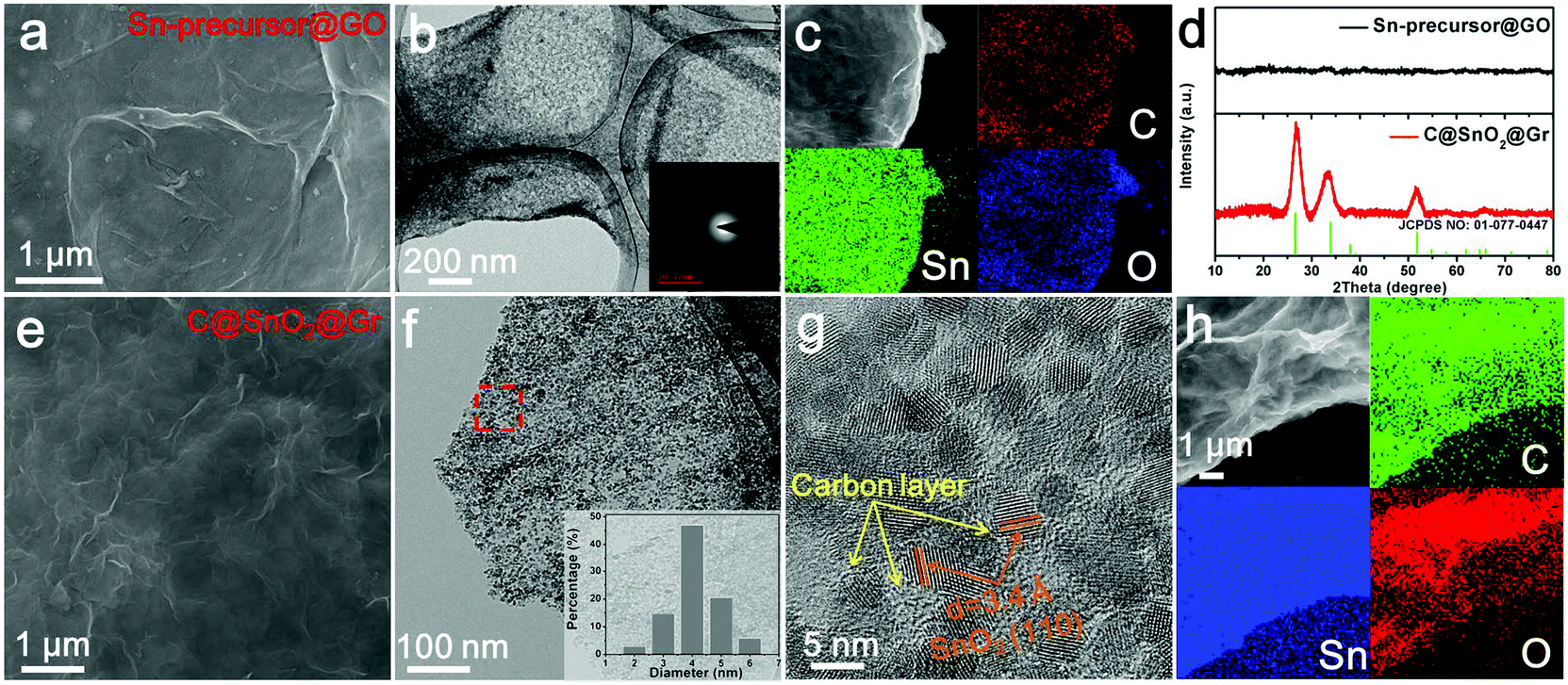 | ||
| Fig. 2 Characterization of uniform Sn-precusor@GO and C@SnO2@Gr. SEM image (a), TEM image (b) and EDX mapping image (c) of Sn-precusor@GO. Inset of (b) is the SAED pattern. (d) XRD patterns of Sn-precusor@GO and C@SnO2@Gr. SEM image (e), TEM image (f), HRTEM image (g), and EDX mapping image (h) of C@SnO2@Gr. Inset of (f) is the size distribution of the nanodots. | ||
In this synthesis strategy, the key point to avoid the growth of the precursor nanodots is the mismatched coordination reactions between metal ions and organic ligands. To confirm the occurrence of this reaction, the Sn-based precursor (Sn-precursor) was synthesized and characterized without the addition of graphene oxides (Fig. 3). Small and amorphous Sn-precursor nanodots obviously aggregate together (Fig. 3a). The pyrolysis process of the Sn-precursor was investigated by TG-DSC analysis (Fig. 3b). Three stages of mass loss can be attributed to the solvent volatilization (<250 °C), the carbonization of organic ligands (400–500 °C), and the reduction of SnO2 (>750 °C), respectively. Compared with 1,4-dicarboxybenzene, the Raman spectrum of the Sn-precursor shows the split D band originating from disoriented carbon at 1420 and 1442 cm−1 (Fig. 3c). Meanwhile, the FTIR spectrum of the Sn-precursor shows the obvious intensity decrease and position shift of characteristic bond vibrations (Fig. 3d). These phenomena indicate the occurrence of a coordination reaction in the Sn-precursor. Furthermore, the chemical composition and formed bonds are investigated by X-ray photoelectron spectroscopy (XPS). The typical peaks of C, O and Sn elements exist in the Sn-precursor (Fig. S8c, ESI†). In high-resolution C 1s and O 1s XPS spectra, the existence of new C–O and Sn–O peaks directly confirms the formation of coordination bonds in the Sn-precursor (Fig. 3e and f). The high-resolution Sn 3d spectrum exhibits two obvious bands located at ∼486.8 and 495.3 eV, corresponding to the Sn 3d5/2 and Sn 3d3/2 (Fig. S8d, ESI†). From XPS analysis, the atom ratio of Sn is about 9.02%. After in situ carbonization, the obtained carbon-confined SnO2 nanoparticles (SnO2@C) obviously agglomerate together (Fig. S9, ESI†). The HRTEM image confirms the core-shelled structure of SnO2@C nanoparticles. The carbon content in SnO2@C is about 7.95 wt% from the TG-DSC analysis (Fig. S9f, ESI†). Because the total carbon content in C@SnO2@Gr is 11.39 wt%, the graphene content in C@SnO2@Gr is calculated to be about 3.34 wt%. Furthermore, solid-state nuclear magnetic resonance (NMR) spectroscopy was carried out to reveal the structrue of the amorphous Sn-precursor (Fig. S10, ESI†). In contrast, all individual resonances in the 13C cross-polarization (CP) magic angle spinning (MAS) NMR spectrum of the amorphous Sn-precursor are much broader than those for the crystalline Zn-MOF-5, which is in accordance with the existance of amorphous phases in previous studies.36 Compared with the 1,4-dicarboxybenzene, the NMR results of the Sn-precursor and Zn-MOF-5 confirm the existance of the intact organic ligands.
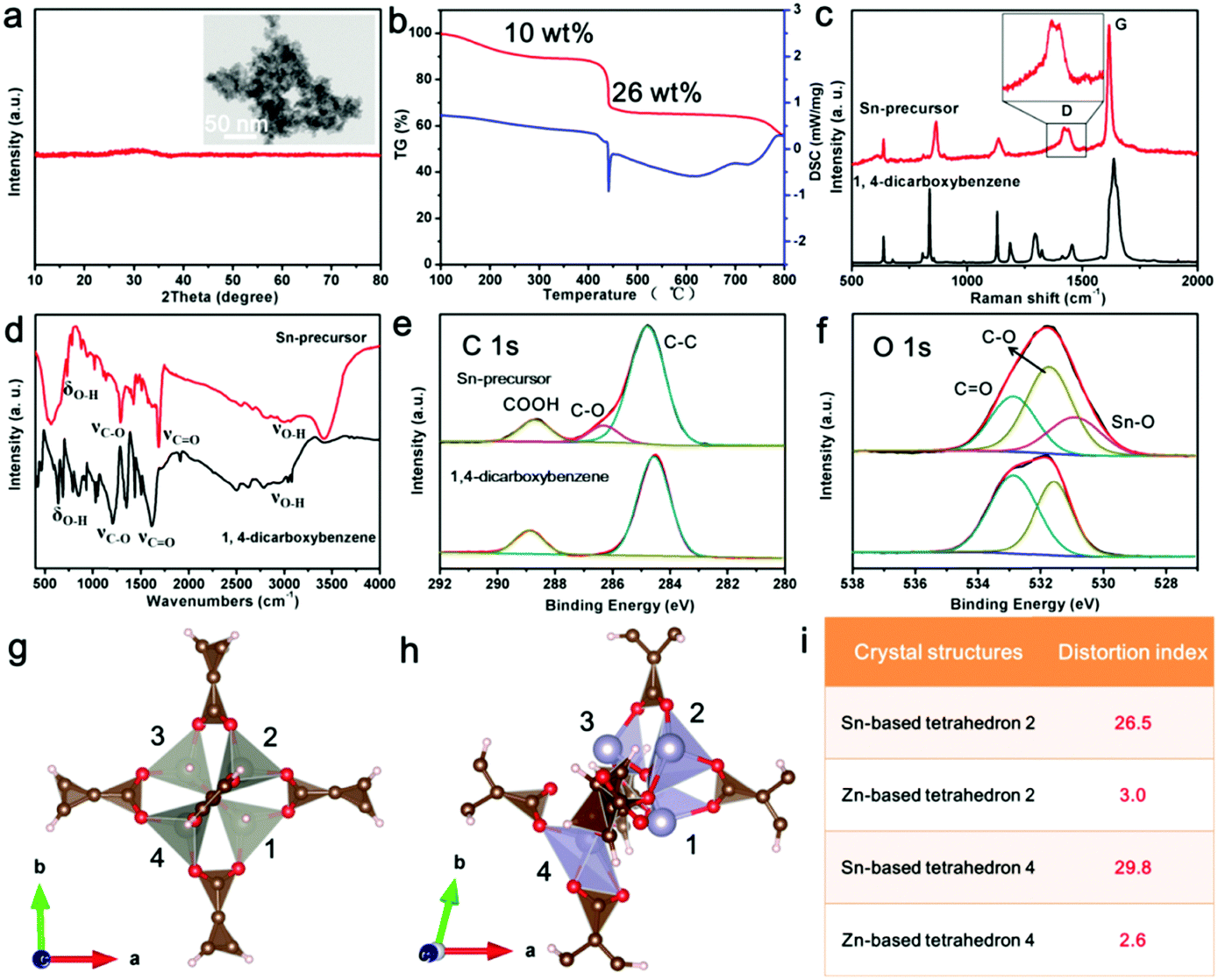 | ||
| Fig. 3 (a) XRD pattern of the Sn-precursor. Inset of (a) is the TEM image. (b) TG and DSC curves of the Sn-precursor. (c and d) Raman and FTIR spectra of the Sn-precursor and 1,4-dicarboxybenzene, respectively. (e and f) High-resolution C 1s and O 1s XPS spectra of the Sn-precursor and 1,4-dicarboxybenzene, respectively. (g) Crystal structure of Zn-MOF-5. (h) DFT calculations on the crystal structure of the Sn-precursor by using Sn2+ to replace Zn2+ of Zn-MOF-5. (i) Comparisons of crystal structures between Zn-MOF-5 and the Sn-precursor. | ||
To get further insights into the coordination reaction, we performed density functional theory (DFT) simulations to investigate the local structure of the Sn-precursor. By using Sn2+ to replace Zn2+ of Zn-MOF-5, the crystal structure of the Sn-precursor was illustrated after DFT calculations (Fig. 3g and h). The distortion indexes of the Sn-based tetrahedra (26.5 and 29.8) are much higher than those in Zn-MOF-5 (3.0 and 2.6) (Fig. 3i and Table S1, ESI†). These severe structural distortions cause the mismatched coordination between Sn2+ ions and the organic ligands, and hinder the further growth of the Sn-precursor, leading to the formation of an amorphous and small-sized product. In constrast, the as-prepared Zn-MOF-5 crystals showed a large size and good crystallinity (Fig. S11, ESI†). Therefore, the introduction of the mismatched coordination reaction plays a significant role in the size control and the narrow size distribution of carbon-confined metal oxide nanodots.
To confirm the versatility of our strategy, various uniform carbon-confined metal oxide nanodots on graphene with controlled dopants and desirable compositions are efficiently fabricated by simply modulating the organic ligands and metal ions, including nitrogen-doped C@SnO2@Gr (NC@SnO2@Gr), C@Cr2O3@Gr, NC@Fe3O4@Gr and NC@Al2O3@Gr (see the ESI† for the detailed synthesis processes). According to the aforementioned mechanism, amorphous metal–ligand complex nanodots on graphene oxide are first synthesized by the mismatched coordination reactions (Fig. S12–S15, ESI†). SEM images and EDX mappings show a homogeneous distribution of the precursor nanodots on graphene oxide. The corresponding XRD patterns show no diffraction peaks, further confirming the amorphous nature. Subsequently, when treated at high temperature in argon, the morphology-preserved architectures are obtained after in situ carbonation. Fig. 4 shows the typical FESEM images, EDX mappings and TEM images of NC@SnO2@Gr, C@Cr2O3@Gr, NC@Fe3O4@Gr and NC@Al2O3@Gr. The corresponding XRD patterns are well indexed to pure metal oxide phases. Further, the broad diffraction peaks also reveal that the obtained metal oxide nanodots have a small size according to the Scherrer equation. Again, TEM images clearly display that the carbon-confined metal oxide nanodots with a narrow size distribution are homogeneously flatted on graphene (Fig. S12–S15, ESI†). The corresponding HRTEM images further reveal the core-shelled structures of the obtained metal oxides@C products. In contrast, without the introduction of graphene oxides, carbon-confined metal oxide nanoparticles as control samples can also be obtained. For N-doped carbon-confined samples, the corresponding EDX mapping images show the homogeneous distribution of nitrogen (Fig. S16–S18, ESI†).
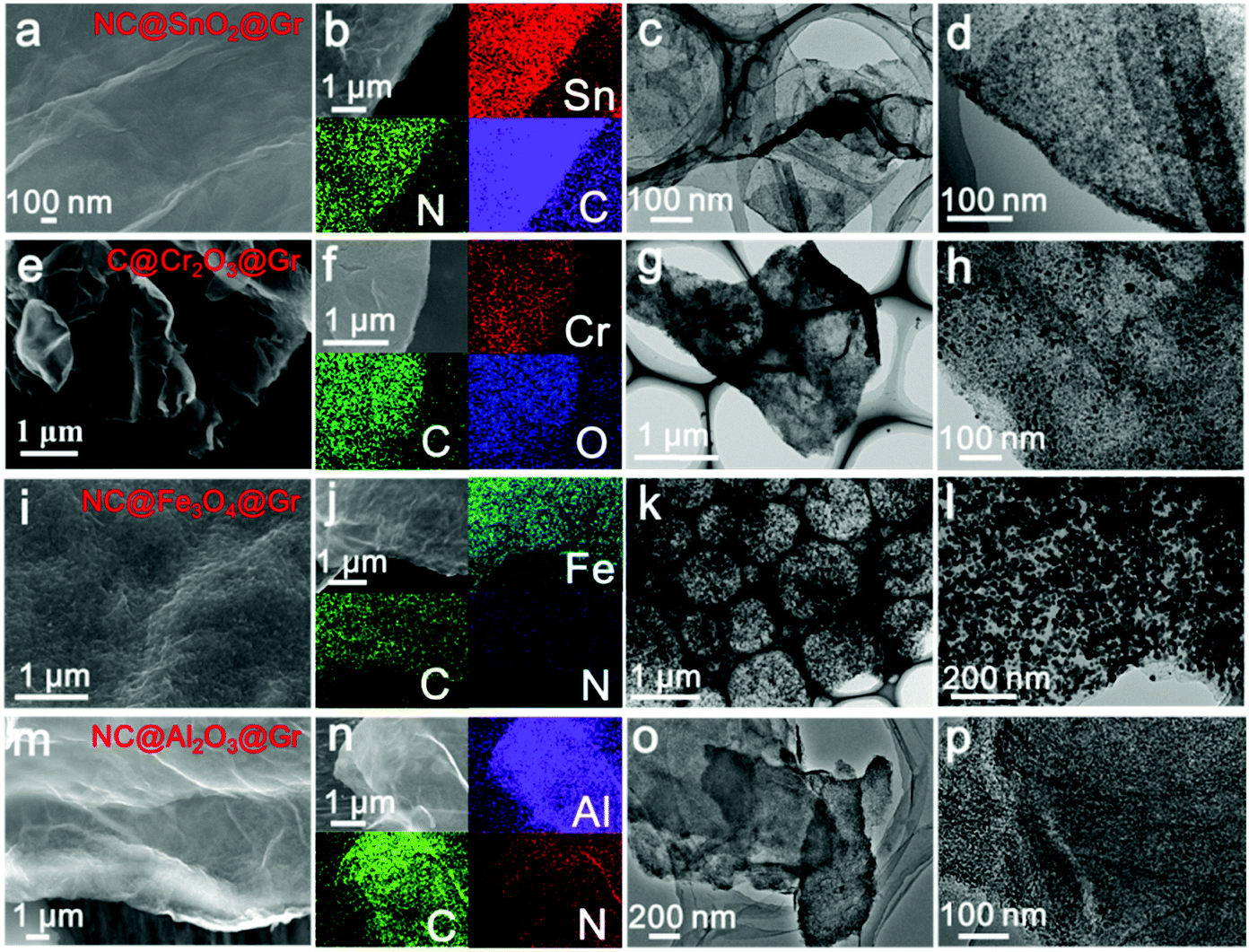 | ||
| Fig. 4 SEM images, EDX mapping images, and TEM images of NC@SnO2@Gr (a–d), C@Cr2O3@Gr (e–h), NC@Fe3O4@Gr (i–l) and NC@Al2O3@Gr (m–p), respectively. | ||
In our strategy, precise control over these synthetic conditions plays a significant role in the successful preparation of delicate architectures. Initially, the control of mismatched coordination reactions between selected organic ligands and metal ions is crucial in the formation of small and complex nanodots (<20 nm) which can be obtained by mismatched coordination reactions. By contrast, large ZIF crystals (ZIF-67) (>300 nm) are formed via a well-defined coordination reaction between 2-methylimidazole and Co2+ in the solution (Fig. S19, ESI†). Furthermore, the carbon layers generated from the in situ carbonization can efficiently restrain the agglomeration of crystallized metal oxides during the high-temperature treatment. In addition, the introduction of PVP and the appropriate content of GO have a great influence on the formation of uniform and monodisperse metal oxide nanodots on graphene. PVP as a bridging agent ensures the homogeneous oriented assembly of selected metal ions on graphene oxides by Coulomb force. While graphene oxide sheets are used as soft substrates for loading the precursor nanodots. Without the addition of PVP and a sufficient content of GO, serious agglomeration and chaotic stacking of the formed metal–ligand complex nanodots on graphene oxides were clearly observed (Fig. S20, ESI†). After in situ carbonization, partial carbon-confined metal oxide nanodots obviously aggregated, resulting in nonuniform carbon-confined metal oxide nanoparticles on graphene (denoted as C@SnO2/Gr). Therefore, compared with previous reports for graphene-metal/metal oxide hybrid materials, this method is a facile, efficient and general strategy for controlling the oriented formation of uniform carbon-confined metal oxides on soft graphene substrates (Table S2, ESI†).
Furthermore, the as-prepared C@SnO2@Gr sample was employed as an anode material for LIBs. First, the cyclic voltammetry (CV) measurements were carried out in a potential range from 0.01 to 3.0 V vs. Li+/Li at a scan rate of 0.2 mV s−1 (Fig. 5a). In the first cycle, there is an unobvious and smooth peak at ∼0.7 V, which is attributed to the formation of a solid electrolyte interface (SEI).37 And the subsequent two CV curves mostly overlap, indicating highly reversible lithium storage. To demonstrate the structural superiority of C@SnO2@Gr, the SnO2@C and C@SnO2/Gr electrodes were also tested as anode materials for LIBs. When tested at a current density of 0.2 A g−1 for 120 cycles, the C@SnO2@Gr electrode exhibits higher specific discharge capacity and higher capacity retention (1090 mA h g−1, 104.5%), than those of the SnO2@C (325 mA h g−1, 37.9%) and C@SnO2/Gr (660 mA h g−1, 69.3%) electrodes, indicating the excellent stability (Fig. 5b). The rate capability of the electrode was tested at various current densities. The C@SnO2@Gr electrode can deliver highly reversible average specific capacities of 905, 782, 737, and 552 mA h g−1 at the current densities of 0.5, 1, 2, and 5 A g−1, respectively (Fig. 5c). When returned to 0.5 A g−1, the specific capacity of C@SnO2@Gr can recover to 910 mA h g−1 with an approximately 100% capacity retention. The corresponding discharge–charge voltage curves of rate performance display a low polarization and high coulombic efficiency (Fig. 5d). Furthermore, when tested at a high current density of 2 A g−1 after 1200 cycles, the C@SnO2@Gr possesses a high specific capacity of 702 mA h g−1 and an excellent capacity retention of over 100%, indicating ultrafast lithium storage and outstanding long-term cycling stability (Fig. 5e). Compared with other nanostructured SnO2 materials in previous reports, our as-prepared C@SnO2@Gr shows outstanding rate capability and excellent structural stability (Table S3, ESI†).22,25,37–44 Notably, the increased capacity of the C@SnO2@Gr is also widely observed for many transition metal oxides, which may be attributed to the formation of an electrochemically gel-like polymer layer.45 For another reason, during the cycling process especially at high rates, the proportion of the total active materials increases and the sizes of the tin oxide nanodots further reduce. This phenomenon can stimulate more electrochemical activity and improve the capacity.
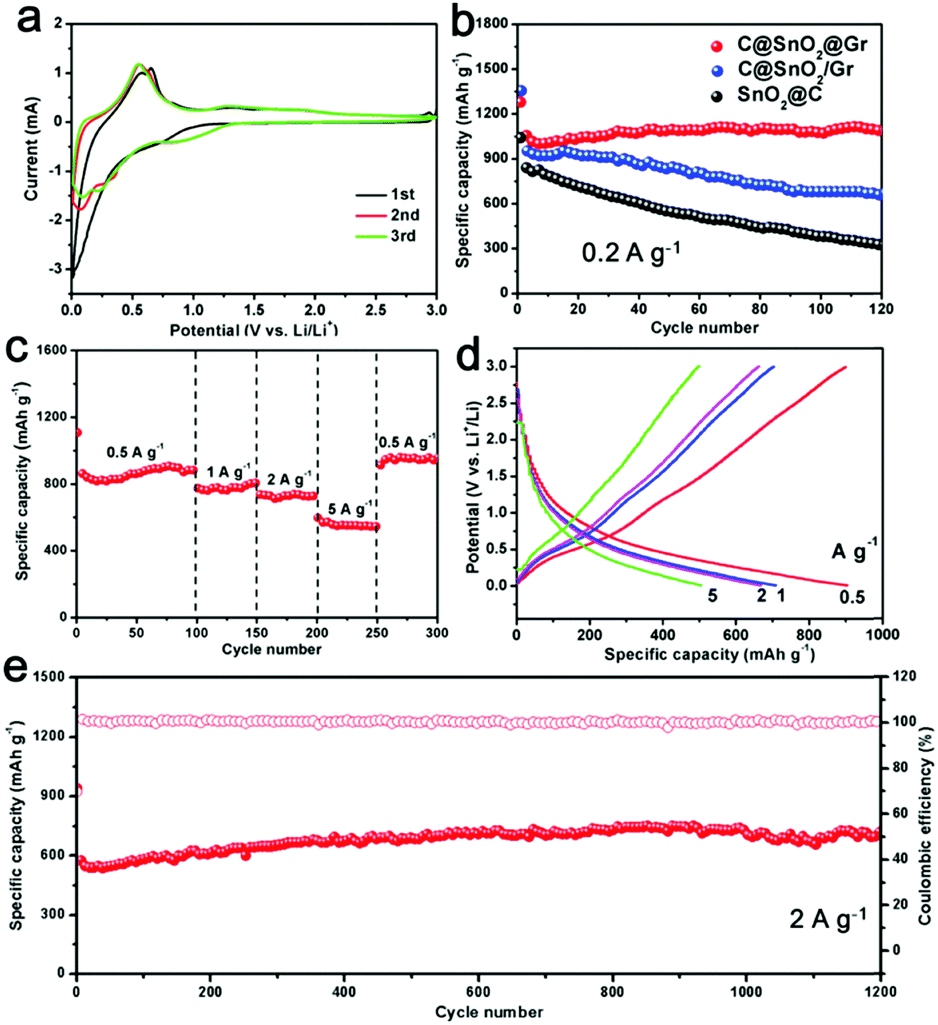 | ||
| Fig. 5 Lithium storage performances of C@SnO2@Gr, C@SnO2/Gr and SnO2@C. (a) The first three CV curves tested at a scan rate of 0.2 mV s−1. (b) Cycling performance of C@SnO2@Gr, C@SnO2/Gr and SnO2@C tested at the current density of 0.2 A g−1. (c) Rate performance tested at current densities varying from 0.5, 1, 2, and 5, back to 0.5 A g−1. (d) The corresponding charge–discharge curves at different current densities. (e) Cycling performance and coulombic efficiency tested at a high current density of 2 A g−1. | ||
To give further insights, the TEM images of C@SnO2@Gr after 120 cycles were obtained (Fig. S21, ESI†). The morphology of C@SnO2@Gr can be maintained after cycling, confirming the structural stability. The carbon shells and graphene substrates not only enhanced the conductivity but also enabled the structural integrity, which are beneficial for stable and ultrafast lithium storage. In addition, the integration of carbon confined SnO2 nanodots and graphene guarantee the formation of a stable SEI layer over the electrode material. The ex situ XRD patterns were carried out to detect the structural evolution of the C@SnO2@Gr electrode during the initial discharge/charge process (Fig. S22, ESI†). The electrode material evolved from the crystalline phase to the amorphous phase, which is in accordance with the results from previously reported studies.46 After discharging to 0.01 V, the formed LixSn is not observed from ex situ XRD due to the poor crystallinity. The kinetic analysis shows obvious lithiation pseudo-capacitive behavior, which is beneficial for ultrafast lithium storage (Fig. S23a–e, ESI†). Moreover, electrochemical impedance spectroscopy (EIS) was measured to evaluate the charge-transfer resistance (Rct) of these samples (Fig. S23f, ESI†). The Rct of the C@SnO2@Gr is 70 Ω, which is smaller than those of C@SnO2/Gr and SnO2@C (105 Ω and 162 Ω), indicating fast electronic mobility. To further demonstrate the structural superiority, the as-prepared NC@Fe3O4@Gr sample as an anode material also exhibits stable and ultrafast lithium storage (Fig. S24, ESI†). Therefore, the remarkable lithium storage can be mainly attributed to the co-contribution between the chemical compositions and delicate architecture.
Conclusions
In summary, we have successfully developed a facile and general strategy for the oriented assembly of uniform carbon-confined metal oxide nanodots on graphene via a nicely-designed process of surfactant-induced assembly, mismatched coordination reaction and subsequent in situ carbonization. The key point of this method is the mismatched coordination reaction between selected metal ions and organic ligands during the solvothermal process, which was clearly verified by experimental analyses and DFT calculations. This simple strategy has been applied to fabricating various carbon-confined metal oxide nanodots on graphene, including C@SnO2@Gr, NC@SnO2@Gr, C@Cr2O3@Gr, NC@Fe3O4@Gr and NC@Al2O3@Gr. These architectures can provide high surface area, short diffusion length, robust structures and high conductivity. Notably, the as-prepared C@SnO2@Gr electrode exhibits remarkable rate capability and cycle stability. Therefore, our work represents a facile and general approach towards the controlled synthsis of functional metal oxide architectures, which will have great potential in many frontier fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFA0202603), the National Basic Research Program of China (2013CB934103), the Programme of Introducing Talents of Discipline to Universities (B17034), the National Natural Science Foundation of China (51521001), the National Natural Science Fund for Distinguished Young Scholars (51425204), and the Fundamental Research Funds for the Central Universities (WUT: 2016III001 and 2016-YB-004). We are grateful to Bichao Xu and Pei Zhang of the Core Facility and Technical Support, Wuhan Institute of Virology for her technical support in transmission electron microscopy. Prof. Liqiang Mai gratefully acknowledges financial support from China Scholarship Council (No. 201606955096).Notes and references
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed.
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- L. Mai, X. Tian, X. Xu, L. Chang and L. Xu, Chem. Rev., 2014, 114, 11828–11862 CrossRef CAS PubMed.
- Y. Jiang, M. Wei, J. Feng, Y. Ma and S. Xiong, Energy Environ. Sci., 2016, 9, 1430–1438 Search PubMed.
- Y. Zhong, M. Yang, X. Zhou and Z. Zhou, Mater. Horiz., 2015, 2, 553–566 RSC.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- C. Niu, J. Meng, X. Wang, C. Han, M. Yan, K. Zhao, X. Xu, W. Ren, Y. Zhao, L. Xu, Q. Zhang, D. Zhao and L. Mai, Nat. Commun., 2015, 6, 7402 CrossRef PubMed.
- Y. Liu, N. Zhang, L. Jiao and J. Chen, Adv. Mater., 2015, 27, 6702–6707 CrossRef CAS PubMed.
- M. R. Palacín, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926–5940 RSC.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502175 CrossRef.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- J. Meng, C. Niu, X. Liu, Z. Liu, H. Chen, X. Wang, J. Li, W. Chen, X. Guo and L. Mai, Nano Res., 2016, 9, 2445–2457 CrossRef CAS.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- J. S. Chen and X. W. Lou, Small, 2013, 9, 1877–1893 CrossRef CAS PubMed.
- J. Lu, Z. Chen, Z. Ma, F. Pan, L. A. Curtiss and K. Amine, Nat. Nanotechnol., 2017, 12, 1031–1038 Search PubMed.
- K. X. Wang, X. H. Li and J. S. Chen, Adv. Mater., 2015, 27, 527–545 CrossRef CAS PubMed.
- C. Peng, B. Chen, Y. Qin, S. Yang, C. Li, Y. Zuo, S. Liu and J. Yang, ACS Nano, 2012, 6, 1074–1081 CrossRef CAS PubMed.
- H. Cheng, M. Ye, F. Zhao, C. Hu, Y. Zhao, Y. Liang, N. Chen, S. Chen, L. Jiang and L. Qu, Adv. Mater., 2016, 28, 3305–3312 CrossRef CAS PubMed.
- Y. Liu, A. A. Elzatahry, W. Luo, K. Lan, P. Zhang, J. Fan, Y. Wei, C. Wang, Y. Deng, G. Zheng, F. Zhang, Y. Tang, L. Mai and D. Zhao, Nano Energy, 2016, 25, 80–90 CrossRef CAS.
- L. Zhang, G. Zhang, H. B. Wu, L. Yu and X. W. Lou, Adv. Mater., 2013, 25, 2589–2593 CrossRef CAS PubMed.
- H. Sun, L. Mei, J. Liang, Z. Zhao, C. Lee, H. Fei, M. Ding, J. Lau, M. Li, C. Wang, X. Xu, G. Hao, B. Papandrea, I. Shakir, B. Dunn, Y. Huang and X. Duan, Science, 2017, 356, 599–604 CrossRef CAS PubMed.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- K. Zhao, L. Zhang, R. Xia, Y. Dong, W. Xu, C. Niu, L. He, M. Yan, L. Qu and L. Mai, Small, 2016, 12, 588–594 CrossRef CAS PubMed.
- B. Li, H. Cao, J. Zhang, M. Qu, F. Lian and X. Kong, J. Mater. Chem., 2012, 22, 2851 RSC.
- P. Lian, J. Wang, D. Cai, L. Ding, Q. Jia and H. Wang, Electrochim. Acta, 2014, 116, 103–110 CrossRef CAS.
- Y. Zheng, T. Zhou, C. Zhang, J. Mao, H. Liu and Z. Guo, Angew. Chem., Int. Ed., 2016, 55, 3408–3413 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- A. Mahmood, W. Guo, H. Tabassum and R. Zou, Adv. Energy Mater., 2016, 6, 1600423 CrossRef.
- J. Meng, C. Niu, L. Xu, J. Li, X. Liu, X. Wang, Y. Wu, X. Xu, W. Chen, Q. Li, Z. Zhu, D. Zhao and L. Mai, J. Am. Chem. Soc., 2017, 139, 8212–8221 CrossRef CAS PubMed.
- R. Wu, D. P. Wang, X. Rui, B. Liu, K. Zhou, A. W. Law, Q. Yan, J. Wei and Z. Chen, Adv. Mater., 2015, 27, 3038–3044 CrossRef CAS PubMed.
- H. X. Zhong, J. Wang, Y. W. Zhang, W. L. Xu, W. Xing, D. Xu, Y. F. Zhang and X. B. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS PubMed.
- J. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. von Elverfeldt, C. E. Hagemeyer and F. Caruso, Angew. Chem., Int. Ed., 2014, 53, 5546–5551 CrossRef CAS PubMed.
- M. A. Rahim, K. Kempe, M. Müllner, H. Ejima, Y. Ju, M. P. van Koeverden, T. Suma, J. A. Braunger, M. G. Leeming, B. F. Abrahams and F. Caruso, Chem. Mater., 2015, 27, 5825–5832 CrossRef CAS.
- T. D. Bennett, Y. Yue, P. Li, A. Qiao, H. Z. Tao, N. G. Greaves, T. Richards, G. I. Lampronti, S. A. Redfern, F. Blanc, O. K. Farha, J. T. Hupp, A. K. Cheetham and D. A. Keen, J. Am. Chem. Soc., 2016, 138, 3484–3492 CrossRef CAS PubMed.
- V. Etacheri, G. A. Seisenbaeva, J. Caruthers, G. Daniel, J.-M. Nedelec, V. G. Kessler and V. G. Pol, Adv. Energy Mater., 2015, 5, 1401289 CrossRef.
- C. Guan, X. Wang, Q. Zhang, Z. Fan, H. Zhang and H. J. Fan, Nano Lett., 2014, 14, 4852–4858 CrossRef CAS PubMed.
- Y. J. Hong, M. Y. Son and Y. C. Kang, Adv. Mater., 2013, 25, 2279–2283 CrossRef CAS PubMed.
- B. Huang, X. Li, Y. Pei, S. Li, X. Cao, R. C. Masse and G. Cao, Small, 2016, 12, 1945–1955 CrossRef CAS PubMed.
- R. Huang, L. Wang, Q. Zhang, Z. Chen, Z. Li, D. Pan, B. Zhao, M. Wu, C. M. L. Wu and C.-H. Shek, ACS Nano, 2015, 9, 11351–11361 CrossRef CAS PubMed.
- A. Jahel, C. M. Ghimbeu, L. Monconduit and C. Vix-Guterl, Adv. Energy Mater., 2014, 4, 1400025 CrossRef.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536–2539 CrossRef CAS.
- X. Zhou, L. Yu and X. W. Lou, Adv. Energy Mater., 2016, 6, 1600451 CrossRef.
- S. Grugeon, S. Laruelle, L. Dupont and J.-M. Tarason, Solid State Sci., 2003, 5, 895–904 CrossRef CAS.
- J. Y. Huang, L. Zhong, C. M. Wang, J. P. Sullivan, W. Xu, L. Q. Zhang, S. X. Mao, N. S. Hudak, X. H. Liu, A. Subramanian, H. Fan, L. Qi, A. Kushima and J. Li, Science, 2010, 330, 1515–1520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, TEM images, calculations of bonding energies, XRD patterns, XPS spectra, Raman spectra, FTIR spectra, BET/BJH curves, and electrochemical performance. See DOI: 10.1039/c7mh00801e |
| This journal is © The Royal Society of Chemistry 2018 |