
C2 adsorption in zeolites: in silico screening and sensitivity to molecular models†

Abstract
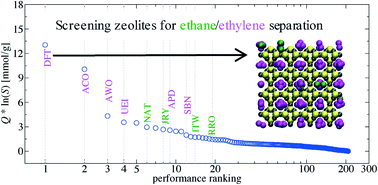
Efficient separation of ethane and ethylene has been a long-standing challenge for the chemical industry. In this study, we use molecular modeling to identify zeolite and zeotype frameworks that have the potential to be the next-generation solution for the separation of these C2 compounds. Using two different united-atom versions of the transferable potentials for phase equilibria (TraPPE) force field, the zeolitic structures in the database of the International Zeolite Association are screened for the separation of ethane and ethylene. A detailed analysis, with regards to accessibility of favorable sites and sensitivity to molecular models (also considering the explicit-hydrogen TraPPE model for ethane), is carried out on the top-performing structures. This study provides insights on the performance and limitations of molecular models for predicting mixture adsorption in zeolites.
 Please wait while we load your content...
Please wait while we load your content...

