
Exploring attachment chemistry with FRET in hybrid quantum dot dye-labeled DNA dendrimer composites†
 cd
Igor L.
Medintz
*a
cd
Igor L.
Medintz
*a
Abstract
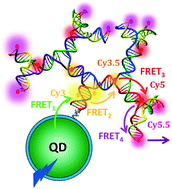
Luminescent semiconductor quantum dots (QDs) and a range of biomolecules are now being routinely co-integrated into functional optical devices in pursuit of creating novel ‘value added’ photonic and energy harvesting/transfer materials. Amongst the biological molecules, structural DNA architectures are particularly useful due to their unrivaled ability to assume almost any desired shape along with allowing fluorophores to be precisely arranged on them with controlled stoichiometry and sub-nanometer positional accuracy. The unique properties available to joint QD–DNA composites suggest them for a host of new applications in light harvesting, biosensing, and molecular computation amongst others. To fully realize the synergistic benefits from such organic–inorganic composites, especially when they constitute complex, multidimensional Förster resonance energy transfer (FRET) networks, a detailed understanding of the mechanisms that govern the individual components is imperative. Here, we demonstrate hybrid FRET systems comprising an initial QD scaffold/donor displaying DNA dendrimers decorated with dyes and which are capable of efficiently capturing UV light and transporting it to spectrally and spatially distant fluorophores via multistep FRET. We evaluate two primary strategies to conjugate the DNA-dendrimers to the QDs, namely covalent attachment of DNA to the termini of the QDs surface ligands and polyhistidine-based metal affinity coordination of modified DNA to the QD's ZnS shell surface. Analysis of the resulting FRET data shows that the dendritic arrangement of the dyes and the ability to place multiple dendrimer copies around the QD's nontrivial surface provides for significant energy transfer efficiencies of 20–25% through these multi-FRET step systems. In analyzing the properties of the conjugates, we further find that each assembly chemistry brings with it a series of benefits and liabilities that serve as mutual trade-offs and potential rules of thumb for designing future nanodevices based on these materials.
 Please wait while we load your content...
Please wait while we load your content...

