
Evaluation of affibody charge modification identified by synthetic consensus design in molecular PET imaging of epidermal growth factor receptor†
Brett A.
Case
 ,
Max A.
Kruziki
,
Lawrence A.
Stern
and
Benjamin J.
Hackel
*
,
Max A.
Kruziki
,
Lawrence A.
Stern
and
Benjamin J.
Hackel
*
Department of Chemical Engineering and Materials Science, University of Minnesota – Twin Cities, Minneapolis, MN 55455, USA. E-mail: hackel@umn.edu
First published on 9th November 2017
Abstract
Tumor overexpression of epidermal growth factor receptor (EGFR) correlates to therapeutic response in select patient populations. Thus, molecular positron emission tomography (PET) imaging of EGFR could stratify responders versus non-responders. We previously demonstrated effectiveness of a “synthetic consensus” design principle to identify six neutralizing mutations within a 58-amino acid EGFR-targeted affibody domain. Herein, we extend the approach to identify additional neutralized variants that vary net charge from −2 to either −4 or +4 while retaining high affinity (1.6 ± 1.2 nM and 2.5 ± 0.7 nM), specific binding to EGFR, secondary structure, and stability (Tm = 68 °C and 59 °C). We radiolabeled the resultant collection of five charge variants with 64Cu and evaluated PET imaging performance in murine models with subcutaneously xenografted EGFRhigh and EGFRlow tumors. All variants exhibited good EGFRhigh tumor imaging as early as 1 h, with EA35S (+3/−5) achieving 7.7 ± 1.4 %ID g−1 tumor at 4 h with 1.5 ± 0.3 %ID g−1 EGFRlow tumor, 34 ± 5 tumor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :muscle and 12 ± 3 tumor:blood ratios. The positively charged EA62S mutant (+6/−2) exhibited 2.2–3.3-fold higher liver signal than the other variants (p < 0.01). The EA68 variant with higher charge density was more stable to human and mouse serum than neutralized variants. In a comparison of radiometal chelators, 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA) exhibited superior physiological specificity to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA). In total, these studies comparatively evaluated a set of EGFR-targeted affibodies varying in net charge and charge density, which revealed functional variations that are useful in engineering an ideal probe for translational studies.
:muscle and 12 ± 3 tumor:blood ratios. The positively charged EA62S mutant (+6/−2) exhibited 2.2–3.3-fold higher liver signal than the other variants (p < 0.01). The EA68 variant with higher charge density was more stable to human and mouse serum than neutralized variants. In a comparison of radiometal chelators, 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA) exhibited superior physiological specificity to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA). In total, these studies comparatively evaluated a set of EGFR-targeted affibodies varying in net charge and charge density, which revealed functional variations that are useful in engineering an ideal probe for translational studies.
Design, System, ApplicationMolecular imaging of biomarkers requires design and optimization of a molecular probe that exhibits sensitive, specific localization to the biomarker in vivo. Effective performance requires broad distribution of the probe to enable binding to biomarkers while also rapidly clearing unbound probe. The current study focused on probe optimization for positron emission tomography (PET) of epidermal growth factor receptor (EGFR), which could empower stratification of select cancer patient populations as responders versus non-responders to molecularly targeted therapies. We engineered a set of small affibody proteins to vary in net charge and charge density using a synthetic consensus design approach and evaluated their PET efficacy in mouse tumor models. The nexus of protein engineering and physiological evaluation in small animal models allows for systematic elucidation of the impact of molecular design parameters on probe performance, which can enable efficient optimization. |
Introduction
Small protein scaffolds are evolvable towards high target affinity and molecular specificity to create valuable imaging,1,2 therapeutic,3,4 and diagnostic agents.5–7 Numerous scaffold characteristics influence ligand efficacy including scaffold structure,8 hydrophilicity,9,10 stability,11,12 and ionic charge.10,13,14Ligand charge modulation has yielded significant impacts on physiological distribution and retention. Cationic charge has increased renal retention in several cases: cystine knots rich in arginine versus serine,15 affibodies with more lysines in the chelator,14 and cationic synthetic polypeptides versus amphoteric and anionic variants.16 Other experimental results implicate anionic charge in renal retention: cystine knots rich in glutamic acid versus serine,15 bombesin peptide analogs with increased glutamic acids,17 and affibodies with increased glutamic acids in the chelator.13 Thus, charge manipulation impacts renal retention with both cationic and anionic residues implicated. Yet charge removal is not systematically sufficient to reduce renal retention. Intermediate charge removal was optimal for some fibronectin domain variants.10 Comparison of cationic and anionic Fab fragments revealed that increased positive charge corresponded to nearly a 2-fold increase in lung localization whereas elevated negative charge resulted in an almost 3-fold higher stomach signal.18 Cationic charge sped plasma clearance kinetics19 for albumin,20 IgG,21,22 Fab,22,23 and dextran.24 Cationic charge improved tumor vascular permeability – a key limitation for solid tumor targeting25 – for albumin,20 IgG20 and nanoparticles.26 Moreover, tumor interstitium is negatively charged27 and there is some evidence of selective retention of positively charged entities, including liposomes,28 though this could hinder molecular selectivity.29 In short, ligand charge can be impactful, though the relationships between charge density and net ligand charge with off-target uptake and physiological performance are not well understood, making a priori predictions difficult and necessitating several mutants for evaluation. One interpretation of the data suggests a reduction in charge density may aid performance. Yet, it is important to modulate charge without diminishing desirable ligand characteristics including high production yield and soluble recovery, strong affinity, and thermal stability. However, these charge modifications can be particularly difficult in the context of a small ligand10,30 where such mutations would encompass a greater portion of the protein's structure.31
A ligand that may benefit from charge modification is the affibody scaffold.32 Affibodies are 58-residue, three-helix bundles based on the B domain of staphylococcal protein A33 with 13 sites on helices 1 and 2 commonly diversified for binding. They have been used extensively to molecularly target clinically relevant biomarkers such as transferrin,34 TNF-α,35 HER2,36 and gp12037 with pM to μM affinities. Affibodies are also broadly applicable having been used for molecular imaging,38,39 therapy,40,41 and biotechnology.42,43
We previously hybridized a known epidermal growth factor receptor (EGFR) targeted paratope region (ZEGFR:1907)44 and an optimized backbone framework containing seven mutations which were incorporated to improve stability and reduce immunoglobulin binding.45 The resultant affibody ligand, EA68, containing 6 positive and 8 negative residues, was engineered for charge reduction.46 Consensus design from natural homologs47–49 and mutation tolerance data generated from high throughput screening (i.e. ‘synthetic consensus’ design) were used to neutralize six charged residues on EA68 creating clones EA35A8E,D37N and Synthetic 2N4P,K7P,A8Q,Q49K,N58Y herein known as ‘EA35C’ and ‘EA35S’. Both charge-reduced clones maintained single-digit nanomolar affinity (6.9 ± 1.4 nM, 1.7 ± 0.5 nM), high thermal stability (71 °C, 68 °C), and sufficient recombinant yield (12.7 ± 0.9 mg L−1, 7.0 ± 0.5 mg L−1).
Epidermal growth factor receptor (EGFR), also known as HER1 or ErbB1, is a glycoprotein in the ErbB family50 that has been shown to play a key role in tumor survival, growth, and metastasis.51 EGFR is overexpressed in several tumor types including head and neck, breast, renal, non-small cell lung, colorectal, ovarian, pancreatic, and bladder cancers52 making it an important prognostic or predictive biomarker and therapeutic target. Surface overexpression and gene copy number are predictors of disease response to receptor (cetuximab) and tyrosine kinase (gefitinib) inhibitor therapies in both primary and metastatic colorectal cancers53–55 as well as non-small-cell lung cancers.56–58 EGFR overexpression alone was associated with poor prognosis for patients with HER2-positive primary breast cancer and predictive of response to trastuzumab therapy.59 Notably, these results were independent of EGFR copy number and inapplicable to metastatic occurrences.
Thus, effective molecular imaging of EGFR would provide an unmet clinical need and allow for appropriate patient stratification to differentiate responders from non-responders. Multiple molecular probes have been preclinically developed for positron emission tomography (PET) of EGFR including small engineered proteins – affibodies,60–64 fibronectin domains,10,65 and Gp266 (and nanobodies for single-photon emission computed tomography67,68) – antibodies,69–73 small molecule inhibitors,74–77 and the native ligand.78,79 Further development of such probes, to advance sensitivity and selectivity, has potential benefit for EGFR imaging.
Herein, we extend use of mutant tolerance data to explore further charge modulation of EGFR-targeted affibody domains. We evaluate the set of mutants – varied in net charge and charge density – for PET molecular imaging of epidermal growth factor receptor in murine tumor models. Effective imaging is achieved, and functional variations between mutants informs probe development.
Materials and methods
Clone production and chelator conjugation
Affibody gene construction and ligand production were performed as described previously.46 EA26S and EA62S genes were created through overlap extension PCR,80 digested using BamHI-HF and NheI-HF restriction enzymes (New England Biolabs, Ipswich, MA), ligated into pET-22b vector containing a C-terminal hexa-histidine (Novagen, EMD Millipore, Billerica, MA) using T4 DNA ligase (New England Biolabs), and transformed into T7 express high efficiency Escherichia coli (New England Biolabs) via heat shock. EA68, EA35C, and EA35S plasmids, created previously46 and stored at −20 °C, were also transformed into T7 express using the heat shock method. Proper transformants were selected on lysogeny broth (LB) based agar plates (15 g L−1 agar, 10 g L−1 tryptone, 10 g L−1 sodium chloride, 5 g L−1 yeast extract in H2O) containing kanamycin antibiotic (50 mg L−1). After overnight growth at 37 °C, individual colonies were added to 5 mL of liquid LB with kanamycin (50 mg L−1) and shaken at 250 rpm, 37 °C until saturated (8–12 h). Upon saturation, 1 mL of culture was added to 100 mL of sterile LB and shaken at 250 rpm, 37 °C until optical density values at 600 nm reached 0.5–1. Cells were induced with 100 μL of 0.5 M isopropyl β-D-1-thiogalactopyranoside (IPTG) for 2 h at 25–28 °C. For instances where larger protein yields were required, production values were scaled to 5 mL saturated culture, added to 1 L LB, and induced with 1 mL of IPTG; all other variables held constant. Production cultures were pelleted, supernatant discarded, and cells resuspended in lysis buffer (50 mM sodium phosphate (pH 8.0), 0.5 M sodium chloride, 5% glycerol, 5 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and 25 mM imidazole). After four freeze-thaw cycles, cells were centrifuged at 12000g for 12 minutes, and soluble fraction recovered. Hexa-histidine-tagged affibodies were isolated by immobilized metal affinity chromatography using 0.2 mL HisPur Ni-NTA resin (Thermo Fisher Scientific, Waltham, MA) spin columns (100 mL production culture) or 2 mL resin drip columns (1 L production culture). Protein concentration was determined using absorbance at 280 nm on a Synergy H1 microplate reader (BioTek, Winooski, VT). Large scale productions were further purified by reversed-phase high-pressure liquid chromatography (HPLC) with a C18 column and 15 min gradient of 25% to 80% elution buffer (90% acetonitrile, 9.9% H2O, 0.1% trifluoroacetic acid) with a running buffer of 99.9% H2O, 0.1% trifluoroacetic acid. Clones were snap frozen in liquid nitrogen and lyophilized with a FreeZone Plus2.5 freeze-dry system (LabConco, Kansas City, MO). Affibodies used for PET/CT imaging were resuspended in dimethyl sulfoxide, 2–3% trimethylamine v/v, and either 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)-ester (Macrocyclics Inc., Plano, TX) or 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODAGA)-ester (CheMatech, Dijon, France) at 3:1–10:1 molar ratios of ester:protein. Upon conjugation, the reaction was quenched with excess 1 M Tris buffer, pH 8.0, isolated via HPLC, freeze-dried, and stored at 4 °C until used experimentally.
Mammalian cell growth
A431 epidermoid carcinoma (2.9 ± 1.4 × 106 EGFR per cell81), MDA-MB-435 melanoma-type (1.5 ± 1.1 × 104 EGFR per cell81), and MCF7 (1.2 ± 1.5 × 102 EGFR per cell81) mammary carcinoma cell lines kindly provided by Dr. Daniel Vallera (University of Minnesota), Dr. Tim Starr (University of Minnesota), and Dr. Deepali Sachdev (University of Minnesota), respectively, were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum and 1% penicillin–streptomycin at 37 °C, 5% CO2.Affinity measurement, target specificity, and circular dichroism
At 70–80% confluence, culture media was removed and 0.25% trypsin-EDTA in Hank's balanced salt solution (HBSS) (Corning, Tewksbury, MA) added for 6–10 minutes to detach cells. Cells were washed once with cold PBSA (phosphate buffered saline: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, and 1 g L−1 bovine serum albumin) to prevent EGFR internalization. Approximately 40000 A431 or MCF7 cells were labeled with each affibody at varying concentrations (0–500 nM for affinity titrations and 500 nM for specificity tests) at 4 °C. Cells were again washed in cold PBSA and incubated with fluorescein-conjugated rabbit anti-His6 antibody (ab1206, Abcam, Cambridge, MA). Median fluorescence of each sample was determined with an Accuri C6 (BD Biosciences, San Jose, CA) and affinity values calculated by least squares regression assuming a 1:1 binding event between ligand and receptor. Equilibrium dissociation constants are presented as the average ± standard deviation of titrations.
Lyophilized affibodies were resuspended to 0.5–1 g L−1 in 1xPBS. 300 μL of each suspension was added to a quartz cuvette with 1 mm path length and ellipticity measured from 200 to 260 nm at 20 °C on a Jasco J-815 spectrophotometer. Thermal denaturation curves were created by monitoring ellipticity at 220 nm as the sample was heated from 20 to 98 °C at a rate of 2 °C min−1. The midpoint of denaturation (Tm) was determined using a two-state protein unfolding model. After heating, suspensions were cooled to 20 °C and scanned from 200 to 260 nm to ascertain the degree to which each affibody refolds.
Tumor xenotransplantation
Six week old female (Foxn1nu/Foxn1nu) mice (Jackson Labs, Bar Harbor, ME) were anesthetized with 2.0% isoflurane in 0.8–1 mL min−1 oxygen and subcutaneously injected in their right shoulder with approximately 10 million MDA-MB-435 cells in a 50:50 v/v Matrigel Matrix/DMEM suspension. After 4.5 weeks, this method was repeated with approximately 4 million A431 cells injected subcutaneously into left shoulders. At six weeks after the MDA-MB-435 inoculation, xenografts from both tumor lines had grown to 4–11 mm in diameter. All animal studies were approved by the University of Minnesota Institutional Animal Care and Use Committee (Protocol 1411-32007A).
64Cu chelation of DOTA or NODAGA/purification
10–12 mCi of 64CuCl2 (University of Wisconsin) was suspended in 100 μL of 100 mM NaAc, pH 5.0 for buffering and titrated to pH 6.0 using 0.1 M HCl and 0.1 M NaOH (final volume: 150–225 μL). 1–2 mCi of buffered 64CuCl2 was added to 500 μL of DOTA-affibody or 500 μL of NODAGA-affibody at 70–120 μM in PBS pH 7.4 or 10 mM sodium acetate pH 6.0 respectively, and placed at 37 °C for 1 h to elicit complexation. 64Cu-DOTA-affibody and 64Cu-NODAGA-affibody were separated from free copper using a PD-10 size exclusion column and PBS or 10 mM sodium acetate elution buffer.RadioTLC/enzymatic degradation
64Cu-NODAGA-affibody purity levels and enzymatic degradation were ascertained with an AR-2000 radio-thin layer chromatography scanner (Eckert and Ziegler, Berlin, Germany). For determination of purity, 2 μL of PD-10 elution volume 3.5–4.0 mL was spotted on filter paper and a PBS mobile phase was used for migration. Area under the curve analysis was used to determine the fraction of unconjugated copper found in the solvent front peak compared to the relatively immobile 64Cu-NODAGA-affibodies. One μL from each 64Cu-NODAGA-affibody PD-10 elution volume 3.5–4.0 mL was added to 20 μL of normal human serum (Jackson ImmunoResearch), normal mouse serum (Jackson ImmunoResearch), or 0.25% trypsin-EDTA in HBSS and incubated at 37 °C for 4 hours. Two μL samples were taken from each mixture at time points 1 and 4 h, spotted on filter paper (PBS mobile phase), and scanned to determine the relative percentage of affibody metabolites by means of area under the curve integration.PET imaging/quantification
Mice were anesthetized under 2% isoflurane at 1 mL min−1 oxygen until no longer ambulant. Approximately 2.2 to 3.7 MBq of 64Cu-NODAGA- or 64Cu-DOTA-affibody, as determined by an Atomlab 100 dosimeter (Biodex, Shirley, NY) calibrated for 64Cu measurements, was injected through lateral tail veins. Five minute PET scans were performed at 1 and 4 h post-injection on an Inveon microPET/CT (Siemens, Malvern, PA) and reconstructed with 2D ordered-subset expectation maximization (OSEM2D). Four iterations of Fourier rebinning were performed. Photopeak energy cutoffs were set to 350–650 keV with a time window of 3.438 ns. PET images were smoothed with a 1 × 1 × 1 voxel Gaussian filter. CT scans were executed using 340 projections of 80 kV at 500 μA with 4 × 4 binning and an effective pixel size of 85.6 μm. Scans ran for approximately 384 s and were reconstructed with the Feldkamp–David–Kress algorithm with slight noise reduction and Shepp-Logan filtering.PET image quantification was performed with Inveon Research Workplace software (Siemens). CT images were used as guides to determine 3D regional limits to tissues of interest including the liver, kidneys, thigh muscle, A431, and MDA-MB-435 tumors. The maximum average PET signal in a 3 × 3 × 3 spherical voxel (9.1 mm3) was determined for each tissue type.
Tissue quantification
After imaging, mice were euthanized by cervical dislocation while still under isoflurane anesthesia. After verifying death, mice were dissected and blood, heart, lung, liver, pancreas, spleen, stomach, intestine, skin, A431 tumor, MDA-MB-435 tumor, brain, bone, muscle, and kidneys were collected and weighed. Sample radioactivity was measured by either a CRC 25 W (Capintec, Florham Park, NJ) gamma counter over all energy ranges or an Automatic Gamma Counter (Hidex, Turku, Finland) between 400 and 600 keV with all reads averaged over 60 s. Counts from both instruments were calibrated using serial dilutions based on the dose reported by the Atom Lab 100 dosimeter.Statistics
Comparisons for statistical significance were performed using two-tailed student's t-tests assuming unequal variance. Values are reported as mean ± standard error unless otherwise stated.Results and discussion
Further charge modulation of EA35S -- guided by sequence tolerance data from high throughput selections -- is well tolerated with regards to secondary structure, stability, affinity, and recombinant yield.In a previous study,46 3 basic and 3 acidic residues from the EGFR-targeted affibody EA68 were neutralized via mutation yielding 19 mutants of which clones ‘EA35S’ and ‘EA35C’ retained the greatest combination of target affinity, thermal stability, and recombinant yield. While these clones enable the evaluation of charge density reduction on physiological distribution, additional variations of net charge were desired. Based on the success of the ‘synthetic consensus’ strategy for identifying mutants with three basic and five acidic residues (+3/−5)46 the approach was used to identify alternative mutations. The most frequently observed neutralizing mutations – within the previously enriched affibody mutants – were chosen to mutate EA35S to create clones with +2/−6 (EA26S) and +6/−2 (EA62S) charge (Fig. 1) with the exception of position 47 in EA62S. Although not tested in the synthetic approach, site 47 was mutated to glutamine as necessary based on the prevalence of accepted glutamic acid to glutamate replacements at other sites, E14 residing in the paratope, and residue 36 remaining negatively charged (glutamic of aspartic acid) in 98% of natural homologs.
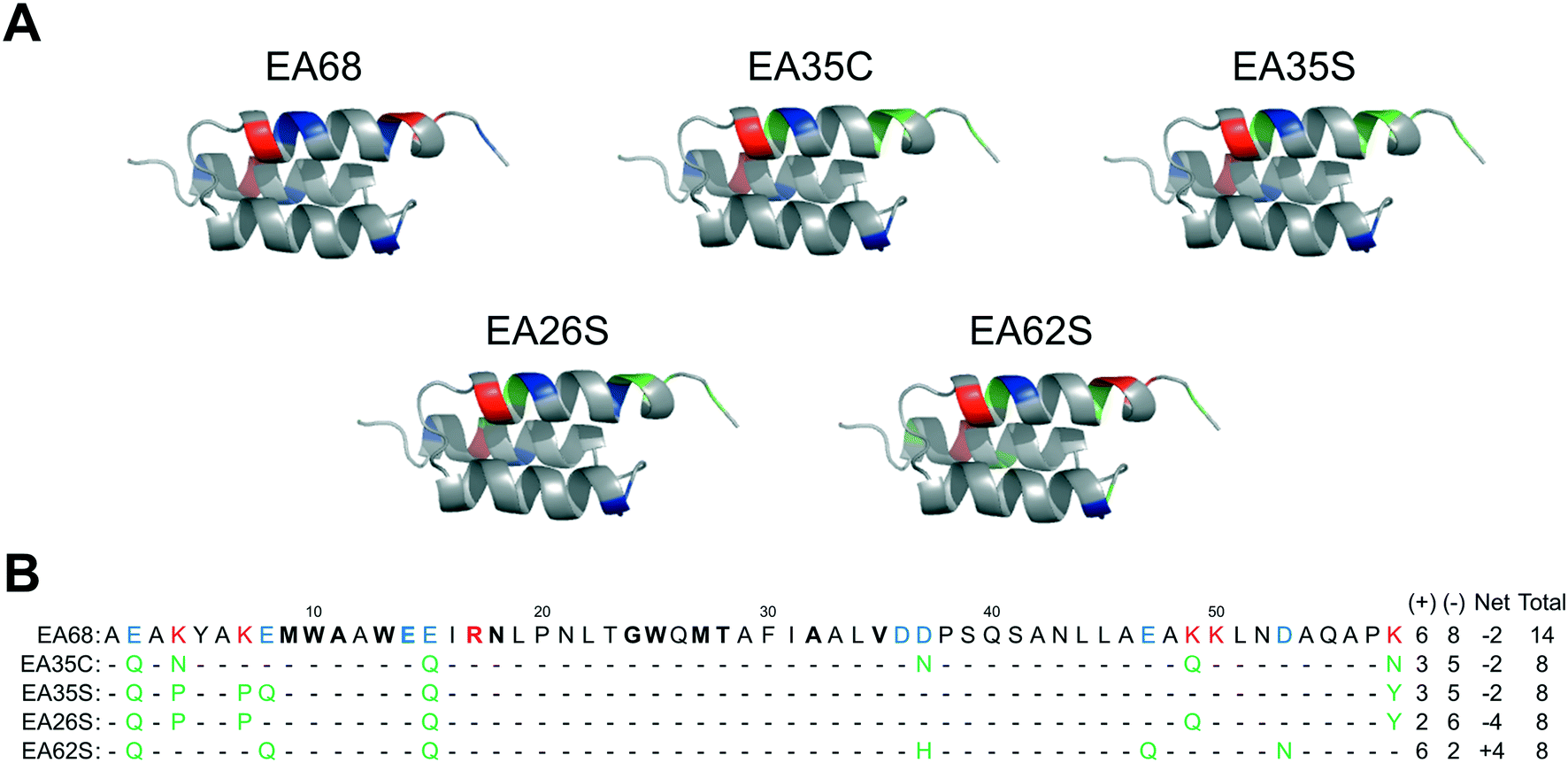 | ||
| Fig. 1 Affibody mutants. (A) Structural schematics of affibody clones EA68, EA35C, EA35S, EA26S, and EA62S. Acidic, basic, and charged-to-neutral mutated residues are designated as blue, red, and green, respectively. Structures are based on PDB 1h0t33. (B) Sequences of the affibody mutants with the same color scheme as in (A). The traditional paratope regions are shown in bold. | ||
EA26S and EA62S were readily produced and purified from the soluble fraction of E. coli culture (0.62 ± 0.02 mg L−1 and 1.9 ± 0.3 mg L−1, respectively). The purified proteins were titrated for binding to EGFR-overexpressing A431 cells and evaluated by flow cytometry. EA26S and EA62S bind with high affinity (1.6 ± 1.2 nM and 2.5 ± 0.7 nM, respectively; Fig. 2A) comparable to EA35S (1.7 ± 0.5 nM) and exceeding the EGFR affinities of both EA68 (5.3 ± 1.7 nM) and EA35C (6.9 ± 1.4 nM). Binding is molecularly specific as demonstrated by the lack of binding to EGFRlow MCF7 cells (Fig. 2B).
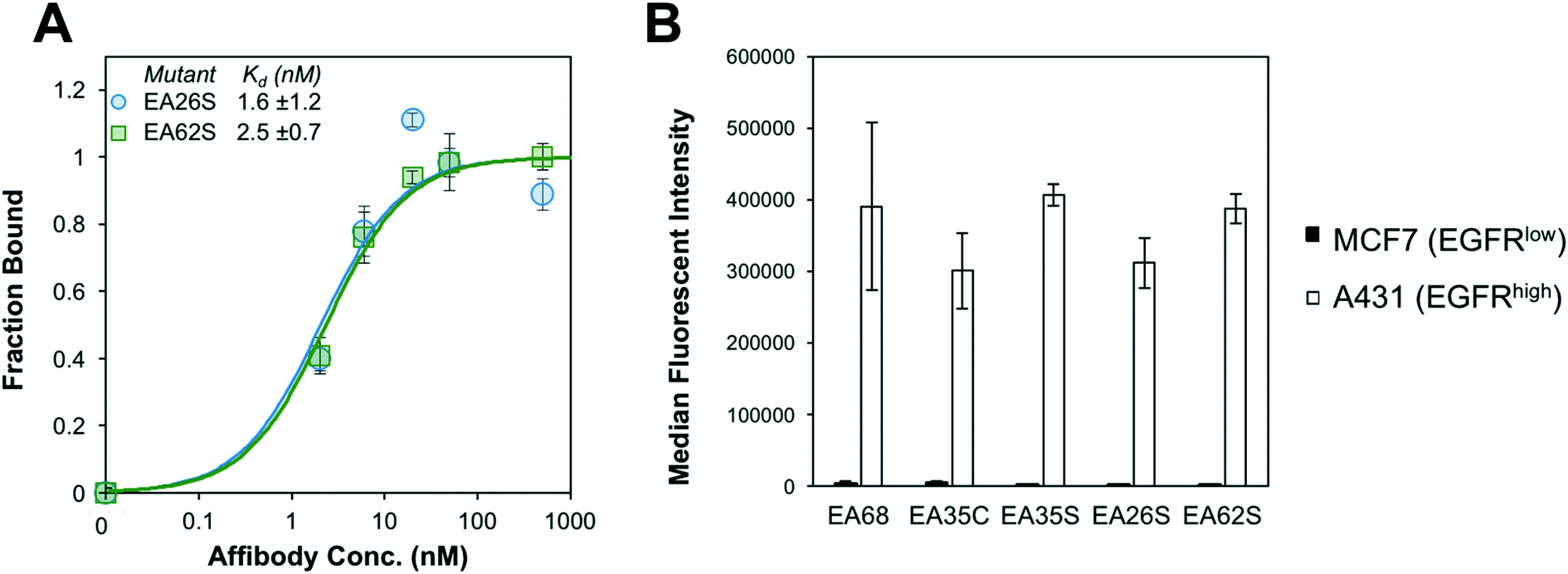 | ||
| Fig. 2 Binding characterization. (A) EGFRhigh A431 cells were mixed with the indicated concentration of affibody (EA26S (blue circles) or EA62S (green squares)). Binding was detected with fluorophore-conjugated anti-His6 antibody via flow cytometry. Equilibrium dissociation constants were calculated assuming a 1:1 binding model. n = 3 measurements per variant. (B) EGFRhigh A431 cells (white) or EGFRlow MCF7 cells (black) were incubated with 500 nM affibody. Binding was detected as in (A). n = 3 measurements per variant. | ||
Like EA35S and EA35C, the new variants exhibit comparable secondary structure and ability to refold after thermal denaturation to the parental EA68 (Fig. 3A and B). EA26S exhibits thermal stability (Tm = 68 °C) comparable to EA68, EA35C, and EA35S (71, 71, and 68 °C, respectively) whereas EA62S is less stable (Tm = 59 °C) (Fig. 3C). Thus, the collection of EGFR-binding affibodies exhibits similar biophysical properties but varied charge distribution (Table S1†). The efficacy of consensus design based on synthetic combinatorial library selections is consistent with previous efforts.46,82 This efficacy may also be supported by the robustness of the previously improved affibody framework.45 The resultant ability to modulate charge yielded a set of affibody variants that enable evaluation of the physiological effects of these mutations, which include changes in net charge and charge density.
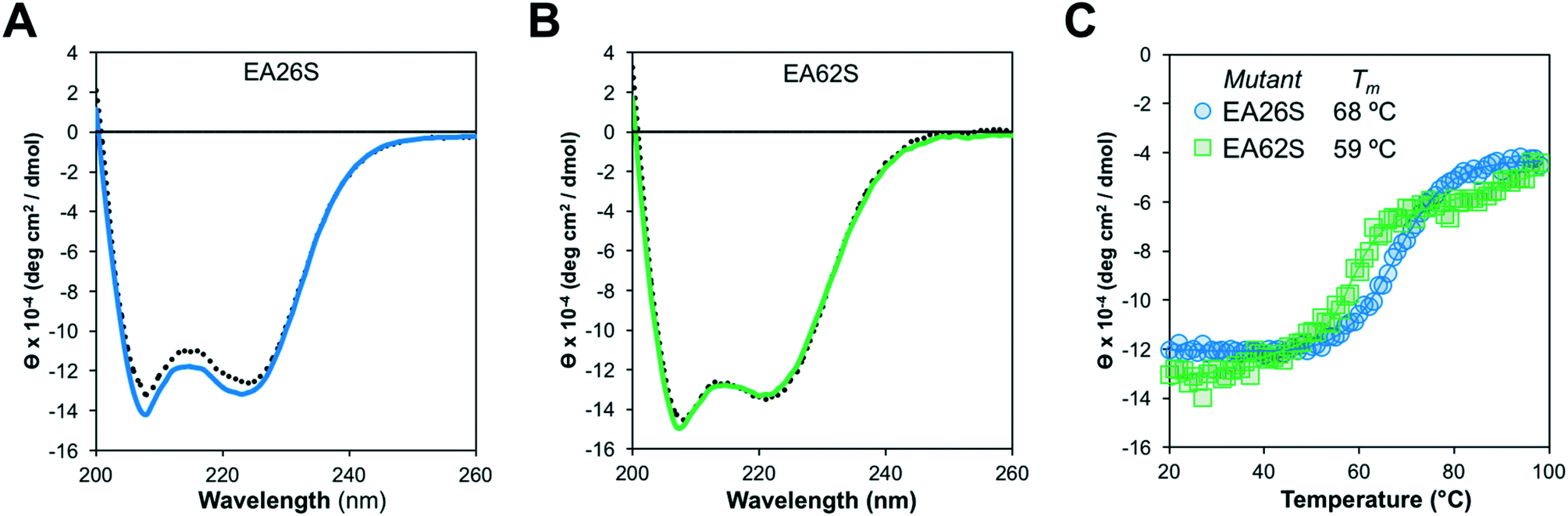 | ||
| Fig. 3 Structural characterization. Purified EA26S and EA62S were evaluated by circular dichroism. (A and B) Wavelength scans from 200 nm to 260 nm before (solid line) or after (dotted line) heating to 98 °C and cooling to 20 °C. (C) Thermal scan from 20 °C to 98 °C at a wavelength of 220 nm. | ||
Affibody variants are functional EGFR imaging probes
The parental EA68 and the four neutralized charge variants with a range of net charge were conjugated with 2,2′-(7-(1-carboxy-4-((2,5-dioxopyrrolidin-1-yl)oxy)-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid, the N-hydroxysuccinimidyl ester of NODAGA. Conjugation was verified by matrix-assisted laser desorption ionization (MALDI) mass spectrometry (Fig. S1†) and showed an average labeling of 1.7, 1.7, 1.5, 0.9, and 1.4 NODAGA per ligand for EA68, EA35C, EA35S, EA26S, and EA62S, respectively. While more site-selective conjugation could have been achieved with unique cysteine incorporation or alternative approaches, the amine-conjugated affibodies exhibited similar target affinities to their unconjugated forms (Fig. S2†). Each NODAGA-conjugated affibody was mixed with 1–1.5 mCi of 64Cu in 100 mM sodium acetate buffer at pH 6.0 to undergo complexation at 37 °C for 1 h. Radiolabeled proteins were separated from free 64Cu through size exclusion chromatography, yielding high radiochemical purity (96–99%) and labeling efficiency (65 ± 6%) for subsequent murine injections. By applying knowledge of previous purifications of their non-radiolabeled forms, approximate specific activities were calculated between 0.4–1.1 MBq nmol−1.Imaging performance and physiological distribution were evaluated in mice harboring dual xenografted tumors: EGFRhigh A431 and EGFRlow MDA-MB-435. 64Cu-complexed affibodies were injected via tail vein, and PET/computed tomography (CT) images were collected at 1 and 4 h post-injection. CT scans were used as locational guides to determine regions of interest including the liver, kidneys, muscle (anterior/posterior thigh), A431, and MDA-MB-435 tumors. All affibody variants exhibited strong EGFR imaging performance at both 1 h and 4 h (Fig. 4 and 5).
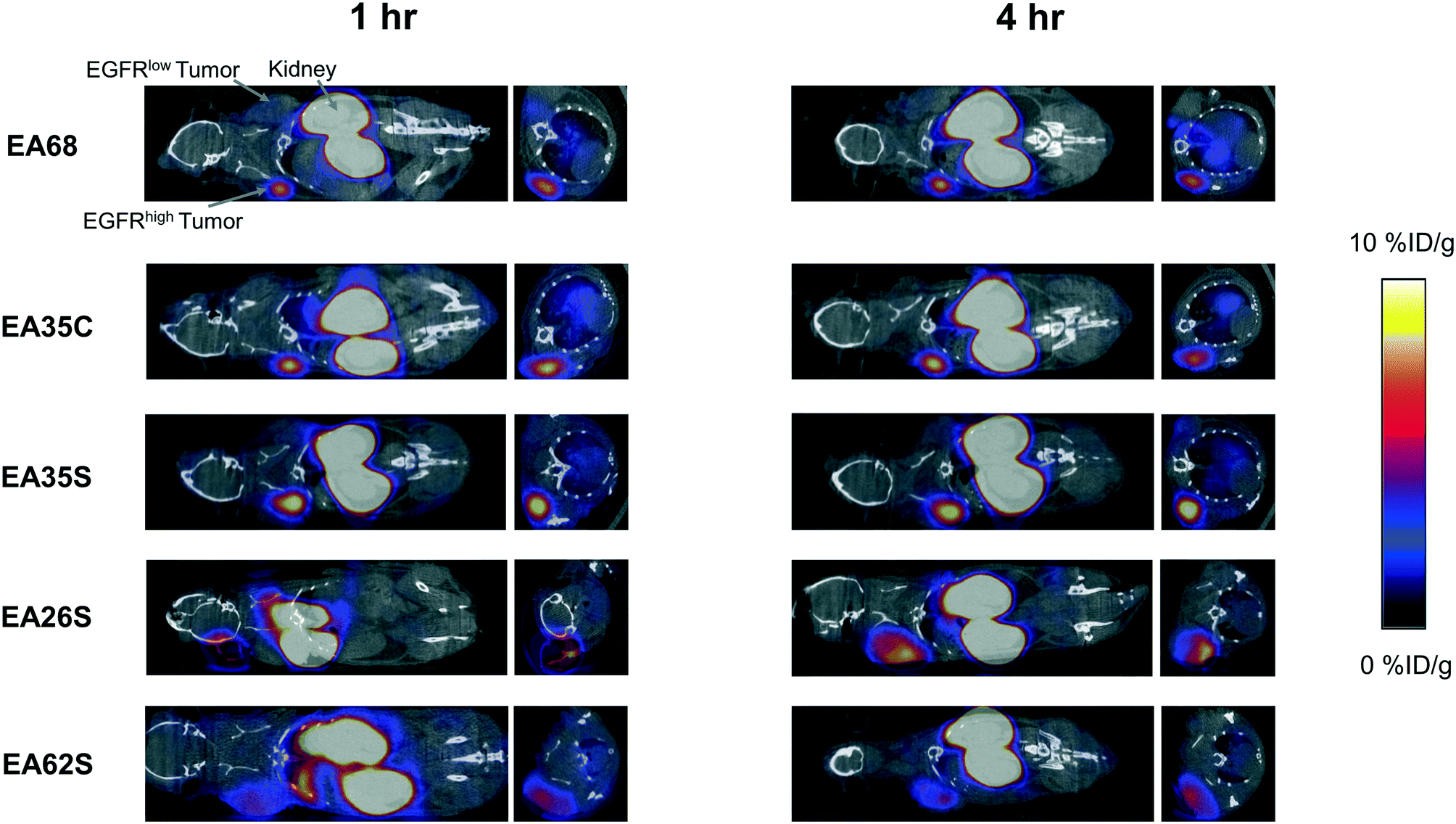 | ||
| Fig. 4 PET/CT imaging. Mice with xenografted A431 (EGFRhigh) and MDA-MB-435 (EGFRlow) tumors in either shoulder were injected intravenously with 64Cu-NODAGA-affibody (five different variants) and imaged via PET/CT at 1 h and 4 h post-injection. Coronal and axial slices through the tumor are shown for representative mice (n = 3–7). | ||
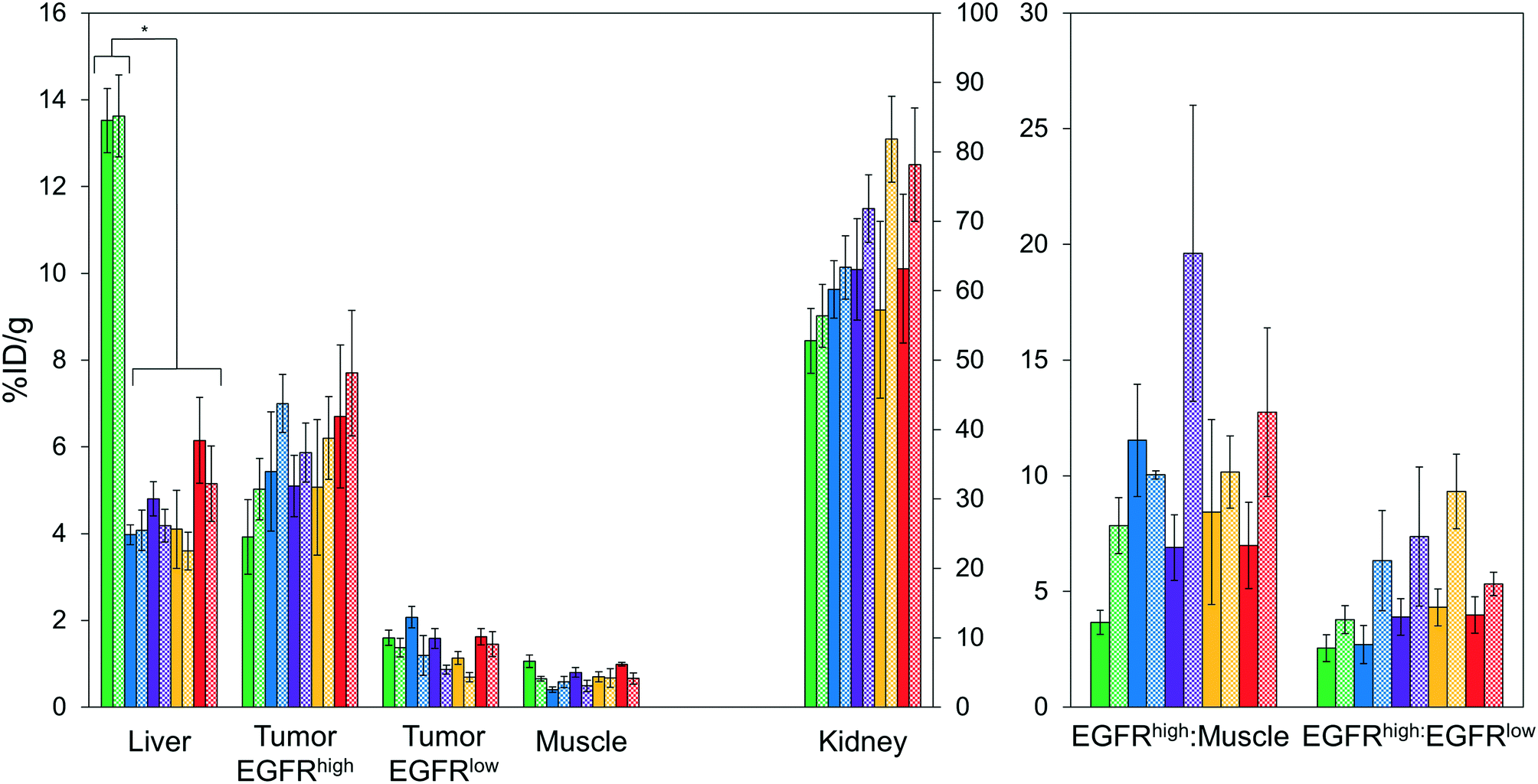 | ||
| Fig. 5 PET imaging quantification. Mice (n = 3–7) with xenografted A431 (EGFRhigh) and MDA-MB-435 (EGFRlow) tumors in either shoulder were injected intravenously with 64Cu-NODAGA-affibody (five different variants) and imaged via PET/CT at 1 h (solid) and 4 h (checkered) post-injection. Signal in the liver, tumors, muscle, and kidney were quantified using Siemens Inveon research workplace software. * designates p < 0.01. | ||
At 1 h, the five affibodies had localized to EGFR-overexpressing A431 tumors with molecular specificity (5.2 ± 0.5 %ID g−1) and cleared from EGFRlow MDA-MB-435 tumors (1.6 ± 0.1 %ID g−1). By 4 h, A431 signal had increased further (6.7 ± 0.5 %ID g−1) while MDA-MB-435 remained low and decreased slightly (1.2 ± 0.1 %ID g−1). Among clones, EA35S displayed the nominally highest EGFR-binding at both 1 h (6.7 ± 1.6 %ID g−1) and 4 h (7.7 ± 1.4 %ID g−1). As is common with renal clearance of small proteins, high levels of kidney signal were prevalent, and average concentration exceeded 50 %ID g−1 for all clones. Note that due to the large number of photons emanating from the kidneys, multiple coincident events may be detected within the allotted time window forcing some events to be discarded to avoid locational ambiguity. This can result in kidney activity measurements being artificially lower than their true values and cross-comparison among clones may not be valid.83 Background clearance was rapid with an average muscle signal of 0.8 ± 0.1 %ID g−1 at 1 h which decreased to 0.6 ± 0.1 %ID g−1 by 4 h. Excluding EA62S, clones exhibited moderate levels of liver signal due to hepatic processing at both 1 h (4.8 ± 0.3 %ID g−1) and 4 h (4.6 ± 0.3 %ID g−1). Conversely, EA62S had significantly greater hepatic retention than all other clones at both 1 h (13.5 ± 0.7 %ID g−1, p < 0.01) and 4 h (13.6 ± 0.9 %ID g−1, p < 0.01).
Imaging results were corroborated by analysis of excised tissues at 4 h post-injection (Fig. 6). The parental protein, EA68, exhibited strong EGFR-specific tumor uptake (7.2 ± 1.5 %ID g−1 in EGFRhighversus 0.9 ± 0.2 %ID g−1 in EGFRlow; p = 0.002; Fig. 6A). Specificity was further supported by 11 ± 4 tumor:blood and 126 ± 56 tumor:muscle ratios (Fig. 6B). Retention is substantial in kidneys (169 ± 12 %ID g−1) and modest in liver (4.5 ± 0.6 %ID g−1). Similarly, EA35C displayed elevated EGFRhigh (7.0 ± 0.9 %ID g−1) and low EGFRlow tumor (1.5 ± 0.4 %ID g−1) signal with substantial tumor:blood (11 ± 4 %ID g−1) and tumor:muscle (151 ± 70 %ID g−1) ratios. EA35S showed the highest level of A431 retention (12 ± 2 %ID g−1) but a nominally elevated hepatic signal (7 ± 1 %ID g−1) versus clones EA35C, EA26S, and EA68 which averaged 4.4 ± 0.3 %ID g−1 in the liver. Compared to all other variants, which are negatively charged, positively charged EA62S has significantly higher liver and spleen retentions of 14 ± 1 %ID g−1 (p ≤ 0.01) and 3.2 ± 0.3 %ID g−1 (p ≤ 0.01) respectively.
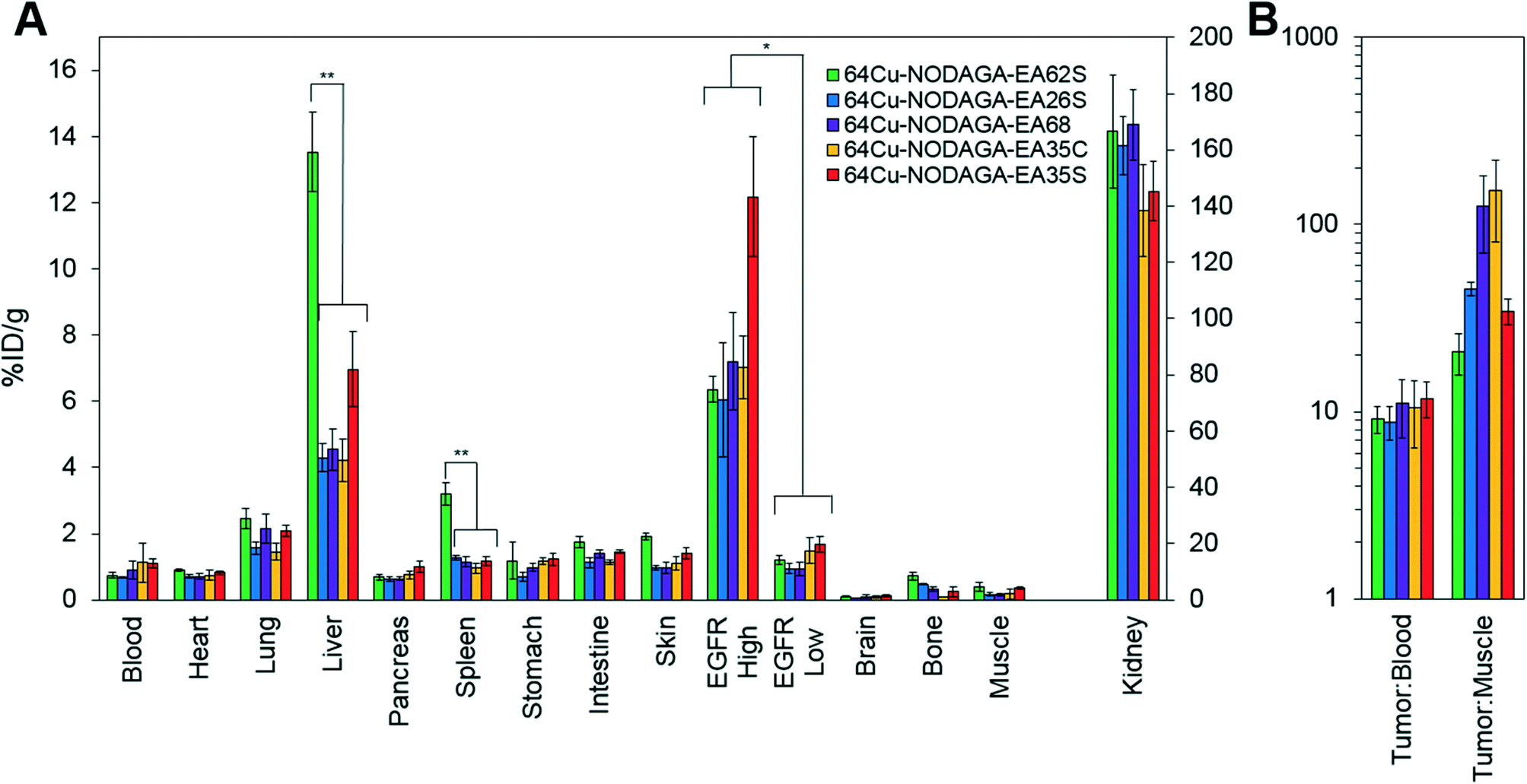 | ||
| Fig. 6 Excised tissue analysis. Mice with xenografted A431 (EGFRhigh) and MDA-MB-435 (EGFRlow) tumors in either shoulder were injected intravenously with 64Cu-NODAGA-affibody (five different variants) and euthanized at 4 h post-injection. Tissues or fluid was excised and weighed; radioactivity was measured (n = 3–7). **: p < 0.01, *: p < 0.05. (A) %ID g−1 in tissues and fluid. (B) Tumor:background ratios. | ||
The rapid distribution of the affibody probes, as well as clearance of unbound probe, enables effective imaging at early time points. While not requisite for diagnostic imaging, additional studies could be used to evaluate probe localization at later times.
Chelator comparison
To determine the effect that the chelator may have on biodistribution, EA68 and EA35S were conjugated with DOTA by reaction of N-hydroxysuccinimidyl-DOTA ester with primary amines on the N-terminus or lysine residues of each protein. MALDI-MS was used to verify an average labeling of 1.1 DOTA per EA68 and 2.0 DOTA per EA35S affibody (Fig. S3†). DOTA labeled EA68 and EA35S were mixed with 1.9–2.0 mCi of 64Cu in PBS at pH 7.4 to undergo complexation at 37 °C for 1 h. Size exclusion chromatography was used to separate complexed affibodies from free 64Cu providing a labeling efficiency of 43% and 60% for 64Cu-NODAGA-EA35S and 64Cu-NODAGA-EA68. The approximate specific activities of EA35S and EA68 were 0.25 and 0.33 MBq nmol−1.Chelator performance was evaluated in six nude mice with human EGFRhigh A431 and EGFRlow MDA-MB-435 tumors xenografts. A431 xenotransplantation and subsequent growth was successful in all murine models. However, four of six mice showed no measureable MDA-MB-435 tumor progression. The remaining two mice displaying both cancer lines were split to provide one negative control tumor for each DOTA-conjugated affibody. Mice were injected intravenously through lateral tail veins with 2.3–3.7 MBq of 64Cu-NODAGA-EA35S and 64Cu-NODAGA-EA68. At 4 h post-injection mice were euthanized, tissues excised, and blood collected.
The use of a NODAGA chelator yields lower signal in all background (i.e. non-A431 tumor) tissues in comparison to DOTA (Fig. 7). 64Cu-NODAGA-EA35S maintained significantly lower blood, heart, liver, and stomach uptake than 64Cu-DOTA-EA35S. Similarly, 64Cu-NODAGA-EA68 had substantially lower signal in the lungs and intestine than 64Cu-DOTA-EA68. EGFRhigh A431 tumor localization was comparable (and nominally greater) for both NODAGA-labeled EA35S and EA68 than their DOTA-conjugated counterparts. These results are consistent with previous data indicating imperfect stability of the 64Cu-DOTA complex in vivo.84
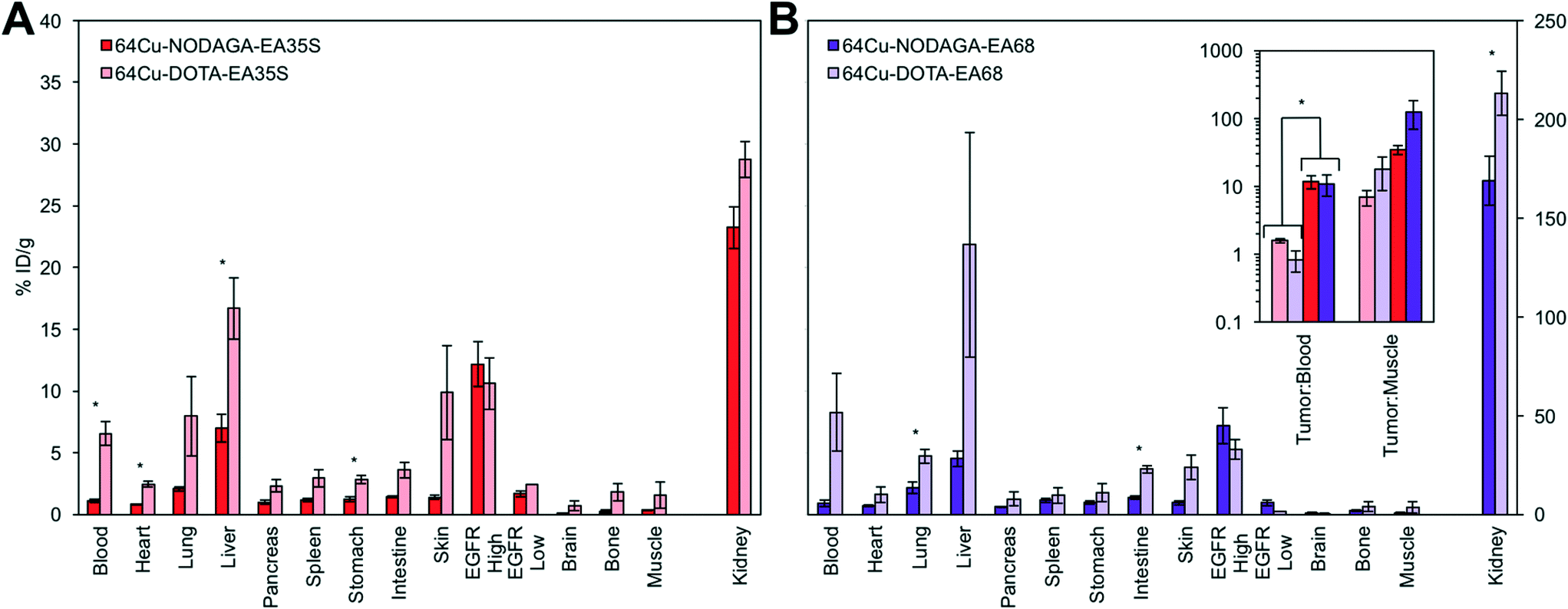 | ||
| Fig. 7 Chelator comparison. Mice with xenografted A431 (EGFRhigh) and MDA-MB-435 (EGFRlow) tumors in either shoulder were injected intravenously with 64Cu-NODAGA-affibody or 64Cu-DOTA-affibody and euthanized at 4 h post-injection. Tissues or fluid was excised and weighed; radioactivity was measured (n = 3–7). * designates p < 0.05. Note that kidney signals correspond to the secondary axis. (A) EA35S. (B) EA68 with inset of tumor:background ratios for both EA35S and EA68. | ||
As numerous EGFR PET probes are in preclinical development, performance comparison is desirable. Strict quantitative comparisons have moderate utility because of differences in tumor models and experimental procedures, but can provide guidance when such differences are limited and acknowledged. The current 64Cu-NODAGA-EA35S exhibits comparable tumor uptake and tumor:muscle specificity but superior tumor:blood (12 ± 3 vs. 1.0 ± 0.1 without blocking) and liver specificity (5.2 ± 0.8 %ID g−1vs. 19 ± 3 %ID g−1) relative to a Miao et al. study85 with 64Cu-DOTA-affibody1907. The chelator comparison experiments (Fig. 7) reveal that this benefit derives predominantly from the use of NODAGA as the 64Cu-DOTA-affibody results are comparable between the current study and Miao et al.85 and similarly matched by an additional study with a Hep3B xenograft.86 Other alternative radiochemistries, with 89Zr-DFO63 or 18F-CBT,87 also reduce hepatic signal albeit without benefit to tumor:blood specificity. Strong tumor:blood specificity was achieved with the 64Cu-DOTA-affibody via a small dose of unlabeled affibody.85 The previous 64Cu-DOTA-affibody1907 and the current 64Cu-DOTA-EA68 only differ in the use of the engineered protein framework, optimized by Feldwisch et al.45 for enhanced stability and reduced immunoglobulin binding. Thus, by historical comparison, these mutations enable these benefits without substantially altering physiological delivery; yet direct comparison in the same experimental system would be needed to discern potential differences more precisely. The current 64Cu-NODAGA-affibodies yield comparable PET imaging and physiological delivery performance to a 64Cu-DOTA-Gp2 probe66 and higher tumor sensitivity and tumor:muscle specificity relative to a 64Cu-DOTA-fibronectin.65
Labeling and serum stability
In vitro enzymatic stability was assessed through radio-thin-layer chromatography. Radiolabeled charge variants were added to murine serum, human serum, or trypsin–EDTA (0.25%) in HBSS at a 1:20 volumetric ratio and incubated for 4 hours at 37 °C. Area under the curve analysis shows that parental EA68 was most resistant to degradation in both human and murine serum to similar degrees. Approximately 2% and 4% of sample signal contained 64Cu-labeled metabolites at both 1 and 4 h in human and murine serum, respectively (Fig. S4†). Similarly, EA35C and EA26S were stable in human serum with 6% and 4% of total signal area due to proteolysis after 4 hours. EA35S and EA62S degraded more rapidly, with 10% and 11% of cleaved 64Cu-peptide in human serum at 4 h. Excluding EA68, clones were more susceptible to enzymatic cleavage in murine serum than human serum. This difference was most pronounced in clones EA26S and EA62S, where 16% and 44% of radioactivity was attributed to metabolic product in mouse serum versus 4% and 11% for human serum. In all cases, trypsin digestion resulted in hydrolysis products displaying the furthest positional shift indicating nearly complete proteolysis.
Conclusions
Molecular imaging of EGFR is poised to address an important clinical need: to stratify a subset of cancer patients as responders vs. non-responders to several molecularly targeted therapies to enable effective treatment planning. While numerous probes have undergone preclinical development, further molecular advances are needed as no clinical imaging probe is yet available for EGFR. Charge has been shown to play an important role in physiological distribution of proteins, but system complexity precludes a priori design and necessitates experimental evaluation. We have further demonstrated the efficacy of ‘synthetic consensus design’ in identifying functional neutralizing mutations to yield a set of small affibody scaffolds varied in net charge and charge density. Evaluation of this set of probes, radiolabeled with 64Cu, for PET imaging of EGFR in mouse tumor models reveals sensitive, specific molecular imaging for multiple variants with notable differences in liver retention, specificity, and probe stability for different charge and chelator designs. Elucidation of these differential efficacies aids engineering of an ideal probe for translational studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health (R21 EB021511 to B.J.H.) and the University of Minnesota's Interdisciplinary Doctoral Fellowship (to M.A.K.).References
- M. L. James and S. S. Gambhir, A molecular imaging primer: modalities, imaging agents, and applications, Physiol. Rev., 2012, 92, 897–965 CrossRef CAS PubMed.
- L. A. Stern, B. A. Case and B. J. Hackel, Alternative non-antibody protein scaffolds for molecular imaging of cancer, Curr. Opin. Chem. Eng., 2013, 2, 425–432 CrossRef PubMed.
- M. Gebauer and A. Skerra, Engineered protein scaffolds as next-generation antibody therapeutics, Curr. Opin. Chem. Biol., 2009, 13, 245–255 CrossRef CAS PubMed.
- S. Banta, K. Dooley and O. Shur, Replacing antibodies: engineering new binding proteins, Annu. Rev. Biomed. Eng., 2013, 15, 93–113 CrossRef CAS PubMed.
- S. Straw, P. K. Ferrigno, Q. Song, D. Tomlinson and F. Galdo, Del. Proof of concept study to identify candidate biomarkers of fibrosis using high throughput peptide aptamer microarray and validate by enzyme linked immunosorbant assay, J. Biomed. Sci. Eng., 2013, 6, 32–42 CrossRef.
- A. Johnson, Q. Song, P. Ko Ferrigno, P. R. Bueno and J. J. Davis, Sensitive affimer and antibody based impedimetric label-free assays for C-reactive protein, Anal. Chem., 2012, 84, 6553–6560 CrossRef CAS PubMed.
- K. Škrlec, B. Štrukelj and A. Berlec, Non-immunoglobulin scaffolds: A focus on their targets, Trends Biotechnol., 2015, 33, 408–418 CrossRef PubMed.
- P. Holliger and P. J. Hudson, Engineered antibody fragments and the rise of single domains, Nat. Biotechnol., 2005, 23, 1126–1136 CrossRef CAS PubMed.
- D. Rosik, et al. Incorporation of a triglutamyl spacer improves the biodistribution of synthetic affibody molecules radiofluorinated at the n-terminus via oxime formation with 18F-4-fluorobenzaldehyde, Bioconjugate Chem., 2014, 25, 82–92 CrossRef CAS PubMed.
- B. J. Hackel, A. Sathirachinda and S. S. Gambhir, Designed hydrophilic and charge mutations of the fibronectin domain: towards tailored protein biodistribution, Protein Eng., Des. Sel., 2012, 25, 639–647 CrossRef CAS PubMed.
- M. Whitlow, et al. An improved linker for single-chain fv with reduced aggregation and enhanced proteolytic stability, Protein Eng., Des. Sel., 1993, 6, 989–995 CrossRef CAS.
- J. Willuda, et al. High thermal stability is essential for tumor targeting of antibody fragments, Cancer Res., 1999, 59, 5758–5767 CAS.
- T. Tran, et al. Tc-99m-maEEE-Z(HER2 : 342), an affibody molecule-based tracer for the detection of HER2 expression in malignant tumors, Bioconjugate Chem., 2007, 18, 1956–1964 CrossRef CAS PubMed.
- T. A. Tran, et al. Effects of lysine-containing mercaptoacetyl-based chelators on the biodistribution of 99mTc-labeled anti-HER2 Affibody molecules, Bioconjugate Chem., 2008, 19, 2568–2576 CrossRef CAS PubMed.
- R. H. Kimura, et al. Pharmacokinetically Stabilized Cystine Knot Peptides That Bind Alpha-v-Beta-6 Integrin with Single-Digit Nanomolar Affinities for Detection of Pancreatic Cancer, Clin. Cancer Res., 2012, 18, 839–849 CrossRef CAS PubMed.
- M. V. Pimm, et al. Gamma scintigraphy of 111In-labelled branched chain polypeptides (BCP) with a poly(L-lysine) backbone in mice with mammary carcinoma: effect of charge on biodistribution and tumour imaging potential, Ann. Nucl. Med., 1995, 9, 247–251 CrossRef CAS PubMed.
- E. García Garayoa, et al. Influence of the molecular charge on the biodistribution of bombesin analogues labeled with the [99mTc(CO)3]-core, Bioconjugate Chem., 2008, 19, 2409–2416 CrossRef PubMed.
- H. Kobayashi, et al. The pharmacokinetic characteristics of glycolated humanized anti-Tac Fabs are determined by their isoelectric points, Cancer Res., 1999, 59, 422–430 CAS.
- C. A. Boswell, et al. Effects of charge on antibody tissue distribution and pharmacokinetics, Bioconjugate Chem., 2010, 21, 2153–2163 CrossRef CAS PubMed.
- M. Dellian, F. Yuan, V. S. Trubetskoy, V. P. Torchilin and R. K. Jain, Vascular permeability in a human tumour xenograft: molecular charge dependence, Br. J. Cancer, 2000, 82, 1513–1518 CrossRef CAS PubMed.
- T. Igawa, et al. Reduced elimination of IgG antibodies by engineering the variable region, Protein Eng., Des. Sel., 2010, 23, 385–392 CrossRef CAS PubMed.
- G. Hong, M. I. Bazin-Redureau and J. M. G. Scherrmann, Pharmacokinetics and organ distribution of cationized colchicine- specific IgG and Fab fragments in rat, J. Pharm. Sci., 1999, 88, 147–153 CrossRef CAS PubMed.
- G. Hong, M. I. Bazin-Redureau and J. M. Scherrmann, Pharmacokinetics and organ distribution of cationized colchicine-specific IgG and Fab fragments in rat, J. Pharm. Sci., 1999, 88, 147–153 CrossRef CAS PubMed.
- Y. Tabata, T. Kawai, Y. Murakami and Y. Ikada, Electric charge influence of dextran derivatives on their tumor accumulation after intravenous injection, Drug Delivery, 1997, 4, 213–221 CrossRef CAS.
- G. M. Thurber, M. M. Schmidt and K. D. Wittrup, Factors determining antibody distribution in tumors, Trends Pharmacol. Sci., 2008, 29, 57–61 CAS.
- T. Stylianopoulos, K. Soteriou, D. Fukumura and R. K. Jain, Cationic nanoparticles have superior transvascular flux into solid tumors: insights from a mathematical model, Ann. Biomed. Eng., 2013, 41, 68–77 CrossRef PubMed.
- H. Wiig, C. C. Gyenge and O. Tenstad, The interstitial distribution of macromolecules in rat tumours is influenced by the negatively charged matrix components, J. Physiol., 2005, 567, 557–567 CrossRef CAS PubMed.
- S. Krasnici, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels, Int. J. Cancer, 2003, 105, 561–567 CrossRef CAS PubMed.
- M. W. Traxlmayr, et al. Strong enrichment of aromatic residues in binding sites from a charge-neutralized hyperthermostable Sso7d scaffold library, J. Biol. Chem., 2016, 291, 22496–22508 CrossRef CAS PubMed.
- C. N. Pace, Measuring and increasing protein stability, Trends Biotechnol., 1990, 8, 93–98 CrossRef CAS PubMed.
- J. Chen, N. Sawyer and L. Regan, Protein-protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area, Protein Sci., 2013, 22, 510–515 CrossRef CAS PubMed.
- K. Nord, et al. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain, Nat. Biotechnol., 1997, 15, 772–777 CrossRef CAS PubMed.
- E. Wahlberg, et al. An affibody in complex with a target protein: structure and coupled folding, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3185–3190 CrossRef CAS PubMed.
- C. Grönwall, et al. Affibody-mediated transferrin depletion for proteomics applications, Biotechnol. J., 2007, 2, 1389–1398 CrossRef PubMed.
- P.-A. Löfdahl, O. Nord, L. Janzon and P.-A. Nygren, Selection of TNF-alpha binding affibody molecules using a beta-lactamase protein fragment complementation assay, New Biotechnol., 2009, 26, 251–259 CrossRef PubMed.
- A. Orlova, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule, Cancer Res., 2006, 66, 4339–4348 CrossRef CAS PubMed.
- M. Wikman, et al. Selection and characterization of an HIV-1 gp120-binding affibody ligand, Biotechnol. Appl. Biochem., 2006, 45, 93 CrossRef CAS PubMed.
- R. P. Baum, et al. Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules, J. Nucl. Med., 2010, 51, 892–897 CrossRef PubMed.
- V. Tolmachev, I. Velikyan, M. Sandström and A. Orlova, A HER2-binding Affibody molecule labelled with 68Ga for PET imaging: direct in vivo comparison with the 111In-labelled analogue, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 1356–1367 CrossRef CAS PubMed.
- R. Zielinski, et al. Affitoxin—A Novel Recombinant, HER2-specific, Anticancer Agent for Targeted Therapy of HER2-positive Tumors, J. Immunother., 2009, 32, 817–825 CrossRef CAS PubMed.
- A. Orlova, T. A. Tran, T. Ekblad, A. E. Karlström and V. Tolmachev, 186Re-maSGS-ZHER2:342, a potential Affibody conjugate for systemic therapy of HER2-expressing tumours, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 260–269 CrossRef CAS PubMed.
- I. Lyakhov, et al. HER2- and EGFR-specific affiprobes: Novel recombinant optical probes for cell imaging, ChemBioChem, 2010, 11, 345–350 CrossRef CAS PubMed.
- K. Nord, E. Gunneriusson, M. Uhlén and P. A. Nygren, Ligands selected from combinatorial libraries of protein A for use in affinity capture of apolipoprotein A-1(M) and Taq DNA polymerase, J. Biotechnol., 2000, 80, 45–54 CrossRef CAS PubMed.
- M. Friedman, et al. Directed evolution to low nanomolar affinity of a tumor-targeting epidermal growth factor receptor-binding affibody molecule, J. Mol. Biol., 2008, 376, 1388–1402 CrossRef CAS PubMed.
- J. Feldwisch, et al. Design of an optimized scaffold for affibody molecules, J. Mol. Biol., 2010, 398, 232–247 CrossRef CAS PubMed.
- B. A. Case and B. J. Hackel, Synthetic and natural consensus design for engineering charge within an affibody targeting epidermal growth factor receptor, Biotechnol. Bioeng., 2016, 113, 1628–1638 CrossRef CAS PubMed.
- B. Steipe, B. Schiller, A. Pluckthun and S. Steinbacher, Sequence statistics reliably predict stabilizing mutations in a protein domain, J. Mol. Biol., 1994, 240, 188–192 CrossRef CAS PubMed.
- B. Steipe, Consensus-based engineering of protein stability: from intrabodies to thermostable enzymes, Methods Enzymol., 2004, 388, 176–186 CAS.
- Y.-S. Kim, R. Bhandari, J. R. Cochran, J. Kuriyan and K. D. Wittrup, Directed evolution of the epidermal growth factor receptor extracellular domain for expression in yeast, Proteins, 2006, 62, 1026–1035 CrossRef CAS PubMed.
- Y. Yarden, The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities, Eur. J. Cancer, 2001, 37(Suppl 4), S3–S8 CrossRef CAS PubMed.
- R. S. Herbst, Review of epidermal growth factor receptor biology, Int. J. Radiat. Oncol., Biol., Phys., 2004, 59, 21–26 CrossRef CAS PubMed.
- C. Yewale, D. Baradia, I. Vhora, S. Patil and A. Misra, Epidermal growth factor receptor targeting in cancer: A review of trends and strategies, Biomaterials, 2013, 34, 8690–8707 CrossRef CAS PubMed.
- P. Laurent-Puig, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer, J. Clin. Oncol., 2009, 27, 5924–5930 CrossRef CAS PubMed.
- M. Scartozzi, et al. Epidermal Growth Factor Receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: a fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis, BMC Cancer, 2009, 9, 303 CrossRef PubMed.
- M. Moroni, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study, Lancet Oncol., 2005, 6, 279–286 CrossRef CAS PubMed.
- R. Pirker, et al. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: Analysis of data from the phase 3 FLEX study, Lancet Oncol., 2012, 13, 33–42 CrossRef CAS PubMed.
- J.-Y. Douillard, et al. Relationship between EGFR expression, EGFR mutation status, and the efficacy of chemotherapy plus cetuximab in FLEX study patients with advanced non–small-cell lung cancer, J. Thorac. Oncol., 2014, 9, 717–724 CrossRef CAS PubMed.
- F. R. Hirsch, et al. Combination of EGFR gene copy number and protein expression predicts outcome for advanced non-small-cell lung cancer patients treated with gefitinib, Ann. Oncol., 2007, 18, 752–760 CrossRef CAS PubMed.
- H. J. Lee, et al. Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer, Br. J. Cancer, 2015, 112, 103–111 CrossRef CAS PubMed.
- E. Nordberg, et al. In vivo and in vitro uptake of 111In, delivered with the affibody molecule (ZEGFR:955)2, in EGFR expressing tumour cells, Oncol. Rep., 2008, 19, 853–857 CAS.
- V. Tolmachev, et al. Imaging of EGFR expression in murine xenografts using site-specifically labelled anti-EGFR 111In-DOTA-Z EGFR:2377 Affibody molecule: aspect of the injected tracer amount, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 613–622 CrossRef PubMed.
- Z. Miao, et al. PET of EGFR Expression with an 18F-Labeled Affibody Molecule, J. Nucl. Med., 2012, 53, 1110–1118 CrossRef CAS PubMed.
- J. Garousi, et al. PET imaging of epidermal growth factor receptor expression in tumours using 89Zr-labelled ZEGFR:2377 affibody molecules, Int. J. Oncol., 2016, 48, 1325–1332 CrossRef CAS PubMed.
- X. Su, et al. Comparison of two site-specifically 18F-labeled Affibodies for PET imaging of EGFR positive tumors, Mol. Pharmaceutics, 2014, 11, 3947–3956 CrossRef CAS PubMed.
- B. J. Hackel, R. H. Kimura and S. S. Gambhir, Use of 64Cu-labeled fibronectin domain with EGFR-overexpressing tumor xenograft: molecular imaging, Radiology, 2012, 263, 179–188 CrossRef PubMed.
- M. A. Kruziki, B. A. Case, J. Y. Chan, D. Yee and B. J. Hackel, A 64Cu-labeled Gp2 domain for PET imaging of epidermal growth factor receptor, Mol. Pharmaceutics, 2016, 13, 3747–3755 CrossRef CAS PubMed.
- L. Huang, et al. SPECT imaging with 99mTc-labeled EGFR-specific nanobody for in vivo monitoring of EGFR expression, Mol. Imaging Biol., 2008, 10, 167–175 CrossRef PubMed.
- L. O. T. Gainkam, et al. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT, J. Nucl. Med., 2008, 49, 788–795 CrossRef CAS PubMed.
- W. Ping Li, L. A. Meyer, D. A. Capretto, C. D. Sherman and C. J. Anderson, Receptor-binding, biodistribution, and metabolism studies of 64Cu-DOTA-cetuximab, a PET-imaging agent for epidermal growth-factor receptor-positive tumors, Cancer Biother. Radiopharm., 2008, 23, 158–171 CrossRef PubMed.
- W. Cai, et al. Quantitative PET of EGFR expression in xenograft-bearing mice using 64Cu-labeled cetuximab, a chimeric anti-EGFR monoclonal antibody, Eur. J. Nucl. Med. Mol. Imaging, 2007, 34, 850–858 CrossRef CAS PubMed.
- S. Bhattacharyya, et al. Zirconium-89 labeled panitumumab: A potential immuno-PET probe for HER1-expressing carcinomas, Nucl. Med. Biol., 2013, 40, 451–457 CrossRef CAS PubMed.
- R. Chakravarty, et al. Matching the decay half-life with the biological half-life: ImmunoPET imaging with44Sc-labeled Cetuximab Fab fragment, Bioconjugate Chem., 2014, 25, 2197–2204 CrossRef CAS PubMed.
- G. Niu, Z. Li, J. Xie, Q.-T. Le and X. Chen, PET of EGFR antibody distribution in head and neck squamous cell carcinoma models, J. Nucl. Med., 2009, 50, 1116–1123 CrossRef CAS PubMed.
- A. A. Memon, et al. Positron emission tomography (PET) imaging with [ 11C]-labeled erlotinib: A micro-PET study on mice with lung tumor xenografts, Cancer Res., 2009, 69, 873–878 CrossRef CAS PubMed.
- M. R. Zhang, et al. [11C]Gefitinib ([11C]Iressa): Radiosynthesis, In Vitro uptake, and In Vivo imaging of intact murine fibrosarcoma, Mol. Imaging Biol., 2010, 12, 181–191 CrossRef PubMed.
- P. Slobbe, et al. Development of [18F]afatinib as new TKI-PET tracer for EGFR positive tumors, Nucl. Med. Biol., 2014, 41, 749–757 CrossRef CAS PubMed.
- H. Wang, et al. Assessment of 11C-labeled-4-N-(3-bromoanilino)-6,7-dimethoxyquinazoline as a positron emission tomography agent to monitor epidermal growth factor receptor expression, Cancer Sci., 2007, 98, 1413–1416 CrossRef CAS PubMed.
- I. Velikyan, et al. Preparation and evaluation of (68)Ga-DOTA-hEGF for visualization of EGFR expression in malignant tumors, J. Nucl. Med., 2005, 46, 1881–1888 CAS.
- H. Kareem, et al. Blocking EGFR in the liver improves the tumor-to-liver uptake ratio of radiolabeled EGF, Tumor Biol., 2010, 31, 79–87 CrossRef CAS PubMed.
- R. J. Pogulis, A. N. Vallejo and L. R. Pease, In vitro recombination and mutagenesis by overlap extension PCR, Methods Mol. Biol., 1996, 57, 167–176 CAS.
- L. A. Stern, et al. Geometry and expression enhance enrichment of functional yeast-displayed ligands via cell panning, Biotechnol. Bioeng., 2016, 113, 2328–2341 CrossRef CAS PubMed.
- C. Jäckel, J. D. Bloom, P. Kast, F. H. Arnold and D. Hilvert, Consensus protein design without phylogenetic bias, J. Mol. Biol., 2010, 399, 541–546 CrossRef PubMed.
- A. K. Shukla and U. Kumar, Positron emission tomography: An overview, J. Med. Phys., 2006, 31, 13–21 CrossRef CAS PubMed.
- S. Ait-Mohand, et al. Evaluation of 64Cu-labeled bifunctional Chelate-Bombesin conjugates, Bioconjugate Chem., 2011, 22, 1729–1735 CrossRef CAS PubMed.
- Z. Miao, G. Ren, L. Hongguang, L. Jiang and Z. Cheng, Small-animal PET imaging of human epidermal growth factor receptor positive tumor with a 64Cu labeled affibody protein, Bioconjugate Chem., 2010, 21, 947–954 CrossRef CAS PubMed.
- P. Zhao, et al. Molecular imaging of hepatocellular carcinoma xenografts with epidermal growth factor receptor targeted affibody probes, BioMed Res. Int., 2013, 2013, 1–11 Search PubMed.
- X. Su, et al. Comparison of two site-specifically 18F-labeled affibodies for PET imaging of EGFR positive tumors, Mol. Pharmaceutics, 2014, 11, 3947–3956 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental data are available online including a summary of clonal biophysical properties, radio chromatography for serum stability, and mass spectrometry. See DOI: 10.1039/c7me00095b |
| This journal is © The Royal Society of Chemistry 2018 |