
Computational transport analysis of antibody-drug conjugate bystander effects and payload tumoral distribution: implications for therapy†
Eshita
Khera‡
a,
Cornelius
Cilliers‡
a,
Sumit
Bhatnagar
a and
Greg M.
Thurber
 *ab
*ab
aDepartment of Chemical Engineering, University of Michigan, 2800 Plymouth Rd., Ann Arbor, MI 48109, USA. E-mail: gthurber@umich.edu; Tel: +734 764 8722
bDepartment of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA
First published on 17th October 2017
Abstract
Antibody drug conjugates (ADCs) have a proven clinical record with four FDA approved drugs and dozens more in clinical trials. However, a better understanding of the relationship between delivery and efficacy of ADCs is needed to improve the rate of successful clinical development. Recent evidence indicates that heterogeneous distribution can play a large role in the efficacy of these drugs. However, the impact of the drug payload, particularly the ability of the payload to diffuse outside of the original targeted cell into adjacent cells (the bystander effect), is not completely understood. Given the challenges in directly measuring the payload distribution within tumors, we developed a predictive computational model to study payload distribution as a function of antibody dose, payload dose, and payload properties. The computational results indicate that: 1) the heterogeneous tumoral distribution of ADCs impacts efficacy, and increasing the antibody dose improves penetration and efficacy. 2) The increased penetration of payloads with bystander effects can partially compensate for poor antibody penetration, but larger antibody doses still result in further improvement. This occurs because of the higher efficiency of direct cell killing than bystander killing. 3) Bystander effects are important for killing antigen negative cells, and an optimum in physicochemical properties exists. Payloads with a balance in cellular uptake versus tissue diffusion enter cells fast enough to avoid tumor washout but slow enough to reach distant cells. Therefore, optimizing the antibody dose, payload dose, and payload physicochemical properties results in ideal delivery to the site of action and maximum efficacy.
Design, System, ApplicationAntibody-drug conjugates (ADCs) have emerged as sophisticated therapeutics for molecular targeting of cancer, but there is limited understanding of how the heterogeneous, perivascular distribution of ADCs in tumors relates to their overall efficacy. Here we present a computational analysis of payload delivery to the site of action using a partial differential equation model of antibody and payload distribution. The tumor intracellular payload concentration is a function of controllable design parameters: antibody dose, payload dose (drug-antibody ratio, DAR), and payload physicochemical properties that determine its bystander potential. Optimization involves adjusting these parameters based on the target tissue (receptor expression, internalization rate, etc.) to reach the maximum number of cells with a toxic dose. A main constraint is the payload dose, which correlates with toxicity. The results highlight the importance of increasing ADC tissue penetration irrespective of bystander effects, and the payload physicochemical properties can be independently modified to maximize bystander killing for antigen negative cells. When the Damköhler number, a dimensionless number describing payload cellular uptake versus tumor diffusion, is approximately 3, bystander payloads are able to accumulate to their maximum levels throughout the tumor. This work presents strategies for optimizing payload physicochemical properties to match payload potency with tumor distribution. |
Introduction
Antibody-drug conjugates (ADCs) form a promising class of targeted therapeutics, with over 70 in various stages of the clinical pipeline. Until recently, Adcetris and Kadcyla were the only FDA-approved ADCs, with several ADCs failing in phase II or III clinical trials.1,2 In late 2017, the FDA also approved Besponsa (inotuzumab ozogamicin), and re-approved Mylotarg (gemtuzumab ozogamicin), bringing the total number of FDA-approved ADCs up to four. These recent approvals and early stage clinical success of several next-generation ADCs, such as sacituzumab govitecan,3,4 are encouraging, but major challenges in achieving consistently high clinical response rates still exist. The future clinical and commercial success of ADCs requires increasing the efficiency of development, which can be achieved through a better understanding of the pharmacokinetics (PK) and pharmacodynamics (PD) of these complex prodrugs.Structurally, ADCs are composed of three parts: the antibody, linker, and cytotoxic payload. Significant improvements have been made to each part, including more stable linkers,5,6 site-specific conjugation chemistries,7,8 controlled drug loading,9–12 and more toxic payloads.13 Although these advancements in the biophysical properties of ADCs have improved their therapeutic index,6,8,14,15 systemic toxicity from the small molecule payload continues to be problematic,16 and several recent late-stage ADC failures may have been avoided with marginal gains in tolerability.2 Therefore, further optimization of ADCs to better balance potency and toxicity may improve the likelihood of their clinical success.
The heterogeneous, perivascular tumor distribution of antibodies and ADCs is widely known;17–20 however, how this distribution relates to efficacy is not well understood.21 The complex, multi-step process of payload delivery to the site of action includes blood flow to the tumor, extravasation of intact ADC from the blood, diffusion through the interstitium, cellular binding, internalization and degradation, release of the payload into the lysosome, and diffusion/transport from the lysosome into the cytoplasm/nucleus where the payload can exert its cytotoxic effect. Previously, we developed a combined tissue and physiologically-based pharmacokinetic (PBPK) model to describe the cellular and tissue-level transport following systemic distribution of T-DM1.22 This model describes the tumor distribution of an ADC (T-DM1) when co-administered with the unconjugated or ‘naked’ antibody backbone (trastuzumab). We found that the distribution of T-DM1 alone was heterogeneous and perivascular; however, when co-administered with trastuzumab, T-DM1 tumor penetration was dramatically improved.22 Since the toxicity of the payload is typically dose-limiting for ADCs,16 the T-DM1 dose cannot be increased, but adding trastuzumab increases the total antibody dose and tumor penetration of the ADC, resulting in a greater number of targeted cells. However, it lowers the average amount of payload that each targeted cell receives. This is theoretically analogous to lowering the ADC drug to antibody ratio (DAR). To better understand the relationship between ADC distribution and cellular payload delivery, we retrospectively identified and analyzed several tumor growth studies in the literature where the same overall payload dose was given with different combinations of ADC DAR and dose (i.e. DAR2 dosed at 2 mg kg−1vs. DAR4 dosed at 1 mg kg−1). These studies used a variety of ADC systems with different targets, antibodies, linkers, and payloads (with and without bystander effects). Our analysis of these studies suggests that ADC tumoral distribution plays an important role in efficacy for a constant payload dose.
Although ADCs distribute heterogeneously through the tissue, a payload that exhibits bystander effects (subsequently referred to as bystander payloads) would penetrate farther into the tumor, thereby improving its distribution. Once released from the ADC, bystander payloads can permeate out of the cell, diffuse farther into the tumor, and can be taken up in a different cell, meaning that the payload can reach cells that were not targeted by intact ADC (either antigen negative cells or antigen positive cells not reached by the ADC).13,23 Payloads that do not exhibit bystander effects (non-bystander payloads) are either too hydrophilic (often charged) or too large to rapidly diffuse through cell membranes, meaning they are ‘trapped’ in cells targeted by the ADC. Even if some non-bystander payload does escape the targeted cell, its influx rate into adjacent cells is slow, and the payload will be washed out of the tumor before it has a chance to enter another cell. Bystander payloads improve efficacy in mosaic (mixed antigen positive and antigen negative cells) in vitro co-culture models24 and in vivo tumor xenograft models.13,23 The heterogeneous expression of many targets in clinical samples highlights the importance of bystander payloads for killing antigen negative cells. However, little is known about payload transport properties and their resulting cellular and tissue scale distribution. While bystander payloads are theoretically capable of diffusing deeper in tumor tissue even if the delivery vehicle (in this case an antibody) is heterogeneously distributed, several examples from our previous analysis22 showed improved efficacy with better ADC tumor penetration, even when carrying bystander payloads (e.g. MMAE). Thus, we sought to analyze the transport of ADCs and their payloads (with and without bystander effects) to elucidate why ADCs possessing bystander effects still benefit from higher antibody penetration and to help guide the development of next generation ADCs.
Materials and methods
Building on our previous work, we collected additional preclinical studies from the literature and subdivided the results depending on the payload to isolate the impacts of bystander effects and ADC tumoral distribution on efficacy. In this work, we primarily focused on transport simulations for two model payloads: the bystander payload, monomethyl auristatin E (MMAE), and a non-bystander payload, lysine-SMCC-DM1 (DM1).Estimation of payload pharmacokinetic parameters
All antibody specific parameters were selected for trastuzumab as a model antibody and were based on our previously published work.22 Payload transport parameters, specifically the membrane permeability rate and extracellular diffusion rate, were estimated for each payload, as described below. All payloads bound the same target (here tubulin) with the same binding and dissociation rates, obtained from literature.24,25Parallel artificial membrane permeability assay (PAMPA)
Few direct measurements of the cellular uptake rates of small molecules are available (but more are being obtained26). In the absence of these measurements, we estimated the cellular uptake values using permeability data from PAMPA assays. Detailed procedures, calculations and values can be found in the ESI,† but briefly, we used an interpolation of PAMPA data from molecules with known cellular uptake rates to correlate PAMPA permeability with cellular kinetics (Table S1†). For payloads without PAMPA data, we estimated the uptake rate from molecules with similar physicochemical properties (Tables S2 and S3†).Extracellular payload diffusion coefficient
The extracellular diffusion coefficient of each payload was estimated from the physicochemical properties of the payload using a mathematical expression adapted from Pruijn et al.27 (see ESI† for details). Briefly, the molecular weight, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D (pH 7.4), number of hydrogen bond donors and acceptors of each payload were calculated in MarvinSketch (ChemAxon) and are listed in Table S4.† The diffusion coefficient, Dmcl, was calculated by substituting these parameters into the following equation:27
D (pH 7.4), number of hydrogen bond donors and acceptors of each payload were calculated in MarvinSketch (ChemAxon) and are listed in Table S4.† The diffusion coefficient, Dmcl, was calculated by substituting these parameters into the following equation:27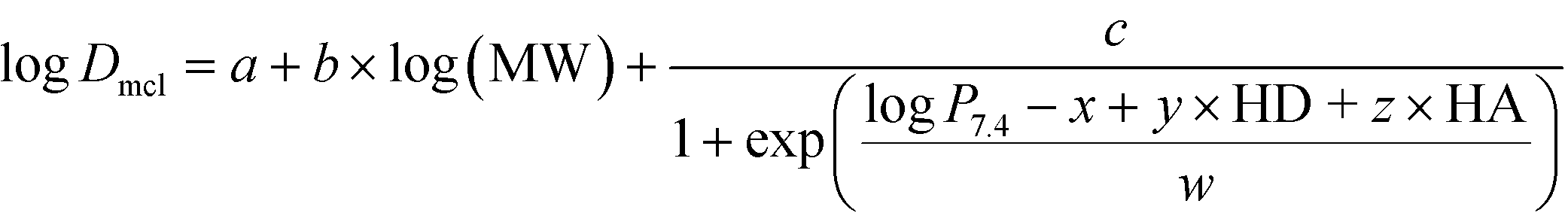
Model specific-parameters (a, b, c, x, y, z, and w) were selected for the SiHa tumor cell line (Table S5†) to account for both transcellular and paracellular diffusion of the payload through the tumor.27 However, the diffusion coefficient obtained from this model represents the rate at equilibrium. Since the diffusion of payload in the tissue occurs transiently, we divided by the partition coefficient, R, of each payload to account for the free fraction in the interstitium, as described previously.28
The antibody and payload distribution model described here is a predictive model as all parameter values (Table 1) are taken from literature or estimated independently, and not fit to data (which currently do not exist for cellular-level bystander concentrations).
| Parameter | Value | Unit | Description | Reference |
|---|---|---|---|---|
| a k out,P = kin,P/(1 + R). | ||||
| R Krogh | 75 | μm | Krogh cylinder (tumor) radius | 67 |
| R Capillary | 8 | μm | Capillary radius | 72 |
| A | 0.43 | ND | Fraction of alpha clearance | 29 |
| k α | 0.0866 | h−1 | Clearance rate for alpha phase | 29 |
| k β | 0.0347 | h−1 | Clearance rate for beta phase | 29 |
| D ADC | 10 | μm2 s−1 | Antibody/ADC diffusivity | 29 |
| P ADC | 3 × 10−9 | m s−1 | Vascular permeability of antibody/ADC | 73 |
| Q | 0.0015 | mL g−1 s−1 | Blood flow rate to tumor | 74 |
| H | 0.45 | ND | Hematocrit | 75 |
| ε | 0.24 | ND | Tumor void fraction | 76 |
| [Ag]0 | 0.833 | μM | Initial antigen concentration | 22 |
| R s | 2.75 × 10−5 | μM s−1 | Antigen recycle rate | 29 |
| k on,Ab | 7.1 × 105 | M−1 s−1 | Antibody/ADC binding rate | 77 |
| k off, Ab | 3.5 × 10−4 | s−1 | Antibody/ADC dissociation rate | 77 |
| K d | 0.5 | nM | Antibody/ADC dissociation constant | 77 |
| k e | 3.3 × 10−5 | s−1 | Antibody/ADC net internalization rate (total internalization·unrecycled fraction) | 20, 31, 32 |
| k deg | 8.0 × 10−6 | s−1 | ADC lysosomal degradation rate | 78 |
| DAR | 1–4 | ND | Drug to antibody ratio | Varied |
| k in,P (DM1) | 5.95 × 10−5 | s−1 | Payload influx rate | Estimated |
| k in,P (MMAE) | 1.41 × 10−3 | s−1 | Payload influx rate | Estimated |
| k out,P (DM1)a | 3.94 × 10−5 | s−1 | Payload efflux rate | Estimated |
| k out,P (MMAE)a | 6.87 × 10−4 | s−1 | Payload efflux rate | Estimated |
| D P (DM1) | 9.8 | μm2 s−1 | Payload diffusion coefficient | 27 |
| D P (MMAE) | 14.8 | μm2 s−1 | Payload diffusion coefficient | 27, 79 |
| P P | 1 × 10−6 | m s−1 | Vascular permeability of free payload | 29 |
| k on,P | 8333 | M−1 s−1 | Payload binding rate | 80 |
| k off,P | 0.003 | s−1 | Payload dissociation rate | 80 |
| ε p | 0.44 | ND | Cell void fraction | 28, 81 |
| P target | 20 | μM | Microtubule concentration | 80 |
Mechanistic tumor distribution model for bystander killing
The current model for ADC distribution in a vascularized tumor builds on previously published work and is based on the Krogh cylinder geometry of tumor blood vessels.22,28,29 The free payload released from degraded antibodies is now explicitly modeled to account for bystander effects (Fig. 1a). The ability of the payload to diffuse out of cells is dependent on the properties of the payload, with smaller and more lipophilic payloads (e.g. MMAE) more readily able to enter adjacent cells than larger/hydrophilic payloads (e.g. Lys-SMCC-DM1), Fig. 1b. A one-dimensional Krogh cylinder was used since ADCs are permeability limited and extravasation is too slow for concentration gradients along the length of the capillary.29 Molecular transport in the model is diffusion-driven due to increased interstitial fluid pressure within the tumor that limits convective transport.30 State variables in the model include free antibody, free ADC, free antigen (Ag), bound antibody, bound ADC, internalized antibody, internalized ADC (IntADC), lysosomal payload (LysP), cytoplasmic payload (CytoP), bound payload (BoundP) and extracellular payload (ExtP), Fig. 1c.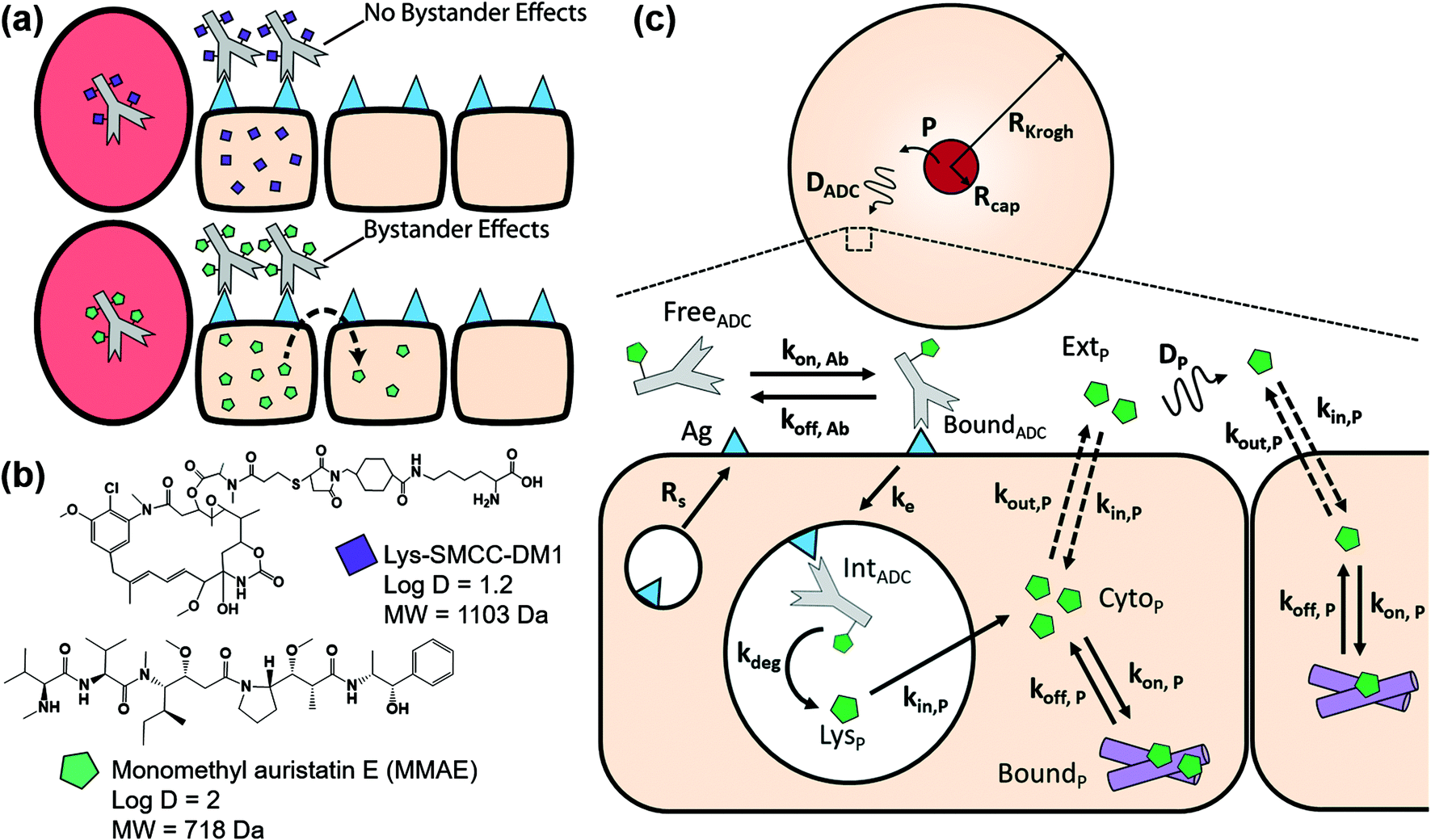 | ||
| Fig. 1 Graphical depiction of bystander effects and Krogh cylinder model. (a) Non-bystander payloads (purple squares) localize to ADC targeted cells while bystander payloads (green pentagons) are able to permeate cell membranes, diffuse farther into the tumor, and are taken up by untargeted cells. (b) Structure and physicochemical properties of non-bystander payload (Lys-SMCC-DM1, or DM1) and bystander payload (MMAE). logD estimated at pH 7.4 using ChemAxon MarvinSketch. MW, molecular weight; Da, Daltons. (c) Graphical depiction of Krogh cylinder model (top) incorporating bystander effects of payload at the cellular level (bottom). ADC in the plasma extravasates from the blood vessel (red circle) into the tumor, diffuses through the tissue (top), binds free receptor, gets internalized, and subsequently degraded (bottom). Upon degradation, free payload is released in the lysosome, which then permeates into the cytoplasm where it either binds microtubules or permeates out of the cell. Extracellular payload can diffuse farther into the tissue before being taken up by other cells or wash out of the tumor. Model definitions are listed in Table 1. Model equations are listed in the ESI.† | ||
Briefly, the antibody and ADC are delivered as bolus doses in the plasma, they extravasate from the blood vessel, diffuse through the tumor interstitium, and bind to free target receptor (e.g. HER2). Both ADC and antibody compete for the same pool of target receptors. Once bound, a fraction of the antibody/ADC internalizes (ke) into the lysosome and is subsequently degraded (kdeg). In these simulations, we used the net internalization rate, which was calculated by multiplying the HER2 internalization rate31 by the fraction not recycled,32 as published previously.20 The internalized target recycles back to the surface to maintain steady state surface receptor expression (Rs).20,22 Lysosomal processing and degradation of the ADC releases cytotoxic payload into the lysosome (LysP). The number of payload molecules released is proportional to the DAR of the ADC. The lysosomal payload irreversibly escapes (kin,P) into the cytoplasm (CytoP), where it can reversibly bind (kon,P, koff,P) to tubulin (BoundP) and mediate cytotoxicity. Cytoplasmic payload can partition into various other membranous organelles (e.g. lysosomes, endosomes, Golgi, mitochondria, etc.) and also efflux out of the cell (kout,P) into the extracellular space (ExtP), diffuse through the tumor (DP), and subsequently influx into surrounding cells (kin,P). The efflux rate of the payload from the cell is scaled by the fraction that is immobilized (e.g. partitioned into lipid membranes), determined by its partition coefficient, R, which was calculated from Poulin and Theil, and is valid for tumors.33,34 In this model, we focused on tubulin binding payloads (e.g. MMAE, DM1), but it is easily adaptable to payloads with alternate intracellular targets (e.g. DNA damage agents, topoisomerase inhibitors). Our simulations focused on a DAR of 4 or less since higher DAR ADCs can exhibit DAR-dependent clearance and/or deconjugation.9 The plasma concentration of free payload is considered to be negligible (i.e. no long-range bystander effects), and the extracellular concentration of payload at the capillary wall is adjusted to account for efflux from the tumor into the blood vessel. Detailed equations and boundary conditions can be found in the ESI.†
Results
Our simulations focused on two common ADC payloads, DM1 (released in the cell as Lys-SMCC-DM1) and MMAE, as models for non-bystander and bystander payloads, respectively (Fig. 1a and b). They also represent the payloads on two of the four FDA-approved ADCs, Kadcyla and Adcetris. Additional simulations were performed for other commonly used payloads with and without bystander effects (Fig. S1†). A graphical representation of the Krogh cylinder model is shown in Fig. 1c. The model captures the heterogeneous perivascular tissue distribution of ADCs, their cellular metabolism and the bystander targeting potential of the released cytotoxic payload.The ADC and antibody distribution model has previously been validated, and the payload distribution model was adapted from previous simulations, histology, and intra-vital microscopy imaging of small molecule distribution.28,29,35,36 In this work, the source of the small molecule is located at the site of ADC metabolism, rather than the blood vessel. Model simulations were used to predict the impact of increasing antibody dose and/or increasing payload dose (through increased DAR) on payload distribution and compared to tumor growth data from the literature. After ADC metabolism and payload release, apoptosis is induced when payload accumulates beyond a threshold intracellular concentration, below which the cancer cell may display some resistance to the cytotoxic effects of the payload.37,38 Since measuring and correlating cell death to the in vivo intracellular distribution of the payload is difficult, we instead established a ‘therapeutic threshold’ as a proxy to help visualize the impact of payload penetration in our simulations. (In reality, there is variability between intracellular drug concentrations and cell death even at the single-cell level due to differences in drug delivery39 and cellular response.40) This ‘therapeutic threshold’ was set to 150 nM based on literature measurements of the intracellular concentration for microtubule-inhibitor payloads at the IC50 value for several cell lines.13,15 The border between red/black and blue/white gradients in the colorbar outlines this threshold.
Increased tumor penetration of non-bystander ADCs improves efficacy
Recently, we analyzed the impact of increasing the antibody dose (which increases ADC tumoral penetration) with a constant total payload dose on efficacy, and demonstrated that increased tumor penetration is a key factor in improving ADC efficacy.22 However, our previous study did not take into account payload transport dynamics and did not explore the impact of bystander effects on ADC efficacy. Here, we include additional examples from the literature to compare results between bystander and non-bystander payloads. For commonly used non-bystander payloads like DM1 and MMAF, the intrinsic potency of the molecules is sufficiently high (sub-nanomolar IC50) that perivascular cells can accumulate the payload to concentrations highly exceeding the required ‘therapeutic threshold’, thereby leading to ‘overkill’ of these cells. If the same payload dose is delivered using a lower DAR, more cells are targeted with a toxic dose (Fig. 2a). For example, doubling the dose of the ADC but simultaneously halving the DAR (e.g. 2 mg kg−1 DAR4 versus 4 mg kg−1 DAR2) will deliver an identical total number of payloads; however, doubling the ADC dose allows the ADC to penetrate farther in the tumor, thereby targeting more cells, albeit with half the number of payloads in each cell (but still greater than the therapeutic threshold). We identified several in vivo tumor growth studies that fit these criteria for non-bystander payloads and found that for the same number of payload molecules delivered, the high ADC dose/low DAR strategy consistently resulted in greater tumor shrinkage and better efficacy (Fig. 2b, blue arrows). Several of these datasets were included in our past analysis,22 but additional examples have been published since that time, and we include all of them for context. It is important to note that all tumor growth studies throughout this work either demonstrated or stated that the xenograft tumors were resistant to treatment with unconjugated antibody alone (i.e. DAR0), indicating that tumor regression was from the cytotoxic payload only.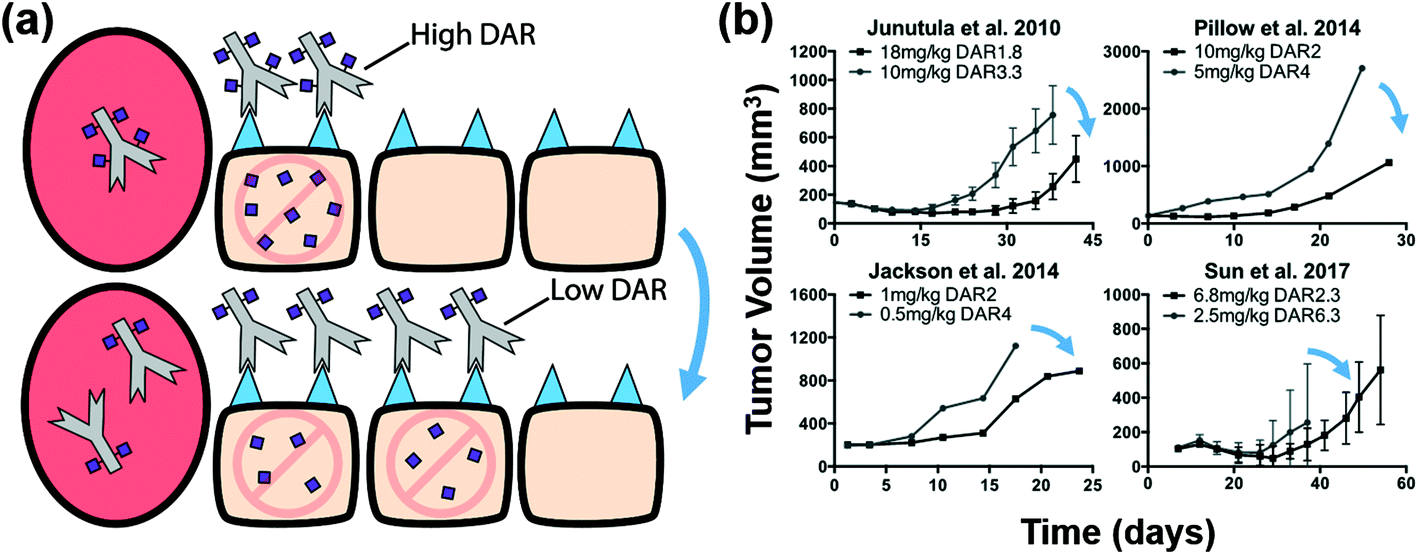 | ||
| Fig. 2 Distribution and efficacy of non-bystander payloads. (a) Graphic depiction of the same payload dose for non-bystander ADCs with a high and low DAR (top and bottom, respectively). Higher antibody doses result in better tumor penetration (bottom) but the lower DAR results in a lower average number of payloads per cell. This is still sufficient for cell death with high potency payloads. (b) Literature review of cases using a non-bystander ADC at the same payload dose but with different DARs/ADC doses. In all these cases the higher antibody dose (lower DAR) improved efficacy despite the same payload dose. Blue arrows indicate cases where the lower DAR improved efficacy at a constant payload dose. Tumor growth data taken from references.10,14,41,43 | ||
We simulated the distribution of the intact, internalized ADC (Fig. 3a) and the catabolized non-bystander payload DM1 (Fig. 3b) over 6 days using the Krogh cylinder model. A higher dose of ADC (10 mg kg−1) penetrates deeper through the tumor relative to a low ADC dose (2.5 mg kg−1) (Fig. 3a). For the same payload dose, deeper penetration of ADC resulted in payload delivery above the therapeutic threshold concentration farther into the tumor (Fig. 3b). A similar trend is observed across varying payload/ADC doses, though the difference in penetration radius is more apparent as the ADC dose increases (Fig. S2†). Since DM1 is a non-bystander payload, it cannot traverse the cell membrane and diffuse into adjacent cells, meaning the increased penetration of the payload (and better efficacy, Fig. 2b) is a direct consequence of better cellular targeting from improved ADC penetration. These results support that increasing tumor distribution of ADCs is a critical factor for improving the efficacy of non-bystander payloads.
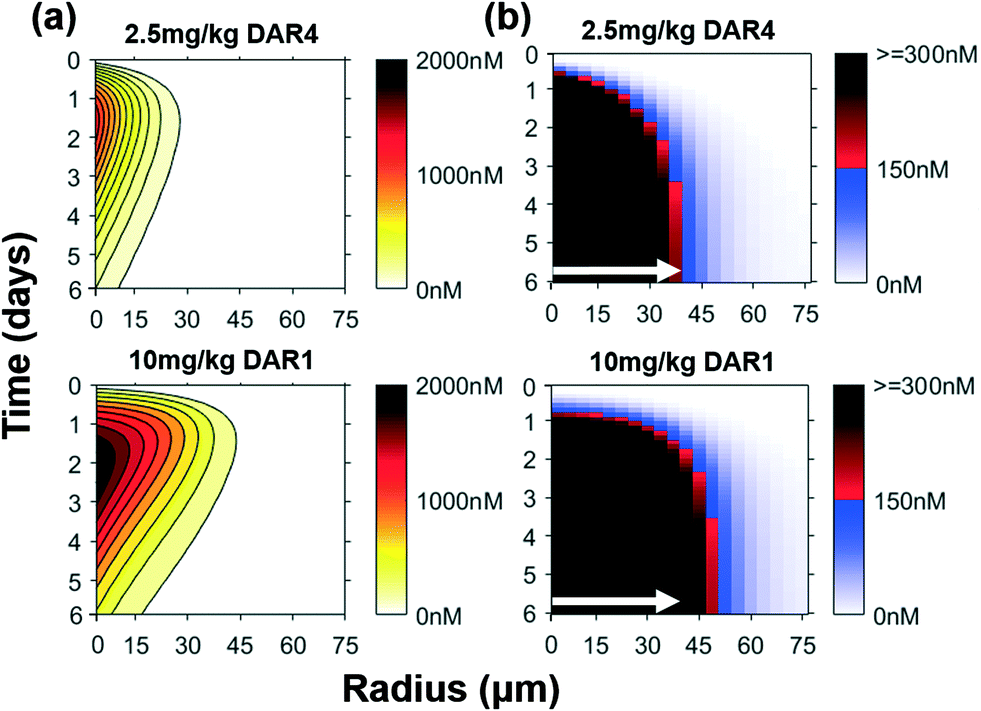 | ||
| Fig. 3 Simulations of improved ADC penetration with constant dose of payload. (a) The concentration of intact internalized ADC over time for a constant payload dose. The higher antibody dose in the 10 mg kg−1 of DAR1 drives ADC penetration farther into the tumor. (b) Cytoplasmic plus bound payload (i.e. intracellular payload) concentration over time for a non-bystander ADC. Despite a constant payload dose, the improved ADC distribution of the DAR1 ADC gives better payload distribution for non-bystander payloads. White arrows indicate distance where a 150 nM intracellular payload concentration is reached for the 2.5 mg kg−1 DAR4 dose (shown in both plots for comparison). | ||
The potency of an ADC is primarily determined by the concentration of payload that accumulates inside the cell (and the mechanism of action). A simple strategy to improve efficacy has been to increase the DAR of the ADC (and therefore total number of payload molecules delivered to the tumor),9,10 but within a moderate range (DAR 2–6).8,41 While this strategy improves the IC50in vitro, in vivo efficacy is more complex. For instance, increasing the DAR can lead to compounding issues such as DAR-dependent deconjugation/clearance that reduce tumor exposure. This provides diminishing returns for improving intratumoral payload concentration,9,10 though novel modifications to overcome these limitations are being explored.15,42 In the next sections, we simulated payload distribution when increasing the DAR for both non-bystander and bystander payloads.
Increased non-bystander payload dose (i.e. higher DAR) without improved ADC tissue penetration does not uniformly improve efficacy
The tumoral distribution of non-bystander payloads is primarily limited by ADC penetration distance, and because of the high potency of these payloads and the high expression level of many ADC targets, the cellular accumulation of payload may be higher than needed for cell death (i.e. “overkill”). Therefore, increasing the DAR for non-bystander payloads (at a constant ADC dose) may only further concentrate payload in perivascular tumor cells, exacerbating the ‘overkill’ of these cells but not necessarily altering the overall efficacy (Fig. 4a). Simulations for this scenario show that for an equivalent dose of ADC, increasing the DAR from 2 to 4 (i.e. doubling the total number of payloads delivered) showed little to no improvement in the penetration distance of the non-bystander payload (white arrows, Fig. 4b and Fig. S3†). Only a few cells at the fringe of the saturation front increase above the threshold. Interestingly, the in vivo data show mixed results when increasing DAR while keeping the antibody dose (i.e. penetration distance) the same (Fig. 4c). Junutula et al. showed that for the given dose of ADC, increasing the DAR nearly two-fold did not significantly decrease the tumor volume,14 which is consistent with our predictions. Jackson et al. found that a higher DAR led to a decrease in the efficacy,43 while Pillow et al. showed a marked increase in efficacy for the same increase in DAR.41 These conflicting studies are also highlighted to avoid potential confirmation bias in the results. We hypothesize that additional factors (DAR-dependent clearance and/or tissue-level effects in vascularization) may be responsible for these deviations when similar efficacy is predicted due to equivalent tissue penetration. Tissue penetration is one factor, albeit an important one, of many properties that impact ADC efficacy.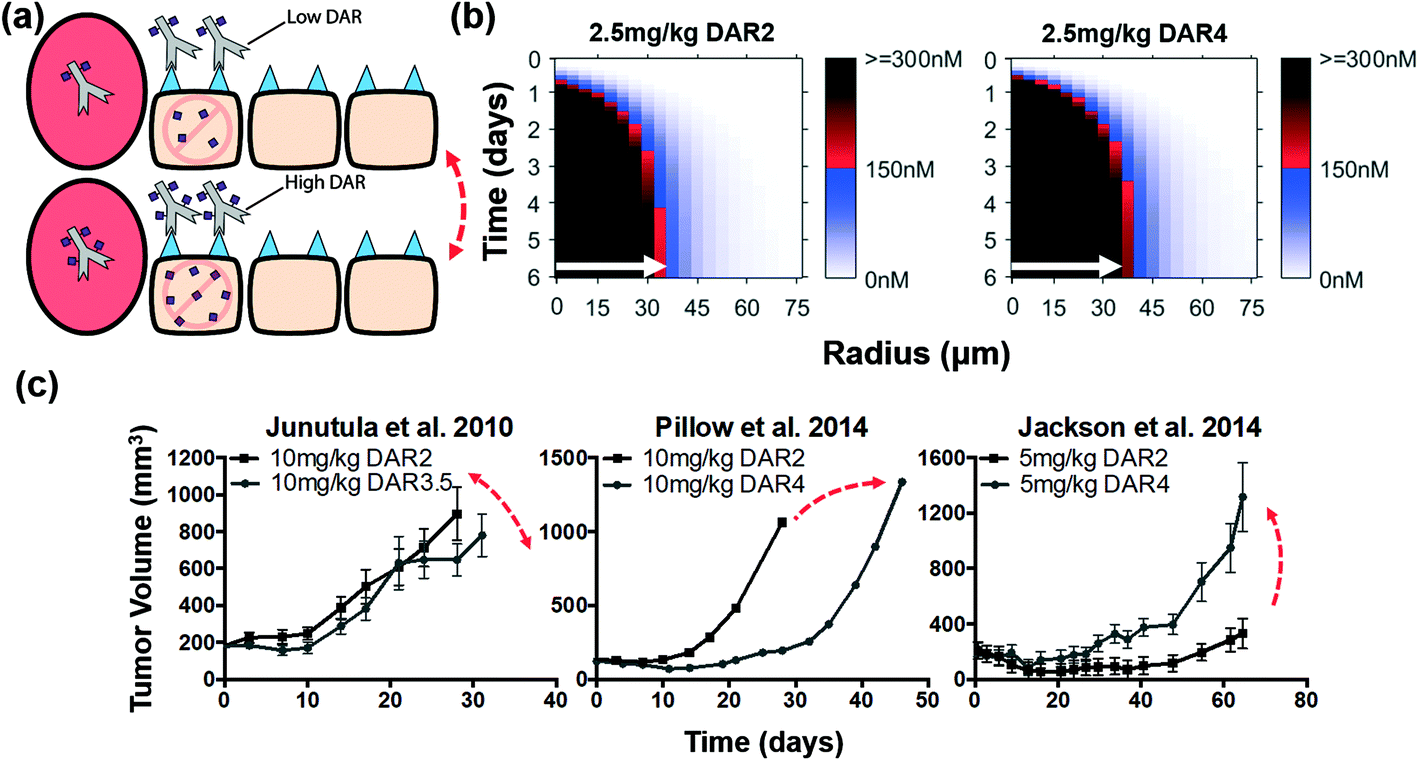 | ||
| Fig. 4 Distribution and efficacy of a constant dose of non-bystander ADCs with increasing DAR. (a) Graphic depiction of the same ADC dose given with increasing DAR. Non-bystander payloads are unable to diffuse farther into the tumor and an equivalent ADC dose targets the same number of cells resulting in similar efficacy between high and low DAR non-bystander ADCs. (b) Intracellular payload concentrations over time for non-bystander ADCs. For non-bystander ADCs, the payload distribution shows little improvement despite doubling the payload dose with the higher DAR. White arrows indicate distance where 150 nM intracellular payload concentration is reached for the 2.5 mg kg−1 DAR2 dose (shown in both plots for comparison). (c) Literature review of cases using a constant non-bystander ADC dose with increasing DAR. Despite delivering twice the payload dose, Junutula et al.14 showed comparable efficacy and Jackson et al.43 showed less efficacy with the higher DAR ADCs. However, Pillow et al.41 showed improved efficacy with the higher DAR, indicating tissue distribution is one factor of many properties impacting ADC efficacy. Red double-sided arrows indicate negligible change in efficacy. Single-sided arrows indicate opposing differences in efficacy. Tumor growth data taken from references.14,41,43 | ||
Increased bystander payload dose (i.e. higher DAR) improves efficacy independent of ADC tissue penetration
Bystander payloads like MMAE are able to efflux from the cell and diffuse into and target adjacent cells to exert their cytotoxic effects, a phenomenon known as the ‘bystander effect’.13,23 While an important role for these payloads is killing antigen negative cells,13,23 they also have the ability to penetrate farther into the tumor beyond ADC-saturated perivascular cells, and potentially improve ADC efficacy. In contrast to non-bystander payloads, bystander payloads that are delivered at a higher dose than needed for cell killing (‘overkill’) can diffuse deeper into the tissue, thus improving the penetration distance of the payload and killing additional cells (Fig. 5a). Literature review of cases using a constant bystander ADC dose with increasing DAR revealed that for an equivalent dose of bystander ADC, higher DAR consistently improves efficacy (Fig. 5b). Our model predictions show a progressively increasing penetration radius (red-black gradient, white arrows) with increasing DAR (Fig. 5c) and higher concentrations beyond the penetration border (blue gradient).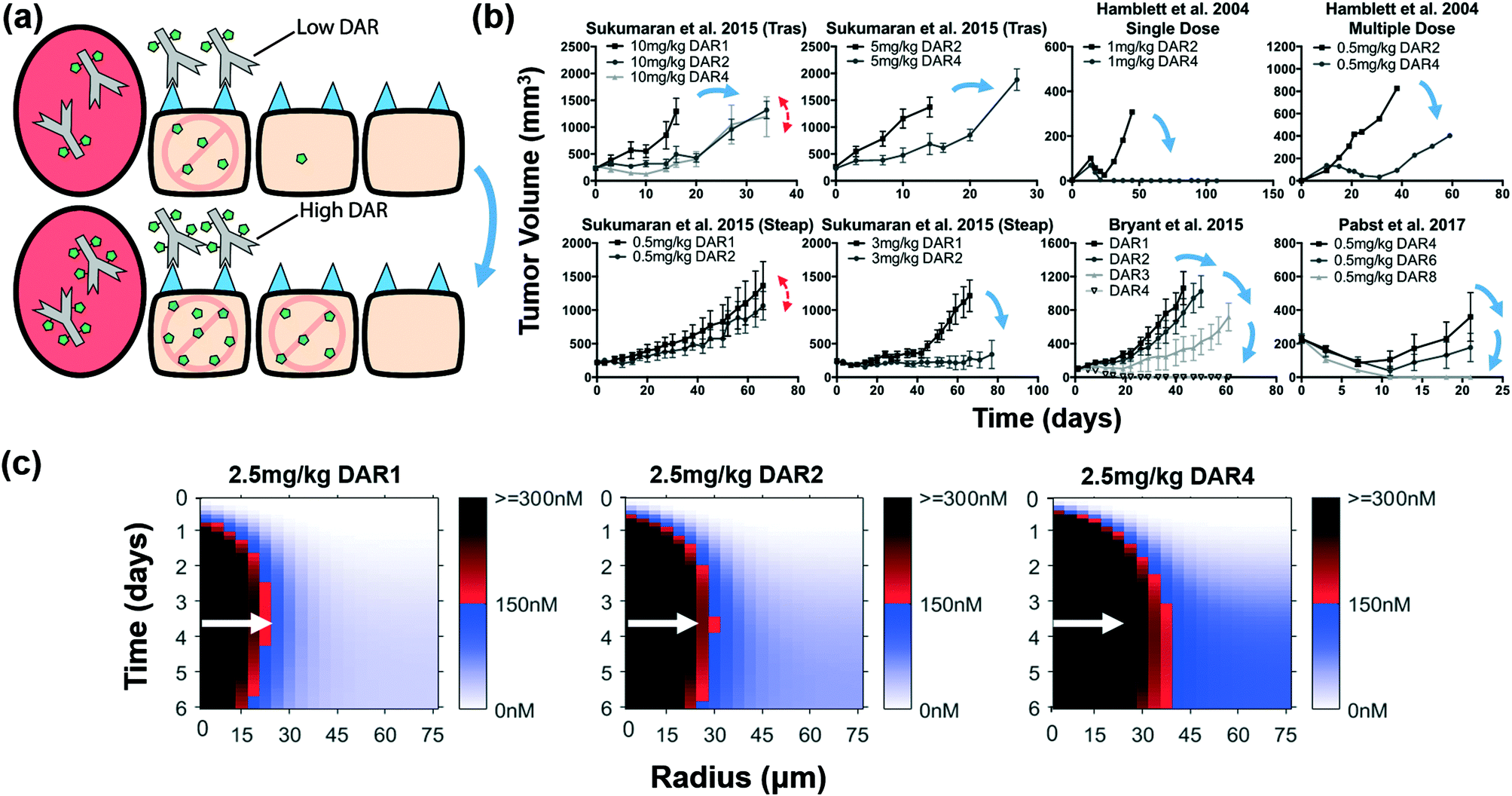 | ||
| Fig. 5 Distribution and efficacy of a constant dose of bystander ADCs with increasing DAR. (a) Graphic depiction of the same ADC dose given with increasing DAR. The same ADC dose targets the same number of cells; however, bystander payloads are able to diffuse farther into the tumor, reaching more cells than initially targeted. (b) Literature review of cases using a constant bystander ADC dose with increasing DAR. Higher DAR bystander ADCs at the same ADC dose gave improved efficacy in ten cases (blue arrows). Two cases showed similar efficacy (red double-sided arrows). (c) Intracellular payload concentrations over time at the same ADC dose with increasing DAR. Despite the ADC dose targeting the same number of cells, the payload reaches more cells with the higher DAR ADCs. White arrows indicate distance where 150 nM intracellular payload concentration is reached for the 2.5 mg kg−1 DAR1 dose (shown in all plots for comparison). Tumor growth data taken from references.9,11,44,82 | ||
The only exceptions in Fig. 5b were, first, when a single, low dose of the ADC (0.5 mg kg−1) in a low DAR range (1–2) was delivered (Sukumaran et al.44), where increased DAR showed little improvement. This could be due to the total number of bystander payloads delivered being too low to exhibit significant bystander killing, resulting in negligible differences in distribution (Fig. S4†). This data highlights an important concept for bystander payloads like MMAE – increasing efficacy by taking advantage of the bystander effect requires consideration of the total payload delivered to each cell (i.e. ADC dose and DAR).9 Bystander payloads can penetrate deeper into the tissue (blue gradient and improved red/black threshold), but they lower the concentration in the directly targeted cells.45 The second exception (Sukumaran et al.44) was when the DAR was doubled from 2 to 4 for a high dose of ADC (10 mg kg−1).44 At these doses, it is possible that the total payload dose is approaching the therapeutic threshold in all cells (Fig. S4†), or the tumor-to-tumor variability was high enough to obscure the improvement seen at earlier time (e.g. 10 days). Two additional cases in Sukumaran et al.44 agreed with our predictions (1 mg kg−1 of DAR1 or DAR2 for the increasing DAR case, and 3 mg kg−1 DAR2 or 6 mg kg−1 DAR1 for the constant payload case); however, we included these exceptions to avoid confirmation bias.
While the results of higher DAR with the same antibody dose are consistent with our predictions (Fig. 4 and 5), these comparisons are not necessarily relevant for clinical translation. Typically, the maximum tolerated dose (MTD) is limited by the payload dose, not the antibody, so increasing the payload dose (by increasing DAR) is often not clinically feasible.16 Excluding other important factors (such as DAR-dependent clearance), a constant antibody dose with higher DAR simply increases the toxic payload dose. An alternative strategy to improve efficacy is conjugating the maximum tolerated payload dose to a large antibody dose to ensure efficient ADC tissue penetration. Fig. 2 demonstrated this result for non-bystander ADCs, so we next examined if this strategy could also benefit ADCs with bystander payloads.
Improved tumor penetration of bystander payloads improves efficacy due to more efficient killing from direct cell targeting
The increased penetration of a bystander payload (Fig. 5) could compensate for poor ADC penetration. This raises the question of whether tumoral distribution of ADCs with bystander payloads is important or whether the payload bystander effect is sufficient to counteract the heterogeneous ADC distribution. We used our quantitative simulations and available literature data to address this question.Similar to a non-bystander payload, when delivering a higher dose of antibody at the same total payload dose, more cells are targeted, but the lower DAR results in a lower average number of payloads per cell. Bystander payloads can penetrate farther in the tumor in either case, but they may not achieve therapeutic concentrations in untargeted cells (Fig. 6a). A comparison of tumor growth studies from literature shows that the high ADC dose/low DAR strategy consistently shows increased efficacy (Fig. 6b), with one exception (reasons for which have been discussed in the previous section; see Fig. S5†). This suggests that directly targeting the cells via the higher ADC dose can achieve greater efficacy than diffusion of the bystander payload into the cell.
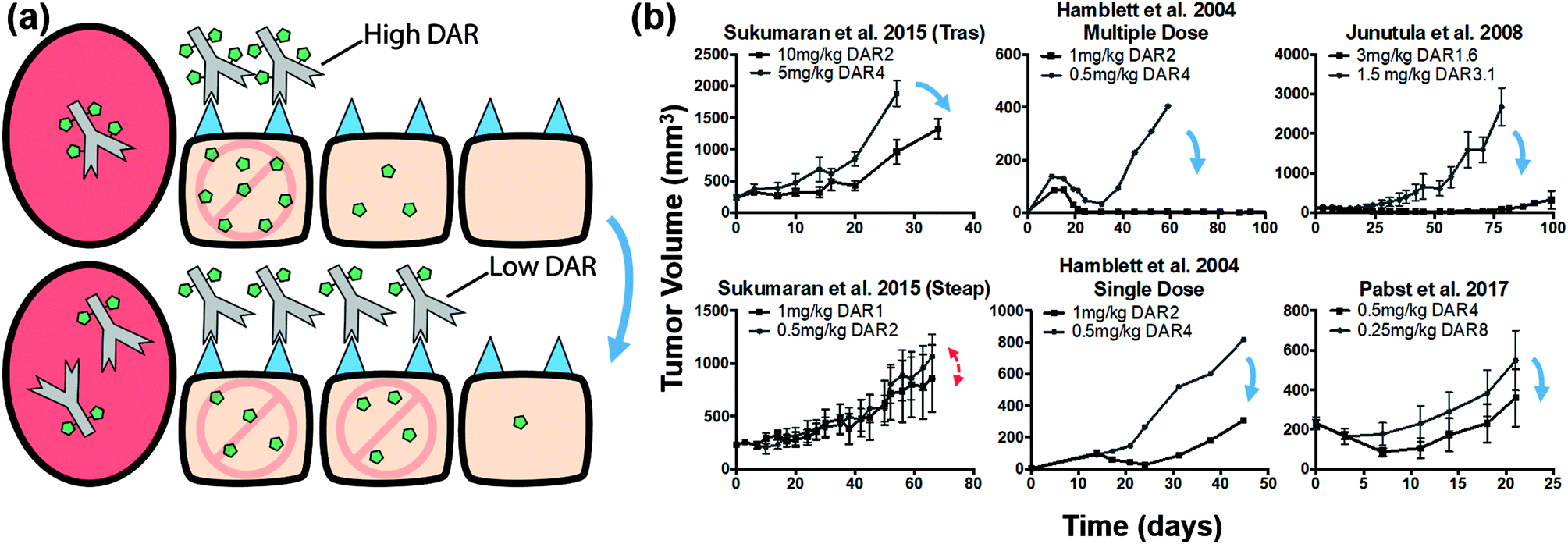 | ||
| Fig. 6 Distribution and efficacy of bystander ADCs given at constant payload dose with different DAR/antibody doses. (a) Graphic depiction of the same payload dose for ADCs with a high and low DAR (top and bottom, respectively). Higher ADC doses target more cells, but the lower DAR results in a lower average number of payloads per cell. This is still sufficient for cell death with high potency payloads. Bystander payloads are able to diffuse farther into the tumor; however, they may not achieve the high therapeutic concentrations in untargeted cells (top) that direct targeting of ADCs can achieve (bottom). (b) Literature review of cases using a bystander payload at the same payload dose. Although bystander payloads are able to diffuse farther into the tumor, in almost all cases the lower DAR ADC had improved efficacy, indicating direct cell targeting by the ADC may improve payload distribution more than bystander effects. Blue arrows indicate cases where the lower DAR (or higher ADC dose) improved efficacy despite a constant payload dose. Red double-sided arrows indicate similar efficacy. Tumor growth data taken from references.8,9,44,82 Tras, trastuzumab; Steap, anti-Steap. | ||
Conceptually, increased tumor tissue penetration of ADCs with bystander payloads improves efficacy by more efficiently delivering the payload via direct targeting (i.e. killing antigen positive cells that internalize the ADC) compared to released payload diffusing into adjacent cells for indirect cell killing as shown in Fig. 6a. Similar to the non-bystander case, our simulations in Fig. 7a show that 10 mg kg−1 of DAR1 bystander ADC results in a greater fraction of the tumor than a 2.5 mg kg−1 DAR4 (same total payload dose), with intracellular payload concentrations above the therapeutic threshold (red-black gradient), even though the payload diffuses through the entire tumor in both cases (blue gradient). Therefore, direct cell targeting enhances ADC payload delivery to the site of action better than bystander effects alone.
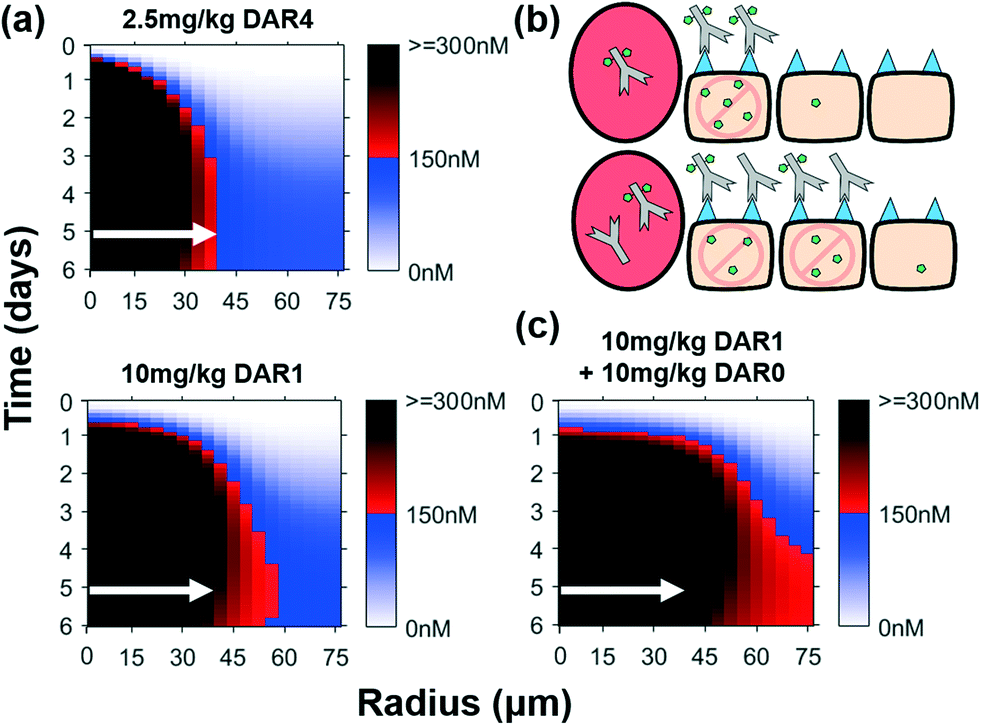 | ||
| Fig. 7 Direct cell targeting by ADC improves payload distribution more than bystander effects alone. (a) Intracellular payload concentrations over time for bystander ADC. Despite a constant payload dose, the improved ADC distribution of the DAR1 ADC improves the payload distribution throughout the tumor. (b) Graphic depiction of the same ADC dose with and without DAR0 (unconjugated) antibody. Adding DAR0 antibody drives ADC penetration farther into the tumor, lowering the average number of molecules per cell but increasing the number of directly targeted cells. (c) Despite the same payload dose, co-administering DAR0 antibody further improves the payload distribution, resulting in therapeutic concentrations of payload throughout the tumor. White arrows indicate distance where 150 nM intracellular payload concentration is reached for the 2.5 mg kg−1 DAR4 dose (shown in all plots for comparison). | ||
Despite the ability of the bystander payload to diffuse deeper into the tumor tissue (Fig. 7), there is still heterogeneity within the tumor (red-black gradient in the perivascular cells, blue-white gradient away from the vessel). Since higher antibody doses improve ADC distribution and increase direct cell targeting, a progressive increase in the dose of ADC delivered will eventually saturate surface receptors in the entire tumor; however, ADCs are clinically administered at, or near, their maximum tolerated dose (which generally does not saturate the receptors), and any increase in dose is not feasible due to systemic toxicity from the payload. Conversely, unconjugated antibodies are often well-tolerated and can be given at much higher doses. To further explore the impact of improving cellular targeting of the ADC, we simulated co-administration of unconjugated antibody (DAR0) with a bystander ADC, which leads to increased payload penetration despite the same payload dose (Fig. 7b and c). Co-administration of a single dose of 10 mg kg−1 DAR0 antibody and 10 mg kg−1 DAR1 ADC (1:1 ratio dose) resulted in the intracellular concentration of the payload above the therapeutic threshold throughout the tumor and markedly improved penetration of the ADC (Fig. S6b†). This effect is similar with non-bystander payloads, although the bystander payloads reach more tumor cells (Fig. S6c†). The addition of unconjugated antibody will indeed lower the in vitro potency of the ADC, but counter-intuitively, our results indicate this improves efficacy of the drug in vivo (Fig. 7c and Fig. S6a†), even in tumors that are resistant to the unconjugated antibody. These data indicate that with transport-guided dosing strategies, direct cell targeting (for antigen positive cells) and bystander effects (for antigen negative cells) can work synergistically to improve ADC distribution and maximize efficacy.
Payload design parameters for optimal bystander effects
We next analyzed the impact of the payload physicochemical properties on bystander penetration distance to determine the theoretically optimal payload properties. Simulations of commonly used payloads with different physicochemical properties resulted in variable bystander effects and penetration distances (Fig. S1†). Although diffusion of the payload helps reduce its distribution heterogeneity, it comes at the expense of progressively greater dilution of the payload within the tissue and faster washout from the tumor interstitium into the capillary. Consequently, the intracellular concentration of payload throughout the tumor, though more homogeneous, can decrease below the therapeutic threshold. Optimizing the pharmacokinetic properties of a payload can improve its tumor distribution while maintaining intracellular concentrations at or above the therapeutic threshold, thereby maximizing the efficacy from bystander effects. Assuming the payload is capable of efflux into the extracellular space, the two most important pharmacokinetic properties that determine its bystander potential are the cell uptake/target binding rate (‘immobilization reaction rate’), described here by kin,P, and the diffusion rate of the payload through the tumor, DP. The two properties can collectively be described by a dimensionless Damköhler number (Da), given by the equation below.29 A detailed derivation can be found in ESI.†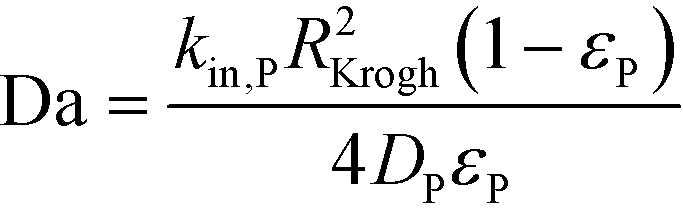
When the Damköhler number is small, the payload rapidly diffuses throughout the tumor extracellular space before entering cells, resulting in a uniform distribution outside the cells. If the uptake rate is too slow, however, it will wash out of the tumor before entering cells. When the Damköhler number is large, the reaction (cell uptake) rate dominates, and adjacent cells take up the payload rapidly before it can diffuse farther, thereby retaining its heterogeneous distribution (Fig. 8a). This maintains a higher concentration within these adjacent cells, but the payload reaches fewer cells. (This assumes that uptake is effectively irreversible46 due to the high payload target concentration within cells, e.g. microtubules and DNA.) We explored payload distribution by using a fixed rate of reaction (kin,P(1 − εP)/εP), but variable Damköhler number (i.e. variable rate of payload diffusion). As seen in Fig. 8b and Fig. S7,† when Da ≤ 0.1 (small Damköhler number), the payload diffuses rapidly through the tumor, diluting the intracellular payload concentration below the therapeutic threshold (blue gradient). When Da ≥ 10 (large Damköhler number), the slower diffusion rate of the payload results in rapid cell uptake by cells closer to the capillary, leading to heterogeneous distribution of the payload. However, the simulations predict that an ‘ideal’ Damköhler number exists where the rate of reaction and the rate of diffusion are optimally balanced such that the payload diffuses fast enough to reach the farthest radius (75 μm) of the tumor (Fig. 8b) but enters cells before it washes out of the tissue. For an ADC dose of 2.5 mg kg−1 DAR4, the simulations indicate an ‘ideal’ Damköhler number of ∼3 (Fig. S7†).
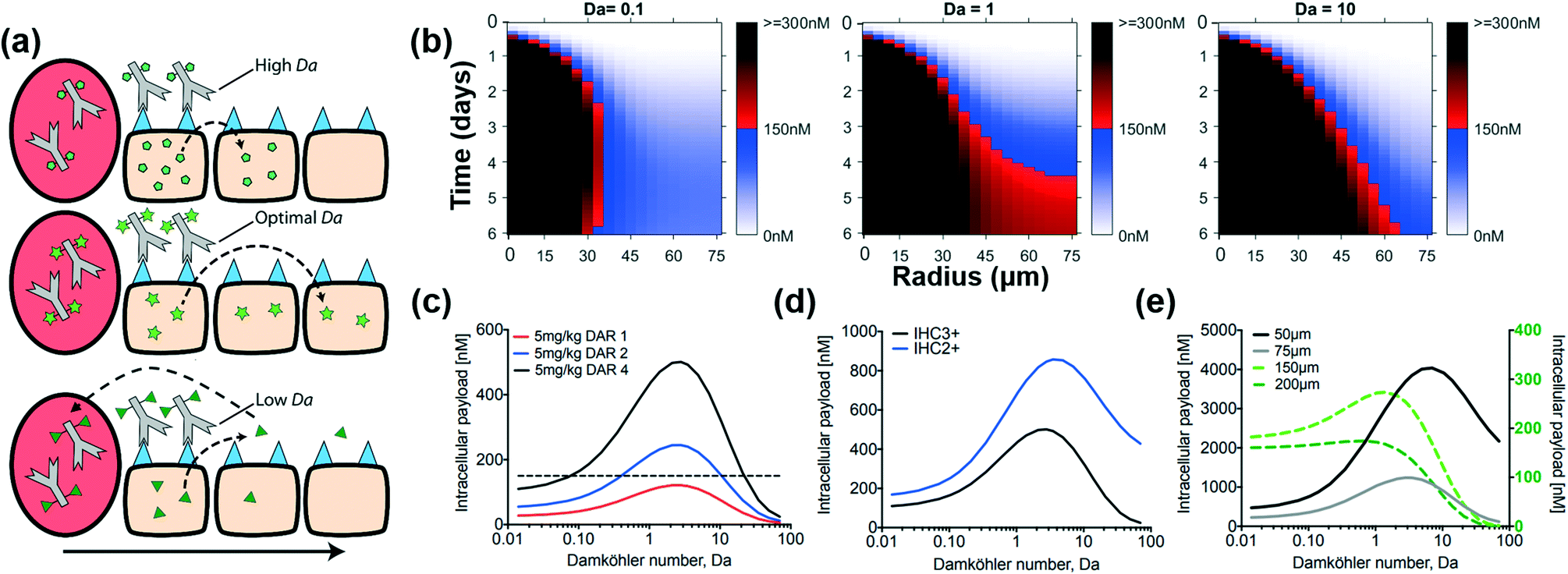 | ||
| Fig. 8 The Damköhler number determines the transport properties of payloads. (a) Graphic depiction of payload distribution with high (pentagons), optimal (stars), and low (triangles) Damköhler numbers. Suboptimal payloads (high and low Da) are still able to exhibit bystander effects in adjacent cells but do not distribute homogeneously through the tissue like optimal payloads. (b) Intracellular payload concentration for payloads with Damköhler numbers of 0.1, 1, and 10. Low Damköhler number payloads diffuse through the tumor but are washed out through the blood vessels before they are taken up by cells. High Damköhler number payloads are taken up quickly, which prevents them from diffusing throughout the tumor. The optimal Damköhler number achieves the highest concentration of payload at the tissue edge (i.e. at RKrogh), independent of the (c) total payload dose, (d) antigen expression (IHC3+ ∼ 106 HER2 receptors/cell, IHC2+ ∼ 3 × 105 HER2 receptors/cell) and (e) tumor vascular density. Solid and dotted lines correspond to left and right axes, respectively. Da, Damköhler number. | ||
Additional simulations show that the range of an efficacious Damköhler number widens (i.e. the value needed to achieve therapeutic concentrations throughout the tumor is less sensitive to payload properties) as more payload is delivered, either by increasing the ADC dose (Fig. S8b†) or increasing the DAR (Fig. 8c). For a constant payload dose, the high dose/low DAR strategy provides a larger ‘ideal’ Damköhler range than a low dose/high DAR strategy (Fig. S8c†). This difference is due to the increased tumor penetration and improved cellular targeting from a higher dose of ADC, which reduces the distance the payload needs to travel. When a sufficient number of payload molecules are delivered, any Da within this range results in the intracellular payload concentration at the farthest radius exceeding the 150 nM threshold. When the minimum number of payload molecules needed to achieve this effect are delivered (here ∼2 mg kg−1 DAR4), the ‘ideal’ Damköhler number converges to Da ∼ 3 (Fig. S8a and S8b†). For all doses that deliver greater than 150 nM concentration of the payload to cells far from the vessel, the ‘ideal’ Damköhler number converged to ∼3, even for nearly an order of magnitude lower surface receptor (Ag) density (Fig. 8d). Furthermore, this range is relatively independent of total tumor radius, which is a function of tumor vascularization. (Note, Da is proportional to the square of the Krogh radius.) In a less vascularized tumor (e.g. double the mean intercapillary distance, i.e. the Krogh cylinder radius), the ‘ideal’ Damköhler number for achieving maximum intracellular payload concentrations at the farthest radius (now 150 μm) is still close to Da ∼ 3. This holds for a wide range of Krogh radii: the maximum intracellular concentration at the farthest point is achieved within a modest Damköhler range of 1 ≤ Da ≤ 5 (Fig. 8e). This optimum value is independent of the payload dose, although the payload dose does impact the absolute intracellular concentration (Fig. 8c).
The Damköhler number of a payload is directly determined by its physicochemical properties. Having identified an optimal Damköhler number range, we predicted the Damköhler number for various ADC payloads used in the clinic or under development to examine how their predicted bystander potential corresponds to observed behavior from the literature. Table 2 lists the estimated kinetics (based on physicochemical properties) of some ADC payloads and the corresponding Damköhler number calculated from these estimates. For payload molecules known to have little to no bystander effects, such as DM1 and MMAF, the estimates for the Da are 0.01 or less. These small values are dominated by the slow cellular uptake rate (half-life on the order of hours). For such molecules, the rate of efflux from the cell is slow (resulting in low concentrations in the interstitium), and the subsequent influx into adjacent cells is slower than the rate of washout from the interstitium. Consequently, the majority of the payload does not enter adjacent cells, making them poor bystander molecules. Nevertheless, the maximum intracellular payload concentration at tumor edge still occurs when Da ∼ 1 (Fig. S9†).
| Payload | clogD |
Molecular weight | R | Estimated payload diffusivity (μm2 s−1) | k in,P half-life (min) | Estimated Damkohler number | Reported bystander effects? |
|---|---|---|---|---|---|---|---|
| a Measurement below PAMPA limit of detection. | |||||||
| Lys-SMCC-DM1 | 1.21 | 1104 | 0.51 | 9.8 | 194 | 0.01 | No23 |
| DM4 | 4.47 | 780 | 184.07 | 1.1 | 5.6 | 3.22 | Yes83 |
| S-methyl DM4 | 4.86 | 794 | 451.24 | 0.5 | 2.9 | 13.16 | Yes83 |
| MMAE | 2.01 | 718 | 1.05 | 14.8 | 8.2 | 0.17 | Yes13 |
| MMAF | 1.22 | 732 | 0.51 | 19.6 | 72 | 0.01 | No13 |
| Dxd1 | 0.55 | 493 | 0.43 | 26.8 | 0.9 | 0.85 | Yes47 |
| Dxd2 | −1.49 | 521 | 0.41 | 17.6 | >10a | <0.12 | No47 |
| PBD | 4.12 | 726 | 82.45 | 3.1 | 3.6 | 1.82 | Yes13 |
| SN-38 | 1.87 | 392 | 0.87 | 153.1 | 2.8 | 0.05 | Yes48 |
| SPP-DM1 | 4.08 | 752 | 75.23 | 3.2 | 7.0 | 0.94 | Yes23 |
Molecules with demonstrated in vivo bystander effects, such as MMAE, Dxd1, and PBD,13,47 have Damköhler numbers ranging from 0.2 to 2. The Damköhler number for MMAE is predicted to be slightly lower than ideal, resulting in some of the molecule washing out of the tumor before efficient cell killing. PBD is a DNA alkylating agent that is predicted to diffuse deep into the tissue, which may explain its effect even at low antibody concentrations.13 The bystander efficiency of SN-38 is surprisingly low given its rapid rate of cellular uptake. Unlike the ‘non-bystander’ payloads, this occurs because of its very high estimated diffusion coefficient. SN-38 is estimated to enter cells faster than MMAE, but it's very rapid diffusion could lower uptake due to washout, potentially reducing its efficiency of bystander killing deep in the tumor. Experimental data could help clarify this result, particularly since in vivo data suggests appreciable bystander effects occur.48,49 Conversely, DM4 has an optimal Damköhler number, but, once in the cytoplasm, it is converted to its methylated variant (S-methyl DM4),45,50 which has a much higher Damköhler number due to a very slow diffusion coefficient, arising from its high lipophilicity, that limits penetration. Taken together, this analysis highlights that while ability of a payload to exhibit bystander effects is determined by its membrane permeability (kin,P), the Damköhler number determines the efficiency of the bystander effects, and a payload with Da between 1 and 3 is predicted to exhibit maximum bystander killing.
In summary, increasing the DAR of non-bystander ADCs at a constant antibody dose resulted in little change in the payload penetration in the tumor (Fig. 4) (with mixed impact on efficacy in the literature), while increasing the DAR for bystander ADCs improved the tumor payload distribution and efficacy (Fig. 5). For both bystander and non-bystander ADCs, improving the ADC distribution by using a higher antibody dose with a lower DAR at a constant payload dose improved payload distribution and had better efficacy (Fig. 2, 3, 6, and 7). Additionally, this could be achieved by co-administering DAR0 (unconjugated) antibody to improve payload distribution in the tumor (Fig. 7). These results suggest that for antigen positive cells, direct cell targeting with the ADC is more efficient at killing cells than bystander effects, and improving ADC tumor penetration increases efficacy more than bystander effects alone. Bystander effects are important for killing antigen negative cells, however, and we outlined design parameters for the payload physicochemical properties to help optimize payload distribution in the tumor (Fig. 8).
Discussion
The heterogeneous distribution of antibodies in tumors has been known for decades and is often referred to as the ‘binding site barrier,51 which is actually a dynamic ‘saturation front’ that develops within the tissue.19 In the preclinical setting relatively large and/or frequent doses of monoclonal antibodies (e.g. 2–15 mg kg−1) can reduce this heterogeneity by saturating the entire tumor, thereby reaching all cells. For ADCs, however, the maximum tolerated doses are much lower, resulting in lower delivered antibody and higher intratumoral drug heterogeneity. The impact of this heterogeneous distribution of ADCs on their efficacy is incompletely understood. Recent data analyses and computational evidence indicates that improved distribution of ADCs plays an important role in overall efficacy.22 For example, a mechanistic ADC tumor penetration model developed by Vasalou et al. predicted that better ADC penetration (either by lower antigen expression or higher vascularization) showed better efficacy.52 The relationship between payload distribution and efficacy is further complicated by the use of bystander payloads (Fig. 1), which are important for killing antigen negative cells and may also mitigate some of the ADC heterogeneity. In this work, we explored the impact of ADC tumoral heterogeneity and bystander effects on efficacy.Multiple strategies have been explored for improving ADC efficacy, such as selection of high expression targets, optimizing affinity, avidity and pharmacokinetics of the antibody backbone, improving the conjugation/linker chemistry for plasma stability and on-target release, selection from a panel of highly cytotoxic payloads, and engineering increasingly potent payloads.2,53 However, it is important to consider all these improvements in the context of limited ADC penetration and heterogeneous tumoral distribution. For example, while potency is an important factor in determining ADC efficacy,54 extremely high payload potency has the potential to have a counter-productive effect on in vivo efficacy. Higher payload potency could further limit the MTD of the ADC, and the lower ADC doses in turn exacerbate distribution heterogeneity in the tumor, reaching fewer cells and lowering efficacy. This is in contrast to uniformly distributed drugs (e.g. small molecules) where a more potent compound may require dose reduction, but the higher potency (evenly distributed throughout the tumor) compensates for the lower, but uniform, tumor concentration. The effect of distribution heterogeneity on ADC efficacy has not received as much attention as structural features (antibody, linker, payload) during the design and optimization of ADCs, but many cases in literature suggest that ADC distribution appears to be a major contributing factor to the overall ADC efficacy (Fig. 2 and 6). Therefore, we used a combination of predictive computational modeling and retrospective analysis of tumor growth studies from the literature to examine the role of bystander effects in the relationship between ADC distribution and efficacy.
In this study, we have built on our previous modeling work to develop a mechanistic Krogh cylinder model to describe the tumor distribution of ADCs and their non-bystander or bystander payloads. Our simulations, combined with retrospective analysis of in vivo tumor growth data from the literature, show that increasing ADC tumor penetration improves efficacy for an ADC with non-bystander payloads (Fig. 2 and 3), since the distance traversed by the ADC determines the maximum number of cells reached by the payload. Interestingly, an increase in the payload dose (the active compound for these antibody-resistant tumors) does not necessarily increase efficacy when the antibody dose is constant (Fig. 4). This is consistent with simulations showing higher DAR on the same antibody dose primarily delivers more non-bystander payload to cells already receiving a therapeutic payload dose. Conversely, bystander payloads are able to diffuse and reach untargeted cells in the tumor, so increasing the payload dose (through a higher DAR) at a constant antibody dose improves efficacy (Fig. 5).
The improved efficacy from better ADC penetration without bystander effects is conceptually straightforward for ADCs with high potency payloads (Fig. 2). However, the situation is more complex with bystander effects, since the payload distribution can be improved either by direct cell targeting with the ADC or diffusion of the payload (Fig. 6a). The simulations show direct cell targeting by the ADC is more efficient at payload delivery than diffusion of payload to bystander cells (Fig. 7). Therefore, similar to non-bystander payloads, spreading the bystander payload over more antibodies ensures more cells farther from the blood vessels receive therapeutic intracellular concentrations of payload. Additionally, for the same total payload dose, co-administration of unconjugated antibody (DAR0) with the ADC (at a 1:1 ratio) showed more homogeneous accumulation of payload above the therapeutic threshold concentration throughout the tumor, analogous to lowering the DAR (Fig. 7b and c). One argument for the improvement in efficacy in the high ADC dose/low DAR tumor growth studies is simply from the therapeutic effect of the antibody alone due to the larger antibody doses. However, in all tumor growth studies reported here, the tumor models were either demonstrated or stated to be resistant to the unconjugated antibody, suggesting payload distribution (not antibody-mediated effects) drives efficacy in these preclinical models. Combined, these data provide evidence that increased penetration and direct cell targeting of the ADC is critical for improving its overall efficacy, regardless of whether the payload exhibits bystander effects or not.
The concept that direct cellular targeting is more efficient than indirect (bystander) targeting appears to be consistent with other targeting systems as well, such as doxorubicin-loaded immunoliposomes.55,56 Despite similar total tumor uptake of HER2-targeted and non-targeted immunoliposomes loaded with doxorubicin,56 the efficacy of the anti-HER2 targeted immunoliposomes was found to be significantly better.55 Deeper investigation showed that anti-HER2 immunoliposomes accumulated inside tumor cells, while non-targeting immunoliposomes accumulated in the tumor stroma and in macrophages. Although doxorubicin (often dosed as a small molecule) can diffuse across cell membranes to exhibit bystander effects, the more efficient tumor cell targeting of the anti-HER2 immunoliposomes significantly improved efficacy. From a drug transport perspective, direct cell targeting, which releases payload from the ADC inside the cell, results in higher intracellular concentrations than diffusion from the interstitium into a bystander cell.
Although bystander payloads cannot fully compensate for antibody distribution heterogeneity, the importance of bystander effects should not be understated. In the preclinical setting, xenograft tumors are grown from cell lines typically expressing surface antigen relatively homogeneously. However, in the clinical setting, tumors can be highly heterogeneous in antigen presentation with inter-/intra-patient and intratumoral variability. When a significant fraction of the tumor cells does not express the antigen, the ADC cannot directly target them, and bystander effects become crucial for killing these antigen negative (Ag−) cancer cells. Several in vivo studies using mosaic tumor models have shown that bystander payloads can eradicate tumors with heterogeneous expression while non-bystander payloads cannot.13,23
The importance of bystander payloads in killing antigen negative cells motivated us to study their transport to help establish design parameters for payloads to maximize bystander killing. In order for a bystander payload to accumulate in Ag− cells, it must efflux from an ADC targeted (Ag+) cell, diffuse through the tumor stroma and influx into the Ag− cell. If the influx of the payload occurs too fast relative to its diffusion rate, payload molecules will rapidly be immobilized inside cells immediately adjacent to the originating cell before they can reach more untargeted cells in the same tumor. On the contrary, if payload diffusion occurs faster than it can influx into the cell, it may be able to diffuse throughout the tumor but will wash out of the tumor before it can accumulate in adjacent cells (Fig. 8a and b). These two pharmacokinetic parameters establish the ‘bystander potential’ of a payload and can be described mathematically by a dimensionless Damköhler number. Damköhler numbers are dimensionless groups that describe the fundamental property of a system by relating a reaction rate (in this case the immobilization rate of the payload) to a transport rate (in this case diffusion through the tumor interstitium). Our simulations show a Damköhler number of ∼3 is ideal for achieving optimal payload distribution, capable of reaching even the edge of the Krogh cylinder (cells farthest from vessels in the tumor) with therapeutic intracellular concentrations (Fig. 8c–e and S8†). In fact, the ‘ideal’ Damköhler number of ∼3 results in the most efficient distribution of payload to the tumor edge regardless of the payload dose (Fig. 8c), antigen expression level (Fig. 8d), intercapillary distance (Fig. 8e), and payload potency (which determines the intracellular therapeutic threshold). These results are for the reversibly bound microtubule inhibitors, where some payload that enters a bystander cell can diffuse back out and deeper into the tissue (i.e. it is close to irreversible due to rapid rebinding, but not completely irreversible). For complete irreversible binding (such as a DNA alkylating agent), the optimal Damköhler number shifts (slightly) to ∼1 (Fig. S10†).
Although a Damköhler number of ∼3 is most efficient at achieving optimal payload distribution, the total payload dose and potency are important considerations for overall ADC efficacy. The Damköhler number is independent of the payload dose/potency, but the overall efficacy is dependent on both the payload dose/potency (i.e. the absolute amount of payload delivered to the tumor) and the Damköhler number (i.e. the relative distribution of this payload throughout the tumor). The intracellular concentration of the payload decreases farther away from the vasculature, so if the total payload dose is too low, the intracellular concentration at the edge of the tumor will not be above the therapeutic threshold (even with a Da ∼ 3). For drugs with lower potency, it may be advantageous to have a larger Damköhler number to ‘concentrate’ the payload on fewer cells to reach a therapeutic level. For example, a payload requiring >300 nM drug concentrations (shown in black in Fig. 8b; Fig. S8a,† 1 mg kg−1 DAR4), the Da = 10 would have higher cell killing than Da = 1 because the drug is diluted less in the tissue. This could be important with non-linear pharmacodynamic effects from the payload: for example, to concentrate DNA damaging agents above the level that can be handled by DNA repair enzymes to kill the cell. Matching the ‘ideal’ Damköhler range with payload potency and predicted payload distribution can help guide development of payloads with optimal bystander effects for efficient targeting of heterogeneous tumors in the clinic.
There are several model limitations and additional considerations for this analysis. One of the biggest limitations in directly confirming model predictions is the current lack of quantitative data on tumoral bystander payload distribution at the tissue and cellular level. Radiolabeled payloads do not have the sensitivity to image the distribution at sub-lethal doses of ADC, and mass spectrometry imaging has not yet provided the resolution needed to address intratumoral heterogeneity, though significant strides have been made recently to overcome these issues.57,58 This limitation, in fact, motivated the use of computational modeling, but ideally, improvements in imaging equipment and techniques will be able to validate (or refute) these predictions. The computational models used here have been primarily developed and validated using fluorescence data28,35,36,59 and in vitro results.27,60
Linker stability and DAR-dependent clearance and/or deconjugation are important for lowering payload release in systemic circulation while maintaining efficient delivery and release to the target tissue.44,61,62 Although DAR-dependent clearance (faster clearance of higher DAR ADCs) or DAR-dependent deconjugation (conversion of high DAR into lower DAR) are important, our previous analysis indicates that this alone is not sufficient to explain the concept of higher efficacy with low DAR/high ADC dose treatment presented here.22 These effects, albeit important, were not considered in the current simulations to isolate the impact of bystander effects and focus on these implications for therapy.
The simulations here assume a high potency payload where saturation of cell surface antigen delivers more payload than needed for efficacy. Many ADCs have IC50 values well below their Kdin vitro, which means there is significant cell death with only a fraction of their receptors bound (high potency). However, lower potency payloads may require higher DAR to ‘concentrate’ the payload on fewer cells to induce toxicity.63 Our previous publication argued for ‘matching’ the potency with the delivery to improve efficacy.22 This concept is analogous to the Damköhler number for the payload. A very high Da (>10) concentrates the bystander payload uptake in only a few cells (possibly many more payloads per cell than are needed for cytotoxicity) while a low Da (<0.1) reaches more cells but potentially at sub-therapeutic concentrations.
It is well documented that increasing antibody dose uniformly improves penetration.64–68 We assume that this also occurs in the literature cases reported here, but since these studies were not directly evaluating ADC distribution, measurements of heterogeneity were not included (a limitation of retrospective analysis). Future prospective studies that directly evaluate heterogeneity of ADC distribution can confirm (or refute) these computational results.
Although the cells used in the literature studies here were resistant to the antibody alone, the antibody could be sensitizing the cells to the payload (e.g. by altering cell signaling69). However, in vitro data indicate the payload is dominating cell death given the increased potency of higher DAR antibodies. If higher antibody concentrations were needed for increased death from the payload, it would also be inconsistent with the in vivo data in Fig. 5, where the same antibody dose achieves higher tumor growth suppression. Therefore, these data do not agree with the antibody sensitizing the cell to the payload. Rather, the data are consistent with the interpretation here: the antibody helps improve the tissue penetration in vivo (but is not required in vitro where there are no transport limitations).
Another major factor not considered in this model (and less relevant for immunocompromised preclinical animal models) is the role of the immune system. Fc-effector functions, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and CDC are known to be important for clinical responses. For example, ADCC by natural killer (NK) cells has been implicated in complete responses from neo-adjuvant trastuzumab therapy.70 The payload from ADCs also appears to be associated with increased immune cell infiltration.71 A majority of the ADCs in clinical development are composed of IgG1 antibodies, the most abundant antibody isotype present in serum and highly active at initiating ADCC. The improved ADC penetration with high antibody/low DAR treatment or the addition of unlabeled antibody to drive ADC deeper into the tissue (e.g.Fig. 7) can also bolster immune effector functions by covering more tumor cells with antibody. Therefore, improved tissue penetration of ADCs not only helps deliver bystander and non-bystander payloads but also has the potential to improve other mechanisms of action.
In summary, these computational results demonstrate that increased ADC tumor penetration and direct cell targeting by ADCs improves payload tumor distribution and efficacy for both non-bystander and bystander payloads. Additionally, these data indicate bystander effects cannot completely compensate for poor tumor tissue penetration of ADCs. However, in clinical settings, the use of bystander payloads is critical for targeting Ag− cancer cells that are unable to be targeted by intact ADCs. The simulations identified an optimum Damköhler range (1 ≤ Da ≤ 3) necessary for maximizing the bystander effect of high potency payloads, which can serve as a guiding tool for the design of novel bystander payloads.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided in part by the Teh-Hsun Lee Fellowship from the Rackham Graduate School (EK) and NSF CAREER award (GMT).References
- R. V. J. Chari, ACS Med. Chem. Lett., 2016, 7, 974–976 CrossRef CAS PubMed
.
- P. Polakis, Pharmacol. Rev., 2016, 68, 3–19 CrossRef CAS PubMed
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed
- J. M. Reichert, mAbs, 2016, 8, 197–204 CrossRef CAS PubMed
- J. F. Ponte, X. Sun, N. C. Yoder, N. Fishkin, R. Laleau, J. Coccia, L. Lanieri, M. Bogalhas, L. Wang, S. Wilhelm, W. Widdison, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2016, 27, 1588–1598 CrossRef CAS PubMed
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed
- X. Sun, J. F. Ponte, N. C. Yoder, R. Laleau, J. Coccia, L. Lanieri, Q. Qiu, R. Wu, E. Hong, M. Bogalhas, L. Wang, L. Dong, Y. Setiady, E. K. Maloney, O. Ab, X. Zhang, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2017, 28, 1371–1381 CrossRef CAS PubMed
- P. Bryant, M. Pabst, G. Badescu, M. Bird, W. McDowell, E. Jamieson, J. Swierkosz, K. Jurlewicz, R. Tommasi, K. Henseleit, X. Sheng, N. Camper, A. Manin, K. Kozakowska, K. Peciak, E. Laurine, R. Grygorash, A. Kyle, D. Morris, V. Parekh, A. Abhilash, J. W. Choi, J. Edwards, M. Frigerio, M. P. Baker and A. Godwin, Mol. Pharmaceutics, 2015, 12, 1872–1879 CrossRef CAS PubMed
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC
- F. Li, K. K. Emmerton, M. Jonas, X. Zhang, J. B. Miyamoto, J. R. Setter, N. D. Nicholas, N. M. Okeley, R. P. Lyon, D. R. Benjamin and C.-L. Law, Cancer Res., 2016, 76, 2710–2719 CrossRef CAS PubMed
- J. R. Junutula, K. M. Flagella, R. A. Graham, K. L. Parsons, E. Ha, H. Raab, S. Bhakta, T. Nguyen, D. L. Dugger, G. Li, E. Mai, G. D. L. Phillips, H. Hiraragi, R. N. Fuji, J. Tibbitts, R. Vandlen, S. D. Spencer, R. H. Scheller, P. Polakis and M. X. Sliwkowski, Clin. Cancer Res., 2010, 16, 4769–4778 CrossRef CAS PubMed
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–736 CrossRef CAS PubMed
- H. Donaghy, mAbs, 2016, 8, 659–671 CrossRef CAS PubMed
- G. P. Adams, R. Schier, A. M. McCall, H. H. Simmons, E. M. Horak, R. K. Alpaugh, J. D. Marks and L. M. Weiner, Cancer Res., 2001, 61, 4750–4755 CAS
- J. J. Rhoden and K. D. Wittrup, J. Pharm. Sci., 2012, 101, 860–867 CrossRef CAS PubMed
- G. M. Thurber, M. M. Schmidt and K. D. Wittrup, Adv. Drug Delivery Rev., 2008, 60, 1421–1434 CrossRef CAS PubMed
- G. M. Thurber, S. C. Zajic and K. D. Wittrup, J. Nucl. Med, 2007, 48, 995–999 CrossRef CAS PubMed
-
G. M. Thurber, in Pharmaceutical Sciences Encyclopedia, John Wiley & Sons, Inc., 2010, DOI:10.1002/9780470571224.pse551
- C. Cilliers, H. Guo, J. Liao, N. Christodolu and G. M. Thurber, AAPS J., 2016, 18, 1117–1130 CrossRef CAS PubMed
- Y. V. Kovtun, C. A. Audette, Y. Ye, H. Xie, M. F. Ruberti, S. J. Phinney, B. A. Leece, T. Chittenden, W. A. Blättler and V. S. Goldmacher, Cancer Res., 2006, 66, 3214–3221 CrossRef CAS PubMed
- A. P. Singh, S. Sharma and D. K. Shah, J. Pharmacokinet. Pharmacodyn., 2016, 43, 567–582 CrossRef CAS PubMed
- M. Lopus, E. Oroudjev, L. Wilson, S. Wilhelm, W. Widdison, R. Chari and M. A. Jordan, Mol. Cancer Ther., 2010, 9, 2689–2699 CrossRef CAS PubMed
- A. P. Singh and D. K. Shah, Drug Metab. Dispos., 2017, 45, 1120–1132 CrossRef PubMed
- F. B. Pruijn, K. Patel, M. P. Hay, W. R. Wilson and K. O. Hicks, Aust. J. Chem., 2008, 61, 687–693 CrossRef CAS
- S. Bhatnagar, E. Deschenes, J. Liao, C. Cilliers and G. M. Thurber, J. Pharm. Sci., 2014, 103, 3276–3286 CrossRef CAS PubMed
- G. M. Thurber and R. Weissleder, PLoS One, 2011, 6, e24696 CAS
- R. K. Jain and L. T. Baxter, Cancer Res., 1988, 48, 7022–7032 CAS
- B. S. Hendriks, L. K. Opresko, H. S. Wiley and D. Lauffenburger, Cancer Res., 2003, 63, 1130–1137 CAS
- C. D. Austin, A. M. De Maziere, P. I. Pisacane, S. M. van Dijk, C. Eigenbrot, M. X. Sliwkowski, J. Klumperman and R. H. Scheller, Mol. Biol. Cell, 2004, 15, 5268–5282 CrossRef CAS PubMed
- P. Poulin, Y.-H. Chen, X. Ding, S. E. Gould, C. E. Hop, K. Messick, J. Oeh and B. M. Liederer, J. Pharm. Sci., 2015, 104, 1508–1521 CrossRef CAS PubMed
- P. Poulin and F. P. Theil, J. Pharm. Sci., 2000, 89, 16–35 CrossRef CAS PubMed
- G. M. Thurber, K. S. Yang, T. Reiner, R. H. Kohler, P. Sorger, T. Mitchison and R. Weissleder, Nat. Commun., 2013, 4, 1504 CrossRef PubMed
- G. M. Thurber, T. Reiner, K. S. Yang, R. H. Kohler and R. Weissleder, Mol. Cancer Ther., 2014, 13, 986–995 CrossRef CAS PubMed
- M. Barok, H. Joensuu and J. Isola, Breast Cancer Res., 2014, 16, 209–221 CrossRef PubMed
- Y. C. Chung, J. F. Kuo, W. C. Wei, K. J. Chang, W. T. Chao and M. Tan, PLoS One, 2015, 10, 1–15 CAS
- K. F. Maass, C. Kulkarni, M. A. Quadir, P. T. Hammond, A. M. Betts and K. D. Wittrup, J. Pharm. Sci., 2015, 104, 4409–4416 CrossRef CAS PubMed
- S. L. Spencer, S. Gaudet, J. G. Albeck, J. M. Burke and P. K. Sorger, Nature, 2009, 459, 428–432 CrossRef CAS PubMed
- T. H. Pillow, J. Tien, K. L. Parsons-Reponte, S. Bhakta, H. Li, L. R. Staben, G. Li, J. Chuh, A. Fourie-O'Donohue, M. Darwish, V. Yip, L. Liu, D. D. Leipold, D. Su, E. Wu, S. D. Spencer, B.-Q. Shen, K. Xu, K. R. Kozak, H. Raab, R. Vandlen, G. D. Lewis Phillips, R. H. Scheller, P. Polakis, M. X. Sliwkowski, J. A. Flygare and J. R. Junutula, J. Med. Chem., 2014, 57, 7890–7899 CrossRef CAS PubMed
- A. V. Yurkovetskiy, M. Yin, N. Bodyak, C. A. Stevenson, J. D. Thomas, C. E. Hammond, L. L. Qin, B. Zhu, D. R. Gumerov, E. Ter-Ovanesyan, A. Uttard and T. B. Lowinger, Cancer Res., 2015, 75, 3365–3372 CrossRef CAS PubMed
- D. Jackson, J. Atkinson, C. I. Guevara, C. Zhang, V. Kery, S. J. Moon, C. Virata, P. Yang, C. Lowe, J. Pinkstaff, H. Cho, N. Knudsen, A. Manibusan, F. Tian, Y. Sun, Y. Lu, A. Sellers, X. C. Jia, I. Joseph, B. Anand, K. Morrison, D. S. Pereira and D. Stover, PLoS One, 2014, 9, e83865 Search PubMed
- S. Sukumaran, K. Gadkar, C. Zhang, S. Bhakta, L. Liu, K. Xu, H. Raab, S. F. Yu, E. Mai, A. Fourie-O'Donohue, K. R. Kozak, S. Ramanujan, J. R. Junutula and K. Lin, Pharm. Res., 2015, 32, 1884–1893 CrossRef CAS PubMed
- H. K. Erickson, W. C. Widdison, M. F. Mayo, K. Whiteman, C. Audette, S. D. Wilhelm and R. Singh, Bioconjugate Chem., 2010, 21, 84–92 CrossRef CAS PubMed
- V. S. Goldmacher, C. A. Audette, Y. Guan, E.-H. Sidhom, J. V. Shah, K. R. Whiteman and Y. V. Kovtun, PLoS One, 2015, 10, e0117523 Search PubMed
- Y. Ogitani, Y. Abe, T. Iguchi, J. Yamaguchi, T. Terauchi, M. Kitamura, K. Goto, M. Goto, M. Oitate, H. Yukinaga, Y. Yabe, T. Nakada, T. Masuda, K. Morita and T. Agatsuma, Bioorg. Med. Chem. Lett., 2016, 26, 5069–5072 CrossRef CAS PubMed
- T. M. Cardillo, S. V. Govindan, R. M. Sharkey, P. Trisal, R. Arrojo, D. Liu, E. A. Rossi, C. H. Chang and D. M. Goldenberg, Bioconjugate Chem., 2015, 26, 919–931 CrossRef CAS PubMed
- T. M. Cardillo, S. V. Govindan, R. M. Sharkey, P. Trisal and D. M. Goldenberg, Clin. Cancer Res., 2011, 17, 3157–3169 CrossRef CAS PubMed
- X. Sun, W. Widdison, M. Mayo, S. Wilhelm, B. Leece, R. Chari, R. Singh and H. Erickson, Bioconjugate Chem., 2011, 22, 728–735 CrossRef CAS PubMed
- K. Fujimori, D. G. Covell, J. E. Fletcher and J. N. Weinstein, Cancer Res., 1989, 49, 5656–5663 CAS
- C. Vasalou, G. Helmlinger and B. Gomes, PLoS One, 2015, 10, e0118977 Search PubMed
- E. G. Kim and K. M. Kim, Biomol. Ther., 2015, 23, 493–509 CrossRef CAS PubMed
- N. Diamantis and U. Banerji, Br. J. Cancer, 2016, 114, 362–367 CrossRef CAS PubMed
- J. W. Park, K. Hong, D. B. Kirpotin, G. Colbern, R. Shalaby, J. Baselga, Y. Shao, U. B. Nielsen, J. D. Marks, D. Moore, D. Papahadjopoulos and C. C. Benz, Clin. Cancer Res., 2002, 8, 1172–1181 CAS
- D. B. Kirpotin, D. C. Drummond, Y. Shao, M. R. Shalaby, K. Hong, U. B. Nielsen, J. D. Marks, C. C. Benz and J. W. Park, Cancer Res., 2006, 66, 6732–6740 CrossRef CAS PubMed
- Y. Fujiwara, M. Furuta, S. Manabe, Y. Koga, M. Yasunaga and Y. Matsumura, Sci. Rep., 2016, 6, 24954 CrossRef CAS PubMed
- E. Pienaar, J. Sarathy, B. Prideaux, J. Dietzold, V. Dartois, D. E. Kirschner and J. J. Linderman, PLoS Comput. Biol., 2017, 13, e1005650 Search PubMed
- A. J. Primeau, A. Rendon, D. Hedley, L. Lilge and I. F. Tannock, Clin. Cancer Res., 2005, 11, 8782–8788 CrossRef CAS PubMed
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed
- S. O. Doronina, B. A. Mendelsohn, T. D. Bovee, C. G. Cerveny, S. C. Alley, D. L. Meyer, E. Oflazoglu, B. E. Toki, R. J. Sanderson, R. F. Zabinski, A. F. Wahl and P. D. Senter, Bioconjugate Chem., 2006, 17, 114–124 CrossRef CAS PubMed
- N. Jain, S. W. Smith, S. Ghone and B. Tomczuk, Pharm. Res., 2015, 32, 3526–3540 CrossRef CAS PubMed
- D. M. Goldenberg, T. M. Cardillo, S. V. Govindan, E. A. Rossi and R. M. Sharkey, Oncotarget, 2015, 6, 22496–22512 Search PubMed
- R. D. Blumenthal, I. Fand, R. M. Sharkey, O. C. Boerman, R. Kashi and D. M. Goldenberg, Cancer Immunol. Immunother., 1991, 33, 351–358 CrossRef CAS PubMed
- G. M. Thurber, J. L. Figueiredo and R. Weissleder, PLoS One, 2009, 4, e8053 Search PubMed
- G. M. Thurber and K. D. Wittrup, Cancer Res., 2008, 68, 3334–3341 CrossRef CAS PubMed
- J. H. E. Baker, K. E. Lindquist, L. A. Huxham, A. H. Kyle, J. T. Sy and A. I. Minchinton, Clin. Cancer Res., 2008, 14, 2171–2179 CrossRef CAS PubMed
- M. E. Ackerman, D. Pawlowski and K. D. Wittrup, Mol. Cancer Ther., 2008, 7, 2233–2240 CrossRef CAS PubMed
- K. Liang, Y. Lu, W. Jin, K. K. Ang, L. Milas and Z. Fan, Mol. Cancer Ther., 2003, 2, 1113–1120 CAS
- E. Muraro, E. Comaro, R. Talamini, E. Turchet, G. Miolo, S. Scalone, L. Militello, D. Lombardi, S. Spazzapan, T. Perin, S. Massarut, D. Crivellari, R. Dolcetti and D. Martorelli, J. Transl. Med., 2015, 13, 204 CrossRef CAS PubMed
- P. Müller, M. Kreuzaler, T. Khan, D. S. Thommen, K. Martin, K. Glatz, S. Savic, N. Harbeck, U. Nitz, O. Gluz, M. von Bergwelt-Baildon, H. Kreipe, S. Reddy, M. Christgen and A. Zippelius, Sci. Transl. Med., 2015, 7, 315ra188 CrossRef PubMed
- D. Hilmas and E. Gillette, Cancer, 1974, 33, 103–110 CrossRef CAS PubMed
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS
- G. Z. Ferl, V. Kenanova, A. M. Wu and J. J. DiStefano, Mol. Cancer Ther., 2006, 5, 1550–1558 CrossRef CAS PubMed
-
E. L. Green, Biology of the laboratory mouse, Dover Publications & The Jackson Laboratory, New York, 2nd edn, 1975 Search PubMed
- M. M. Schmidt and K. D. Wittrup, Mol. Cancer Ther., 2009, 8, 2861–2871 CrossRef CAS PubMed
- J. Bostrom, L. Haber, P. Koenig, R. F. Kelley and G. Fuh, PLoS One, 2011, 6, e17887 CAS
- K. F. Maass, C. Kulkarni, A. M. Betts and K. D. Wittrup, AAPS J., 2016, 18, 635–646 CrossRef CAS PubMed
- L. Zhang, S. Bhatnagar, E. Deschenes and G. M. Thurber, Sci. Rep., 2016, 6, 25424 CrossRef CAS PubMed
- A. P. Singh, K. F. Maass, A. M. Betts, K. D. Wittrup, C. Kulkarni, L. E. King, A. Khot and D. K. Shah, AAPS J., 2016, 18, 861–875 CrossRef CAS PubMed
- A. Krol, J. Maresca, M. W. Dewhirst and F. Yuan, Cancer Res., 1999, 59, 4136–4141 CAS
- M. Pabst, W. McDowell, A. Manin, A. Kyle, N. Camper, E. De Juan, V. Parekh, F. Rudge, H. Makwana, T. Kantner, H. Parekh, A. Michelet, X. Sheng, G. Popa, C. Tucker, F. Khayrzad, D. Pollard, K. Kozakowska, R. Resende, A. Jenkins, F. Simoes, D. Morris, P. Williams, G. Badescu, M. P. Baker, M. Bird, M. Frigerio and A. Godwin, J. Controlled Release, 2017, 253, 160–164 CrossRef CAS PubMed
- H. K. Erickson, P. U. Park, W. C. Widdison, Y. V. Kovtun, L. M. Garrett, K. Hoffman, R. J. Lutz, V. S. Goldmacher and W. A. Blattler, Cancer Res., 2006, 66, 4426–4433 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00093f |
| ‡ Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2018 |