
Rational design of conjugated side chains for high-performance all-polymer solar cells†
Wei
Huang‡
a,
Meilin
Li‡
a,
Fengyuan
Lin‡
a,
Yang
Wu
b,
Zhifan
Ke
b,
Xing
Zhang
a,
Rui
Ma
a,
Tingbin
Yang
a,
Wei
Ma
*b and
Yongye
Liang
 *a
*a
aDepartment of Materials Science and Engineering, Shenzhen Key Laboratory of Printed Electronics, South University of Science and Technology of China, Shenzhen 518055, P. R. China. E-mail: liangyy@sustc.edu.cn
bState Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an 710049, PR China. E-mail: msewma@xjtu.edu.cn
First published on 24th October 2017
Abstract
With their rapid development in recent years, organic solar cells hold the exciting potential to be a groundbreaking form of solar harvesting technology. Among these, all-polymer solar cells (all-PSCs) stand out owing to various advantages such as complementary absorption and superior stability. However, the advance of all-PSCs has been greatly impeded by energy level mismatches and unfavorable morphology of the active layer. Here, we report a molecular engineering approach featuring asymmetrical 4-methoxythiophene/thiophene as conjugated side chains of the donor polymer to fine-tune the energy level alignment and phase separation. The corresponding polymer, namely, poly{4-[5-(2-ethylhexyl)-4-methoxythiophen-2-yl]-8-[5-(2-ethylhexyl)thiophen-2-yl]benzo[1,2-b:4,5-b′]dithiophene}-alt-[bis(5-thiophene-2-yl)-5,6-difluoro-2-(2-hexyldecyl)-2H-benzo[d][1,2,3]triazole-4,7-diyl] (PMOT32) exhibited a power conversion efficiency that exceeded 8.5% in all-PSCs with poly{[N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} (N2200) as the acceptor. PMOT32:N2200 also maintained good photovoltaic performance when processed as a thick film or from non-halogenated solvents. Detailed comparisons with two other polymers with pure thiophene or 4-methoxythiophene as their side chains revealed how these side chains affected the photovoltaic performance via energy level alignment and phase separation.
Design, System, ApplicationTo optimize the photovoltaic performance of all-polymer solar cells (all-PSCs), it is desirable to achieve an appropriate energy level alignment between the donor polymer and the acceptor polymer to afford a high open-circuit voltage and a sufficient energy offset for charge separation. Control of the morphology of the active layer is also important, as it can significantly affect charge transport. Here, we report the design of efficient donor polymers with suitable energy levels and favorable morphology for all-PSCs via fine-tuning their conjugated side chains. A new asymmetrically substituted benzo[1,2-b:4,5-b′]dithiophene (BDT) monomer with one 4-methoxythiophene (MOT) chain and one thiophene chain (BDT-MOT/T) and the corresponding polymer PMOT32 were synthesized. In comparison with the thiophene chain, the MOT chain lowered the energy levels of the donor polymer. When blended with N2200 as the acceptor polymer, PMOT32 solar cells exhibited a better match of energy levels and thus higher photovoltaic performance than two other polymers with pure thiophene or 4-methoxythiophene as their side chains. The photovoltaic performance of these polymer systems was correlated with the phase separation between the donor and acceptor polymers. This study can provide insights into molecular engineering to further enhance the performance of all-PSCs. |
1 Introduction
All-polymer solar cells (all-PSCs), in which an electron-donor polymer interpenetrates an electron–acceptor polymer to form the active layer, have regained attention in recent years.1–3 In comparison with polymer solar cells based on a fullerene acceptor, all-PSCs possess several advantages. Variation of the organic building blocks enables easy matching of the donor and acceptor polymers in terms of absorption spectra and energy levels.3 Complementary absorption ranges from the visible to the near infrared benefit solar harvesting, and appropriately aligned energy levels can minimize energy loss while providing a sufficient energy offset for exciton separation.4 In addition, entanglements between polymer chains can enable all-PSCs to exhibit superior stability4 and flexibility,5 which are difficult to achieve with fullerene PSCs.Although promising, the development of all-PSCs greatly lags behind that of PSCs based on fullerene6 or non-fullerene small-molecule acceptors (SMAs).7–9 In fact, most all-PSCs have performance inferior to that of their fullerene counterparts.3 Low electron mobility and non-optimal morphology are major bottlenecks in improving the photovoltaic performance of all-PSCs. Extensive efforts have been devoted to the development of efficient acceptor polymers based on electron-deficient building blocks, such as cyanated poly(phenylenevinylene) (CN-PPV),1 benzothiadiazole (BT),10 naphthalenediimide (NDI),4,11–17 perylenediimide (PDI)18–20 and double B ← N bridged bipyridine (BNBP).21 Among these, N2200 copolymerized with NDI and dithiophene and its derivatives are attractive acceptor materials.4,11–13,16 All-PSCs with PTB7-Th as the donor and N2200 as the acceptor have achieved a power conversion efficiency (PCE) of 5.7%.12 By further molecular engineering of acceptor polymers, such as side chain modifications,16 fluorination of thiophene,15 selenophene substitution14 and copolymerization,17 improved photovoltaic performance has been recently demonstrated owing to enhanced electron mobility or improved phase separation in the active layer.14
In contrast to the intensive research into acceptor polymers, donor polymers in all-PSCs have received less attention. PTB7 (ref. 11 and 22) and PTB7-Th12,14–18,21 are widely used as donor polymers in all-PSCs, and a PCE of 7.7% has been reported in a PTB7-Th:PNDIS-HD system.14 Nevertheless, most high-performance all-PSC systems that use PTB7-Th as the donor suffer from a low open-circuit voltage (Voc) and a low fill factor. Recently, Li et al. reported an efficient all-PSC system with an impressively high short-circuit current density (Jsc) and fill factor.4 The donor polymer J51,4 which was constructed from 5,6-difluoro-2H-benzo[d][1,2,3]triazole (FTAZ)23 and benzo[1,2-b:4,5-b′]dithiophene (BDT), exhibited complementary absorption ranges and balanced hole/electron mobility in the active layer when blended with N2200, which afforded a high PCE that exceeded 8%. However, the relatively high-lying highest occupied molecular orbital (HOMO) of J51 causes the system to have a moderate Voc and relatively large energy losses,24 which limits further improvements in the PCE.
Recently, we developed a feasible method of lowering the HOMO of donor polymers by introducing 4-methoxythiophene (MOT) as conjugated side chains on BDT.25,26 This approach has proved to be effective in increasing the Voc and PCE in PSCs based on both fullerene and fused-ring electron acceptors (FREAs). Here, we report two new FTAZ-based p-type polymers with MOT-modified BDT and investigate their application in high-performance all-PSCs. In order to fine-tune the energy levels, we designed and synthesized a new asymmetrically substituted BDT monomer with one MOT chain and one thiophene chain (BDT-MOT/T). When the number of MOT substituents on BDT increased, the polymer exhibited a lower HOMO level and thus a higher Voc in all-PSCs. However, the highest PCE was achieved with PMOT32 prepared from BDT-MOT/T, instead of PMOT34 prepared from BDT-MOT, when blended with N2200 in all-PSCs. The PMOT32:N2200 system demonstrated an acceptable PCE of 8.59% from chloroform (CF), 7.98% from o-xylene and 8.56% from 2-methyltetrahydrofuran (MeTHF), which are among the best performances for all-PSCs. The influence of the MOT substituent on the photovoltaic performance of all-PSCs is correlated with the energy level alignment and blend morphology.
2 Experiments
2.1 Synthesis of monomers and polymers
2.2 Cyclic voltammetry
Cyclic voltammetry (CV) measurements were performed with an electrochemical workstation (CHI 600E) to determine the ionization potential (IP) and electron affinity (EA) of the polymers. Polymer films were dip-coated from chloroform solutions on glassy carbon working electrodes (diameter of 3 mm). CV curves were recorded under an argon atmosphere in CH3CN solutions containing 0.1 M Bu4NPF6 with a Pt wire as the counter electrode and Ag/Ag+ as the reference electrode. The redox potential of ferrocene/ferrocenium (Fc/Fc+) under the same conditions is located at 0.073 V, which is assumed to have an absolute energy level of −4.8 eV with respect to vacuum. The IP and EA values were calculated by the following equations:| EIP = Eox + [4.8 − 0.073] (eV) |
| EEA = Ered + [4.8 − 0.073] (eV) |
2.3 Device fabrication
OPV devices were fabricated with a traditional structure, namely, indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/donor polymer:N2200/perylenediimides functionalized with amine oxide (PDINO)28/Al. The ITO substrates were cleaned in an ultrasonic bath with detergent water, deionized water, acetone, and isopropanol, and then treated with UV-ozone for 15 min. A layer of PEDOT:PSS was spin-coated onto ITO glass in air at 2500 rpm for 30 s to give an average thickness of ∼50 nm. After annealing at 150 °C for 15 min, the substrates were transferred into a N2-filled glove box. The active layer was spin-coated onto PEDOT:PSS and then vacuumed for 3 hours to remove residual DIO. Two kinds of solvent were used. The polymers were dissolved in CF + 0.7% DIO or o-xylene + 1% DIO. The concentrations were 6.5 mg mL−1 for J52, PMOT32 and PMOT34 and 3 mg mL−1 for N2200. The active layers were spin-coated at 2500 rpm for 30 s when CF was used and 1500 rpm for 30 s when o-xylene was used. For the MeTHF solution, the concentration of PMOT32 was 4.5 mg mL−1 and the donor/acceptor ratio was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w). This solution was heated at 80 °C overnight and was then spin-coated at 1500 rpm for 30 s to give a blend film with a thickness of ∼110 nm. Subsequently, the as-cast layers were annealed at 100 °C for 10 min. PDINO was dissolved in ethanol to give a concentration of 1.1 mg mL−1 and was then spin-coated onto the active layers at 3000 rpm for 30 s. Finally, 80 nm Al was evaporated on the top surface under a vacuum of 3 × 10−6 mbar. The active area of the devices was 0.045 cm2.
:1 (w/w). This solution was heated at 80 °C overnight and was then spin-coated at 1500 rpm for 30 s to give a blend film with a thickness of ∼110 nm. Subsequently, the as-cast layers were annealed at 100 °C for 10 min. PDINO was dissolved in ethanol to give a concentration of 1.1 mg mL−1 and was then spin-coated onto the active layers at 3000 rpm for 30 s. Finally, 80 nm Al was evaporated on the top surface under a vacuum of 3 × 10−6 mbar. The active area of the devices was 0.045 cm2.
2.4 Characterization of PSC devices
The thicknesses were measured using an XP-200 stylus profilometer (KLA-Tencor). Absorption spectra were recorded with a UV-vis spectrophotometer (UV-3600, Shimadzu). J–V measurements of the devices under illumination by an AM1.5G solar simulator (Enli Technology, 100 mW cm−2, calibrated by a silicon reference cell) were performed using a computer-controlled Keithley 2400 Source Measure Unit. EQE measurements were performed using an Enli Technology QE-R system with a bromine tungsten lamp as the light source. AFM images of the active layers were acquired with an Asylum MFP-3D Stand Alone microscope in tapping mode. TEM images were acquired with an FEI Helios Nanolab 600i system. PL was recorded with a PerkinElmer LS55 fluorescence spectrometer. GIWAXS and RSoXS measurements were performed at the Advanced Light Source. GIWAXS measurements were performed at beamline 7.3.3.29 Samples were prepared on Si substrates using identical blend solutions to those used in the devices. The 10 keV X-ray beam was incident at a grazing angle of 0.11–0.15°, which was selected to maximize the intensity of scattering from the samples. The scattered X-rays were detected using a Dectris Pilatus 2M photon-counting detector. RSoXS transmission measurements were performed at beamline 11.0.1.2.30 Samples used for RSoXS measurements were prepared on a PSS-modified Si substrate under the same conditions as those used for device fabrication and were then transferred by floating in water to a 1.5 mm × 1.5 mm Si3N4 membrane with a thickness of 100 nm supported by a 5 mm × 5 mm Si frame with a thickness of 200 μm (Norcada, Inc.). 2-D scattering patterns were collected with an in-vacuum CCD camera (Princeton Instrument PI-MTE). The distance of the sample detector was calibrated from the diffraction peaks of the triblock copolymer poly(isoprene-b-styrene-b-2-vinylpyridine), which has a known spacing of 391 Å. The beam size at the sample was approximately 100 μm by 200 μm.2.5 Mobility measured by space-charge-limited current method
To study the charge transport properties, both hole-only and electron-only devices were fabricated with architectures of ITO/PEDOT:PSS/polymer blend/MoOx/Ag and ITO/ZnO/polymer blend/PDINO/Al, respectively. The hole/electron mobility was determined by plotting and fitting current–voltage curves. The SCLC is determined by:| J = (9/8)εrε0μ(V2/L3) |
3 Results
The asymmetric BDT monomer with one thiophene side chain and one MOT side chain, namely, BDT-MOT/T, was prepared using a similar method to BDT-MOT but by subsequently reacting benzo[1,2-b:4,5-b′]dithiophene-4,8-dione with (5-alkylthiophen-2-yl)lithium and (4-methoxy-5-alkylthiophen-2-yl)lithium in a molar ratio of 1:1:1 (see Scheme S1, ESI†). PMOT32 was subsequently synthesized by a Stille polycondensation reaction between the BDT-MOT/T distannous monomer and 4,7-bis(5-bromothiophen-2-yl)-5,6-difluoro-2-(2-hexyldecyl)-2H-benzo[d][1,2,3]triazole. Two other polymers, namely, J52 (ref. 27) with thiophene conjugated side chains and PMOT34 with MOT conjugated side chains, were also synthesized for comparison (ESI†). Scheme 1 illustrates the molecular structures of these three polymers, as well as N2200. The number-average molecular weight (Mn) and polydispersity index (PDI), which were estimated by high-temperature gel permeation chromatography (GPC), were 38.5 kDa and 2.16 for PMOT32, 32.8 kDa and 1.91 for PMOT34, and 31.4 kDa and 1.79 for J52, respectively (Table S1†). Both PMOT32 and PMOT34 exhibited high solubility in CF, chlorobenzene (CB), o-dichlorobenzene (o-DCB) and o-xylene at room temperature.
 | ||
| Scheme 1 Chemical structures of the related materials in this work. | ||
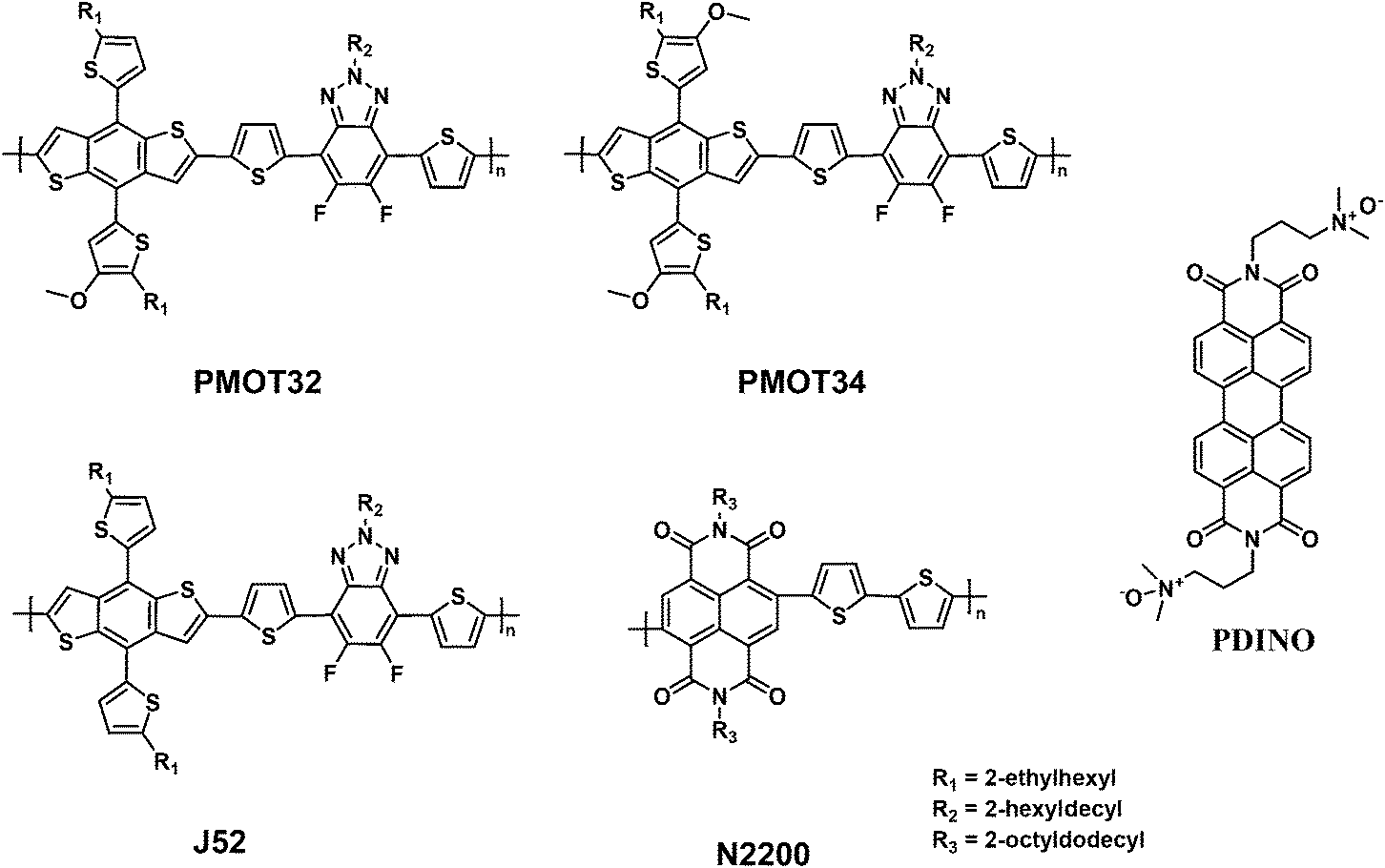
Fig. 1a and S1a† show the absorption spectra of the donor polymers and N2200 in film and solution states, respectively. The donor polymers exhibited similar absorption spectra, which suggested that the MOT substituent had a similar impact on the optical properties when compared with the thiophene substituent. For the pristine films, the replacement of thiophene with MOT side chains resulted in a slight blue shift in the absorption edge, which was 631 nm for J52, 629 nm for PMOT32 and 624 nm for PMOT34, respectively (Fig. 1a). The optical band gap was thus calculated to be 1.96 eV for J52, 1.97 eV for PMOT32 and 1.99 eV for PMOT34, respectively. The absorption ranges of these donor polymers can complement that of the low-band-gap polymer acceptor N2200 (Fig. 1a), which favors solar harvesting from the ultraviolet-visible to the near-infrared region.4
 | ||
| Fig. 1 (a) Normalized absorbance of the pristine polymer films. (b) Schematic diagram of the energy levels of the polymers as determined by cyclic voltammetry. The ionization potential of N2200 was also calculated on the basis of the optical band gap and the measured electron affinity (number in red). | ||
Cyclic voltammetry (CV) was used to determine the energy levels of the polymers. Using ferrocene as an internal standard, the ionization potential (IP) and electron affinity (EA) were 5.12 eV/3.17 eV for J52, 5.18 eV/3.25 eV for PMOT32, 5.26 eV/3.33 eV for PMOT34 and 5.84 eV/3.79 eV for N2200, respectively (Fig. 1b and S2†). It is clear that the IP increases (HOMO is lowered) in proportion to the increase in the ratio of MOT to thiophene in the conjugated side chains from J52 to PMOT32 and PMOT34, which is consistent with our previous work.25,26 In contrast to the donor polymers, the band gap of N2200 measured by CV is much larger than the optical band gap. We deduced that the IP of N2200 might be overestimated by CV, as N2200 is a strong electron acceptor. Therefore, an IP of 5.25 eV was calculated by adding the optical band gap to the EA of N2200 (Fig. 1b). It is noted that the calculated IP of N2200 is very similar to the measured IP of PMOT34, which might cause problems with charge separation and recombination in the blend after photoexcitation. The photophysical properties of the four polymers are summarized in Table S1.†
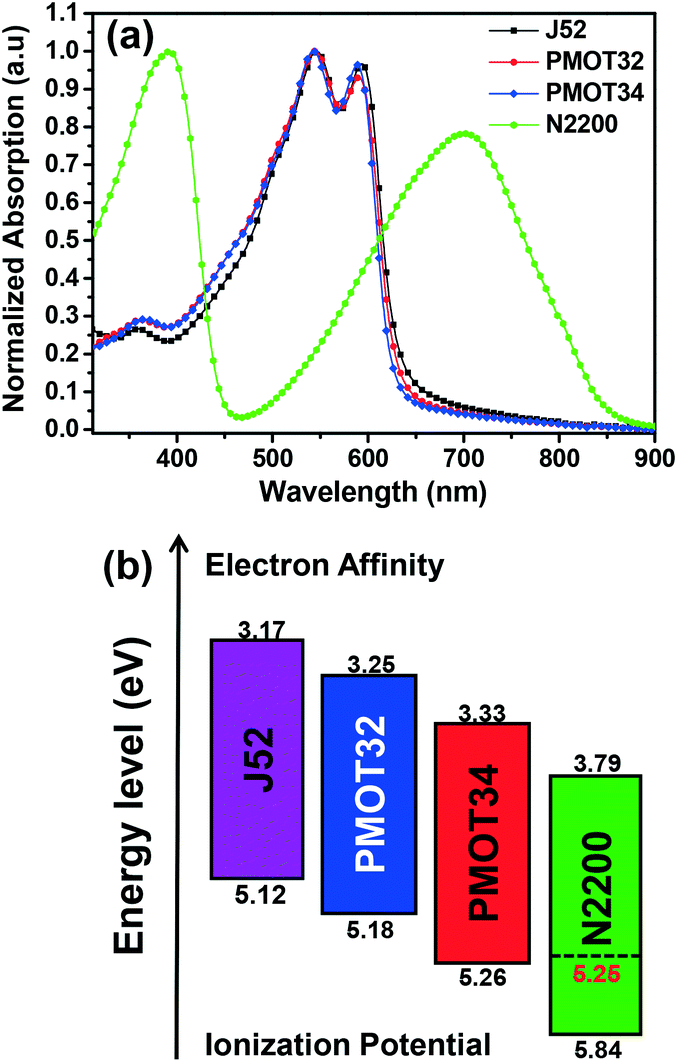
The photovoltaic performance of the all-PSCs was investigated in a conventional structure of ITO/PEDOT:PSS/active layer/PDINO28/Al. Typical current density–voltage (J–V) curves of the all-PSCs are displayed in Fig. 2, and the corresponding device parameters are summarized in Table 1, S2 and S3.† When processed from CF with 0.7% (by volume) 1,8-diiodooctane (DIO) as an additive, PMOT32:N2200 exhibited a Voc of 0.871 V, a Jsc of 13.84 mA cm−2, a fill factor of 71.2% and a PCE of 8.59% in the best-performing device. In contrast, the counterparts displayed inferior performance, with a Voc of 0.798 V and a PCE of 7.51% for J52 and a Voc of 0.912 V and a PCE of 7.25% for PMOT34. The significant increase in the Voc mainly accounted for the superior performance of PMOT32 in comparison with J52, which corresponded to the lower HOMO level of PMOT32. For PMOT34:N2200, however, the highest Voc among these systems did not result in the highest PCE owing to the substantial decline in the Jsc (11.83 mA cm−2 for PMOT34).
 | ||
| Fig. 2 (a) J–V characteristics of polymer:N2200 solar cells fabricated from CF with 0.7% DIO under AM1.5G solar radiation and (b) EQE spectra of the corresponding devices in (a). (c) Power conversion efficiency of all-PSCs with various thicknesses of the active layer. (d) J–V characteristics of polymer:N2200 solar cells fabricated from o-xylene with 1% DIO under AM1.5G solar radiation. | ||
| Donor | Solvents | V oc [V] | J sc [mA cm−2] | FF [%] | PCE [%] |
|---|---|---|---|---|---|
| a Calculated by integrating the EQE spectra. b Average PCEs were obtained from more than 10 devices. | |||||
| PMOT32 | CF + 0.7% DIO | 0.871 | 13.84 [13.27]a | 71.2 | 8.59 [8.50]b |
| o-Xylene + 1.0% DIO | 0.864 | 13.21 [12.47]a | 69.9 | 7.98 [7.87]b | |
| MeTHF | 0.862 | 13.96 [13.38]a | 71.1 | 8.56 [8.41]b | |
| J52 | CF + 0.7% DIO | 0.798 | 14.05 [13.90]a | 67.0 | 7.51 [7.40]b |
| o-Xylene + 1.0% DIO | 0.805 | 13.54 [12.92]a | 67.8 | 7.39 [7.28]b | |
| PMOT34 | CF + 0.7% DIO | 0.912 | 11.83 [11.15]a | 67.2 | 7.25 [7.11]b |
| o-Xylene + 1.0% DIO | 0.915 | 11.10 [10.00]a | 68.4 | 6.95 [6.85]b | |
Fig. 2b shows the corresponding external quantum efficiency (EQE) spectra of the all-PSC devices. PMOT32:N2200 was very efficient in converting photons into electrons with an EQE of greater than 60% in the range of 400–620 nm, but exhibited mediocre performance beyond 650 nm with an EQE of less than 40%. On the basis of the absorption data (Fig. S1b†), the optical absorption of the active layer in the range of 400–650 nm was mainly contributed by the donor polymers, whereas the acceptor polymer N2200 was dominant in the range of 650–800 nm, and both the donor polymer and N2200 made similar contributions in the range of 300–400 nm. With an increase in the acceptor:donor ratio from 1:2.17 to 1:1, the blend exhibited a gradual increase in absorption in the range of 650–800 nm (Fig. S3a†) and an increase in EQE in the corresponding region (Fig. S3b†), which was closely correlated with the slight increase in Jsc at higher acceptor contents (Table S2†). However, an acceptor:donor ratio of greater than 1:2.17 in the blend was detrimental to the fill factor and thus the PCE, which probably resulted from inferior phase separation and charge transport in the active layer.4 The PMOT34 system displayed a significantly lower EQE response when compared with the PMOT32 and J52 systems, and the percentage decrease was more obvious in the ranges of 350–450 nm and 650–800 nm, where the absorption of N2200 played a major role. It should be noted that there were negligible differences between the PMOT34 blend and the PMOT32 and J52 blends in terms of absorption in these ranges (Fig. S1b†). Interestingly, PMOT34:N2200 exhibited an EQE that was almost identical to those of the other two systems at a wavelength of 450 nm, where N2200 has low absorption. These results suggest that the inferior EQE response and thus the low Jsc of the PMOT34:N2200 system may be ascribed to the ineffective conversion of photons by N2200 in the active layer.
It has been proposed that thick active layers can be favorable for printing processes31 and practical applications.32 Thus, we fabricated devices with various thicknesses of the active layer to study their performance in a thick film (Fig. 2c and Table S4†). As can be observed from Fig. 2c, the all-PSCs exhibited a slow decline in PCE when the thickness exceeded the optimal value of ∼120 nm, which was possibly due to the high mobility of FTAZ-based polymers. PMOT32:N2200 still maintained a PCE of around 6% at an active layer thickness of 250 nm. Processing solvents represent another important issue for the practical application of PSCs. The chlorinated solvents that are generally used are toxic and carcinogenic. However, few PSCs can demonstrate high performance when processed from non-halogenated solvents, which is possibly due to their non-optimal morphology.6 Very few all-PSCs with a PCE that exceeded 7% processed from non-halogenated solvents have been reported thus far.31,33,34 When processed from o-xylene, PMOT32:N2200 exhibited an impressive PCE of 7.98% with a Voc of 0.864 V, a Jsc of 13.21 mA cm−2, and a fill factor of 69.9% in the best-performing device (Fig. 2d, Tables 1 and S4†). In comparison, J52:N2200 and PMOT34:N2200 achieved a PCE of 7.39% and 6.95%, respectively (Fig. 2d and Table 1). The significant increase in Voc and the slight enhancement in the fill factor contributed to the better photovoltaic performance of the PMOT32:N2200 system in comparison with J52:N2200, which was similar to that of the systems processed from CF. The corresponding EQE spectra of the respective devices are shown in Fig. S4† and resemble those of the systems processed from CF. Very recently, Huang et al. reported a PTzBI:N2200 system that had an efficiency of ∼9% when processed from MeTHF.34 We also experimented with MeTHF as the processing solvent for the PMOT32:N2200 system, and an acceptable PCE of 8.56% was achieved (Voc = 0.862 V, Jsc = 13.96 mA cm−2 and FF = 71.1%) (Fig. S5†). It should be pointed out that a hot MeTHF solution is required for optimal processing. Preliminary studies of the stability of the PMOT32:N2200 devices were also performed (Fig. S6†). Unencapsulated devices stored in a glove box exhibited relatively high stability, with a decline in efficiency of about 12% over 9 days. Encapsulated devices in air were less stable, with a decline in efficiency of 22% over 9 days. These unsatisfactory stabilities were probably due to the acidic and hygroscopic properties of the hole transport material PEDOT:PSS in the devices. Increases in the stability of these polymer solar cell devices are expected if the device structure can be further optimized. The synthesis of PMOT32 can be less complicated than that of the well-known donor polymer PTB7-TH, and further optimization of synthesis and scale-up will produce these materials at acceptable costs.
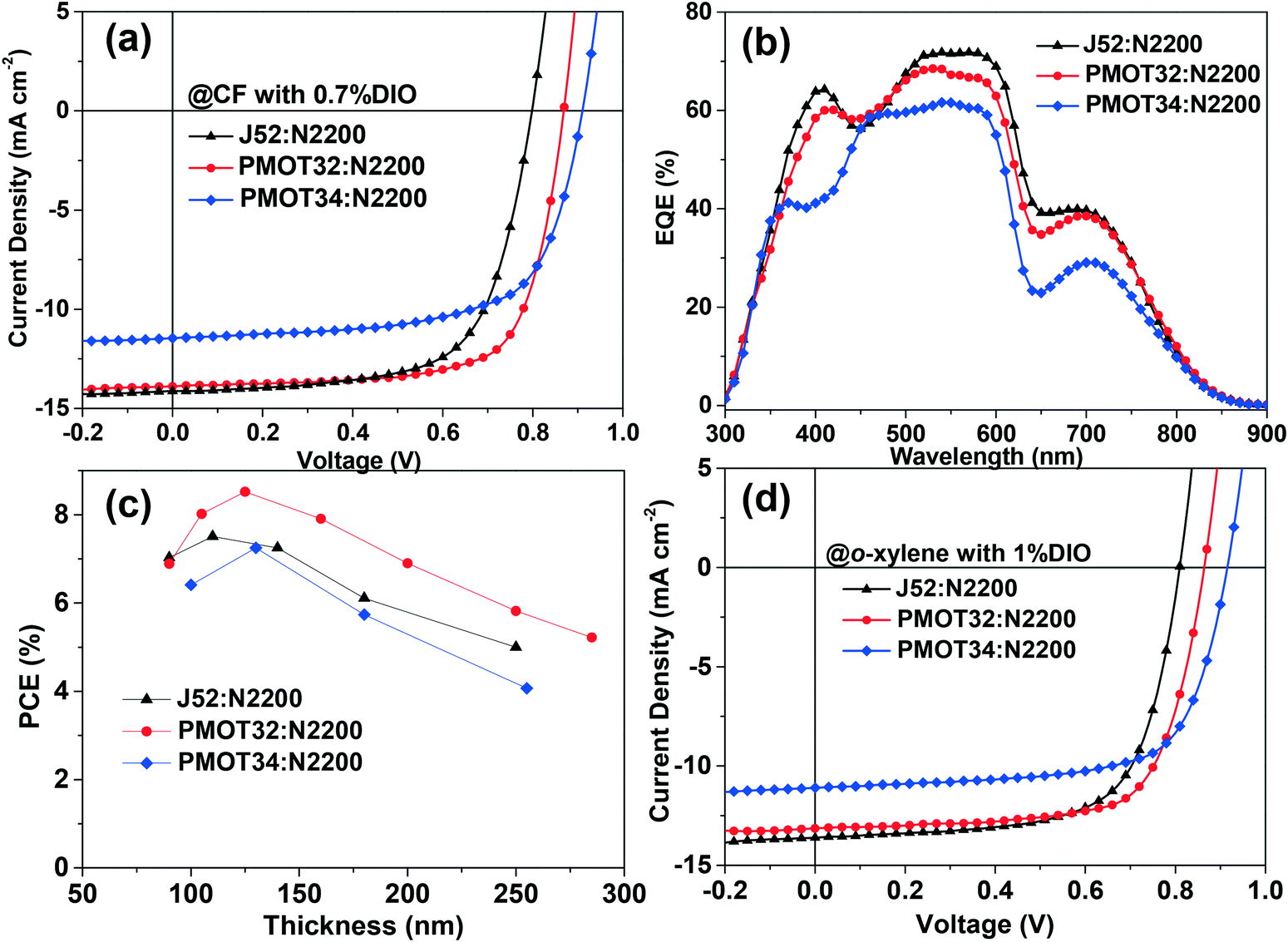
In order to obtain further understanding of how the MOT conjugated side chain affects the photovoltaic properties, atomic force microscopy (AFM) and transmission electron microscopy (TEM) were employed to study the morphology of the active layer of the all-PSCs. Fig. S7† shows AFM topography images of films of the three different blends processed from CF with or without thermal annealing (TA). All the films exhibited a similar topography with a low root mean square (RMS) roughness of around 0.85–0.93 nm. However, a significant difference appeared in the TEM images (Fig. 3). Dark spots were observed for all these polymer blends before TA. These dark spots can be correlated with N2200-rich domains owing to the high electron affinity of N2200.11 The dark spots became smaller from J52 to PMOT32, and the PMOT34 blend displayed the smallest and fewest dark spots, which suggests that the MOT substituent decreased the donor polymer/N2200 phase separation. After TA treatment, both J52 and PMOT32 exhibited more sharply contrasting features and dendritic morphology with an interconnected network of a dark phase,11 which demonstrated greater donor/acceptor phase separation after TA. However, homogeneous fine features were observed in the PMOT34 blend film. Favorable phase separation and aggregation of N2200 in the active layers could improve charge transport and suppress geminate recombination.35 Thus, the improvement in the photovoltaic performance of the J52 and PMOT32 blends after TA was more prominent than that for the PMOT34 blend (Table S3†). A similar trend in phase separation was also observed for films processed from o-xylene after thermal annealing (Fig. S8†).
 | ||
| Fig. 3 TEM images of three different blends prepared under two different film processing conditions: without TA (top row) and TA for 10 minutes (bottom row): (a) and (d) J52:N2200, (b) and (e) PMOT32:N2200 and (c) and (f) PMOT34:N2200, respectively, processed from CF with 0.7% DIO. The scale bars are indicated on the images. | ||
To further investigate the micromorphology of these systems, grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were performed (Fig. 4 and S8a†).36 For pristine films, the lamellar packing peaks of PMOT32 and PMOT34 were located at q ≈ 0.30 Å−1, which corresponded to a lamellar spacing of ∼21 Å, whereas the π–π stacking peaks of PMOT32 and PMOT34 were located at q ≈ 1.7 Å−1 (∼3.7 Å). The lamellar and π–π spacings of PMOT32 and PMOT34 were nearly identical to those of J52, which suggests that the introduction of MOT as a conjugated side chain did not change the packing model of the polymers. When blended with N2200, the lamellar peaks of the three blends shifted to q ≈ 0.27 Å−1 (∼23 Å), whereas a negligible difference was observed in the π–π stacking peaks, which indicated that N2200 did not enter the crystalline phase of the donor polymers but formed a crystalline phase in the lamellar spaces instead.4 Because the GIWAXS results did not reveal any difference between the three polymer blends, resonant soft X-ray scattering (RSoXS)36–38 measurements were also conducted (Fig. S9b†). As can be seen from Fig. S9b,† all these blends exhibited similar scattering profiles, which proved that domains with similar sizes of 20–30 nm were present in the three active layers. As RSoXS provides enhanced contrast between different organic components, the domain size revealed by RSoXS represents phase separation inside the donor-rich phase (bright region in TEM). However, analysis of the purity of the three domains revealed that the PMOT32 blend had higher purity (relative purity of 1) than that of the PMOT34 blend (relative purity of 0.89), which is favorable for charge transport and thus improvements in device performance.31,37
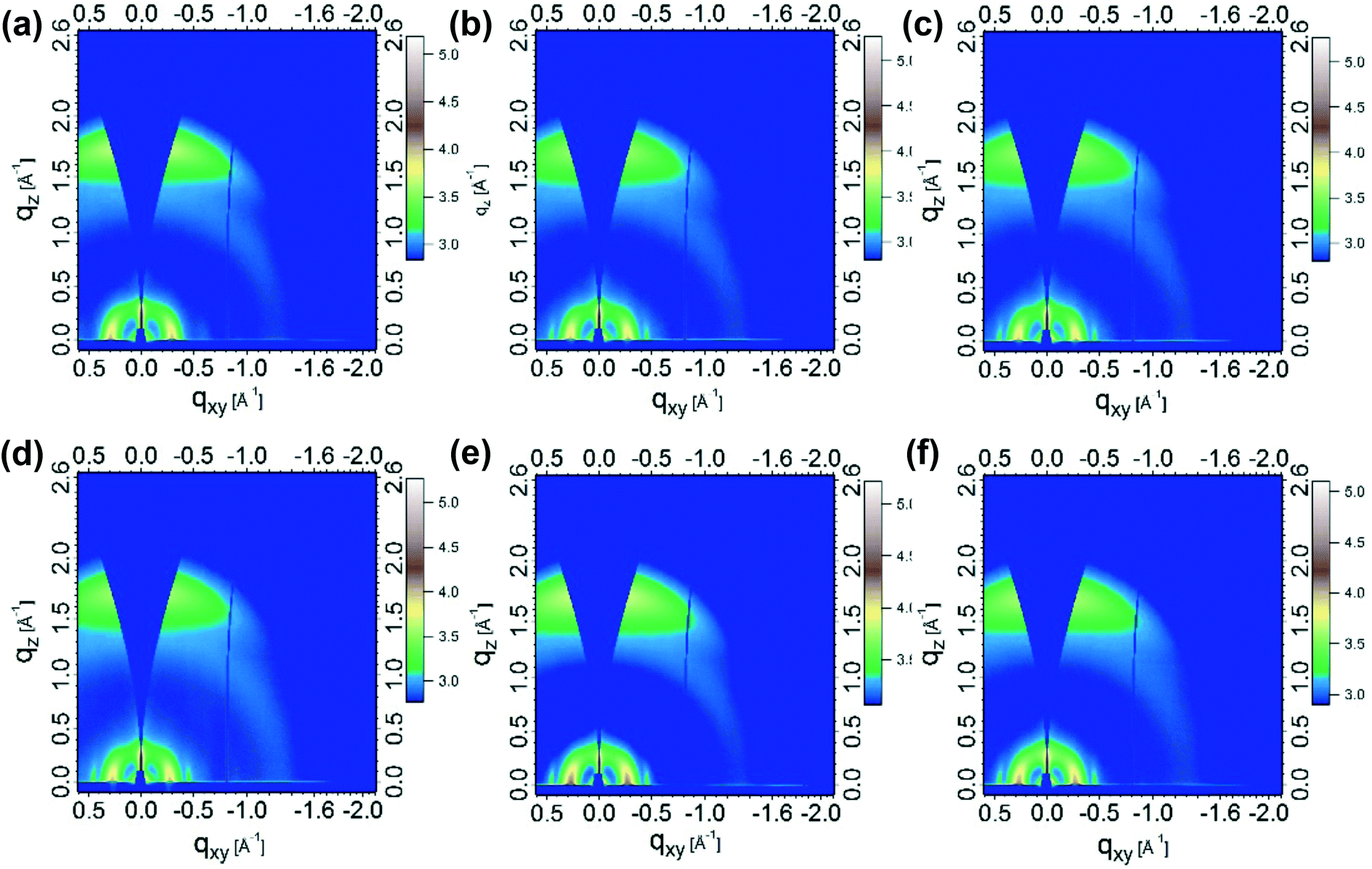 | ||
| Fig. 4 Two-dimensional GIWAXS images of pristine polymers and blends with N2200: (a) J52, (b) PMOT32, (c) PMOT34, (d) J52:N2200, (e) PMOT32:N2200, and (f) PMOT34:N2200. | ||
The space-charge-limited current (SCLC) model was used to investigate the charge transport characteristics (Table S5 and Fig. S10†). The hole-only architecture of ITO/PEDOT:PSS/polymer blend/MoOx/Ag and the electron-only architecture of ITO/ZnO/polymer blend/PDINO/Al were employed to measure the hole and electron mobility, respectively. As can be seen from Fig. S10 and Table S5,† all these systems exhibited high hole/electron mobility, which could account for the high fill factors of these systems. The hole mobility of the three blends was similar (∼8 × 10−4 cm2 V−1 s−1) before TA and increased slightly after TA (∼9 × 10−4 cm2 V−1 s−1). With respect to electron mobility, although all these blends exhibited similar values (∼2.6 × 10−4 cm2 V−1 s−1) before TA, PMOT32 and J52 displayed significant increases after TA (∼3.8 × 10−4 cm2 V−1 s−1), whereas that of PMOT34 remained virtually unchanged, which was probably caused by inferior phase separation in the PMOT34 blend, which was closely correlated with the unfavorable morphology in TEM and lower purity in RSoXS. The lower electron mobility could be partly responsible for the lower Jsc in PMOT34:N2200 PSCs, which was consistent with the EQE spectra. The dependence of the Jsc of the three blend films on the light intensity was also measured to further investigate bimolecular recombination in all-PSCs (Fig. S11†). The relationship between Jsc and light intensity (P) is formulated as Jsc ∝ PS. If all the free charges are swept out and collected without bimolecular recombination, the value of S (the slope in logarithmic coordinates) should be equal to 1. The three all-PSCs gave slopes of 0.984, 0.992 and 0.992 for J52:N2200, PMOT32:N2200 and PMOT34:N2200, respectively, which indicated that bimolecular recombination was negligible in all these systems, which agrees closely with the high charge mobility in the three blends.
Photoluminescence (PL) measurements were conducted to study charge dissociation and recombination in the active layer (Fig. 5a, b and S12†).35,39 The PMOT34 blend displayed more intense PL than the other two blends, which probably resulted from the decrease in the effectiveness of charge separation due to the adjacent HOMO energy levels of PMOT34 and N2200, although homogeneity of the two mixing phases is usually beneficial for the dissociation of excitons at the interface.35
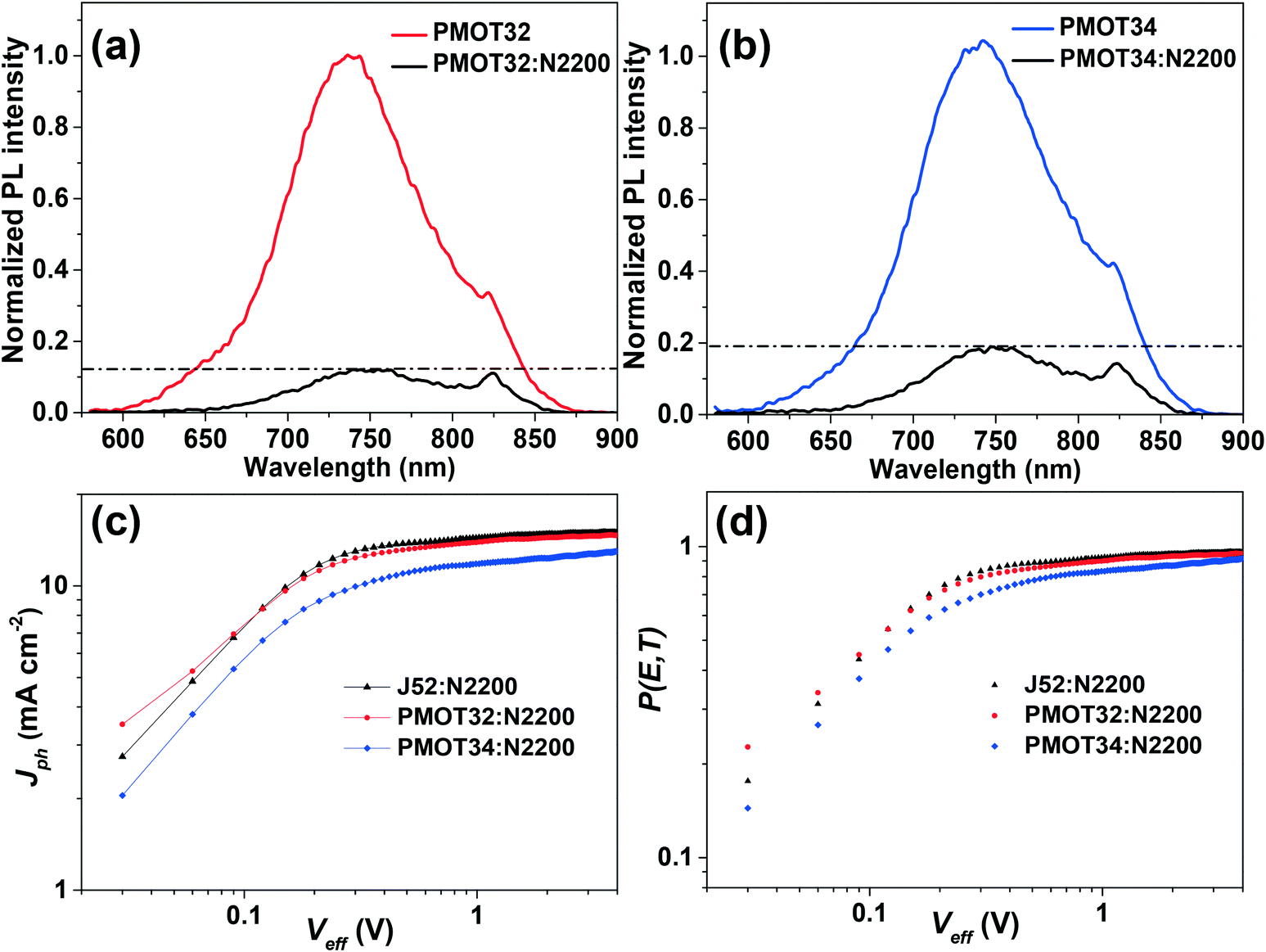 | ||
| Fig. 5 Normalized PL spectra of pristine and blend films excited at a wavelength of 545 nm: (a) PMOT32 system and (b) PMOT34 system. (c) Plots of Jphversus Veff for the three systems and (d) the corresponding curves of P(E, T) versus Veff for the systems in (c). | ||
The maximum exciton generation rates (Gmax) were calculated using the equation Gmax = Jsat/qL, where Jsat is the saturation photocurrent density, q is the electronic charge and L is the thickness of the active layers in the all-PSCs.15Jsat is determined from a curve of effective voltage (Veff) (Veff = V0 − V, where V0 is the voltage when Jph = 0 and V is the applied bias) versus photocurrent density (Jph) (Jph is defined as Jph = JL − JD, where JL and JD are the current densities under illumination and in the dark, respectively). The Jph value was saturated at Veff values close to 1 V for the three all-PSCs (Fig. 5c and Table S6†). The Gmax was determined to be 8.39 × 1027 m−3 s−1 for J52:N2200, 8.23 × 1027 m−3 s−1 for PMOT32:N2200 and 7.39 × 1027 m−3 s−1 for PMOT34:N2200, respectively. Clearly, the PMOT34:N2200 system gave a lower Gmax than those of the other two systems, which corresponds to the fact that the Jsc value was the lowest. On the basis of the Gmax value, the exciton dissociation probability P(E, T) was subsequently obtained by the equation P(E, T) = Jph/qLGmax. As shown in Fig. 5d and Table S6,† unlike the PMOT32 and J52 systems, PMOT34:N2200 exhibited a lower dissociation probability of 0.830. The relatively low value of P(E, T) for the PMOT34 system indicates ineffective charge dissociation and extraction caused by the insufficient energy offset and lower electron mobility, which is in good agreement with the PL results.
4 Conclusions
In summary, we have demonstrated an approach based on fine-tuning side chains on donor polymers for high-performance all-PSCs prepared from both halogenated and non-halogenated solvents. By introducing asymmetric conjugated side chains with one 4-methoxythiophene and one thiophene side chain on a BDT unit, PMOT32 exhibited increases in both Voc and PCE, which exceeded 8.5% in all-polymer solar cells with an N2200 acceptor. In contrast, although the PMOT34 polymer, which was substituted with 4-methoxythiophene, displayed a higher Voc, it suffered from inferior charge separation and unfavorable phase separation, which led to inferior Jsc and PCE values. Our study reveals how engineering a conjugated side chain on donor polymers affects the photovoltaic performance of all-PSCs via tuning the energy level alignment and phase separation. This provides new insights into the optimization of the molecular structure for high-performance all-PSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding support from the Recruitment Program of Global Youth Experts of China and National Science Foundation of China (51503095, 21504066, 21534003). YYL and TBY thank the Shenzhen fundamental research programs (No. JCYJ20150630145302226, JCYJ20150630145302236), Shenzhen Key Lab funding (ZDSYS201505291525382) and Peacock Plan (KQTD20140630110339343) for financial support. The support from the Ministry of Science and Technology (No. 2016YFA0200700) is acknowledged. X-ray data was acquired at beamlines 7.3.3 and 11.0.1.2 at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors thank Chenhui Zhu at beamline 7.3.3 and Cheng Wang at beamline 11.0.1.2 for assistance with data acquisition.References
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- G. Yu and A. J. Heeger, J. Appl. Phys., 1995, 78, 4510–4515 CrossRef CAS.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- L. Gao, Z.-G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS PubMed.
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- Y. Lin, F. Zhao, Y. Wu, K. Chen, Y. Xia, G. Li, S. K. K. Prasad, J. Zhu, L. Huo, H. Bin, Z.-G. Zhang, X. Guo, M. Zhang, Y. Sun, F. Gao, Z. Wei, W. Ma, C. Wang, J. Hodgkiss, Z. Bo, O. Inganäs, Y. Li and X. Zhan, Adv. Mater., 2017, 29, 1604155 CrossRef PubMed.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Rohr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- A. C. Arias, J. D. MacKenzie, R. Stevenson, J. J. M. Halls, M. Inbasekaran, E. P. Woo, D. Richards and R. H. Friend, Macromolecules, 2001, 34, 6005–6013 CrossRef CAS.
- N. Zhou, H. Lin, S. J. Lou, X. Yu, P. Guo, E. F. Manley, S. Loser, P. Hartnett, H. Huang, M. R. Wasielewski, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Adv. Energy Mater., 2014, 4, 1300785 CrossRef.
- D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Energy Environ. Sci., 2014, 7, 2939–2943 CAS.
- K. D. Deshmukh, T. Qin, J. K. Gallaher, A. C. Y. Liu, E. Gann, K. O'Donnell, L. Thomsen, J. M. Hodgkiss, S. E. Watkins and C. R. McNeill, Energy Environ. Sci., 2015, 8, 332–342 CAS.
- Y.-J. Hwang, B. A. E. Courtright, A. S. Ferreira, S. H. Tolbert and S. A. Jenekhe, Adv. Mater., 2015, 27, 4578–4584 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K. Y. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
- C. Lee, H. Kang, W. Lee, T. Kim, K.-H. Kim, H. Y. Woo, C. Wang and B. J. Kim, Adv. Mater., 2015, 27, 2466–2471 CrossRef CAS PubMed.
- Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganäs, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 10935–10944 CrossRef CAS PubMed.
- Y. Guo, Y. Li, O. Awartani, J. Zhao, H. Han, H. Ade, D. Zhao and H. Yan, Adv. Mater., 2016, 28, 8483–8489 CrossRef CAS PubMed.
- E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799–2803 CrossRef CAS PubMed.
- Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed.
- X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- W. Huang, M. Li, L. Zhang, T. Yang, Z. Zhang, H. Zeng, X. Zhang, L. Dang and Y. Liang, Chem. Mater., 2016, 28, 5887–5895 CrossRef CAS.
- F. Lin, W. Huang, H. Sun, J. Xin, H. Zeng, T. Yang, M. Li, X. Zhang, W. Ma and Y. Liang, Chem. Mater., 2017, 29, 5636–5645 CrossRef CAS.
- H. Bin, Z.-G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS PubMed.
- Z.-G. Zhang, B. Qi, Z. Jin, D. Chi, Z. Qi, Y. Li and J. Wang, Energy Environ. Sci., 2014, 7, 1966–1973 CAS.
- H. Alexander, B. Wim, G. James, S. Eric, G. Eliot, K. Rick, M. Alastair, C. Matthew, R. Bruce and P. Howard, J. Phys.: Conf. Ser., 2010, 247, 012007 CrossRef.
- E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Rev. Sci. Instrum., 2012, 83, 045110 CrossRef CAS PubMed.
- Y. Diao, Y. Zhou, T. Kurosawa, L. Shaw, C. Wang, S. Park, Y. Guo, J. A. Reinspach, K. Gu, X. Gu, B. C. K. Tee, C. Pang, H. Yan, D. Zhao, M. F. Toney, S. C. B. Mannsfeld and Z. Bao, Nat. Commun., 2015, 6, 7955 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- S. Li, H. Zhang, W. Zhao, L. Ye, H. Yao, B. Yang, S. Zhang and J. Hou, Adv. Energy Mater., 2016, 6, 1501991 CrossRef.
- B. Fan, L. Ying, Z. Wang, B. He, X.-F. Jiang, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1243–1251 CAS.
- N. C. Miller, E. T. Hoke and M. D. McGehee, in Organic Photovoltaics, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 421–444 Search PubMed.
- J. R. Tumbleston, B. A. Collins, L. Yang, A. C. Stuart, E. Gann, W. Ma, W. You and H. Ade, Nat. Photonics, 2014, 8, 385–391 CrossRef CAS.
- W. Ma, J. R. Tumbleston, M. Wang, E. Gann, F. Huang and H. Ade, Adv. Energy Mater., 2013, 3, 864–872 CrossRef CAS.
- W. Ma, J. R. Tumbleston, L. Ye, C. Wang, J. Hou and H. Ade, Adv. Mater., 2014, 26, 4234–4241 CrossRef CAS PubMed.
- G.-J. A. H. Wetzelaer, L. J. A. Koster and P. W. M. Blom, in Organic Photovoltaics, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 343–376 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00088j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |