
A novel allosteric inhibitor that prevents IKKβ activation†
 *abc
*abc
Abstract
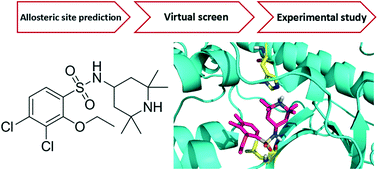
I kappa B kinase β (IKKβ) is one of the primary targets to regulate canonical NF-κB activity. The misregulation of NF-κB is associated with various diseases, including chronic inflammation and cancers. Most of the known IKKβ inhibitors target its active form and suffer from poor selectivity. In the present study, we aim to design inhibitors that can bind to the IKKβ inactive form and block its activation. We identified a potential allosteric site between the kinase domain (KD) and ubiquitin-like domain (ULD) of human IKKβ and used it to virtually screen a chemical library for allosteric inhibitors. Among the 133 compounds tested, 16 inhibited NF-κB activity by over 50% at 50 μM in a reporter gene assay. Further quantitative measurements and cytotoxicity study gave one compound 124 (3,4-dichloro-2-ethoxy-N-(2,2,6,6-tetramethylpiperidin-4-yl)benzenesulfonamide) which specifically targets the IKKβ inactive form. In cells, 124 inhibited IκBα phosphorylation and NF-κB transcriptional activity for the reporter gene with an IC50 of 35 μM by decreasing the phosphorylation level of Ser177/181 on IKKβ and blocking its activation upon TNFα stimulation. Molecular dynamics simulations demonstrated that 124 binds to the pocket between KD and ULD in the inactive conformation of IKKβ rather than the active conformation. As the first allosteric inhibitor that prevents IKKβ activation, 124 provides a good starting point for further inhibitor discovery and a probe for IKKβ enzyme cycle and regulatory mechanism study.
 Please wait while we load your content...
Please wait while we load your content...