DOI:
10.1039/C7MD00471K
(Research Article)
Med. Chem. Commun., 2018,
9, 67-80
Synthesis and pharmaceutical characterization of site specific mycophenolic acid-modified Xenopus glucagon-like peptide-1 analogs†
Received
15th September 2017
, Accepted 5th November 2017
First published on 7th November 2017
Abstract
To develop novel long-acting antidiabetic agents, mycophenolic acid (MPA) was used to modify Xenopus glucagon-like peptide-1 analog (GLP-1) (1) at three Lys residues through a γ-glutamyl linker. Similarly, 6-aminocaproic acid and 12-aminolauric acid with different lengths of fatty chain were used as MPA derivatives which were then conjugated with 1. By using proper protection and deprotection strategies, the synthetic process was completed directly on the resin to minimize the side reactions, and nine MPA-modified 1 derivatives (2a–2i) were obtained. Compounds 2b and 2c, which showed high GLP-1 receptor activation potencies and glucose lowering activities, were selected for further C-terminal modification to improve their stabilities and bioactivities, giving compounds 3a–3d. The receptor activation potencies and hypoglycemic activities of 3a–3d were comparable to that of liraglutide. Physicochemical and in vitro stability tests revealed that MPA conjugation led to enhanced albumin binding abilities as reflected by the improved stabilities of 3a–3d. In particular, at a dose of 25 nmol kg−1, the in vivo antidiabetic and insulinotropic activities of 3d were comparable to those of semaglutide. Finally, long-term administration of 3d achieved beneficial effects on glucose tolerance normalization and glycated hemoglobin (HbA1c) lowering, and no hepatotoxicity was observed. In conclusion, this research demonstrated that MPA derivatization was a practical way to develop long-acting antidiabetic peptides.
Introduction
Type 2 diabetes (T2DM) is a progressive deteriorative metabolic disease characterized by decreased insulin secretion and/or its impaired sensitivity followed by progressive hyperglycemia.1,2 Several conventional antidiabetic agents have been widely utilized for the treatment of T2DM. However, the therapeutic effects of these agents are limited, and there is still demand for improved diabetic therapies.3 Incretins are gut-derived hormones which stimulate insulin secretion and play an important role in overall postprandial insulin release.4 Among the incretins, glucagon-like peptide-1 (GLP-1) is considered to have great therapeutic potential to treat T2DM because of its unique bioactivities such as β cell proliferation and/or differentiation stimulation and glucose-dependent insulin secretion.5 Unfortunately, the biological half-life (t½) of GLP-1 is less than 2 min because of rapid enzymatic degradation by dipeptidyl peptidase-4 (DPP-IV) and neutral endopeptidase 24.11 (NEP 24.11), as well as rapid renal filtration.6 Therefore, numerous research efforts are focused on developing novel, potent and physiologically stable GLP-1 receptor agonists.7 To date, five GLP-1 receptor agonists have been approved, represented by exenatide (short-acting GLP-1 receptor agonists) and Liraglutide (long-acting GLP-1 receptor agonists). However, most of the GLP-1 receptor agonists approved or in clinical trials are based on the peptide backbone of native GLP-1 or exendin-4.8
In a previous report, by encoding the Xenopus proglucagon gene, Irwin et al. successfully identified three Xenopus GLP-1 peptides (xenGLP-1A, xenGLP-1B, and xenGLP-1C) with potent GLP-1 receptor activation and insulinotropic activities.9 Because these Xenopus GLP-1s have different amino acid (AA) sequences to GLP-1 (∼70% homologous), it is possible that they could be developed as novel GLP-1 receptor agonists. In previous research, a novel Xenopus GLP-1 analog (1, Fig. 1) with improved in vitro and in vivo bioactivities was successfully constructed. Furthermore, the t½ of 1 was moderately improved using site specific PEGylation.10
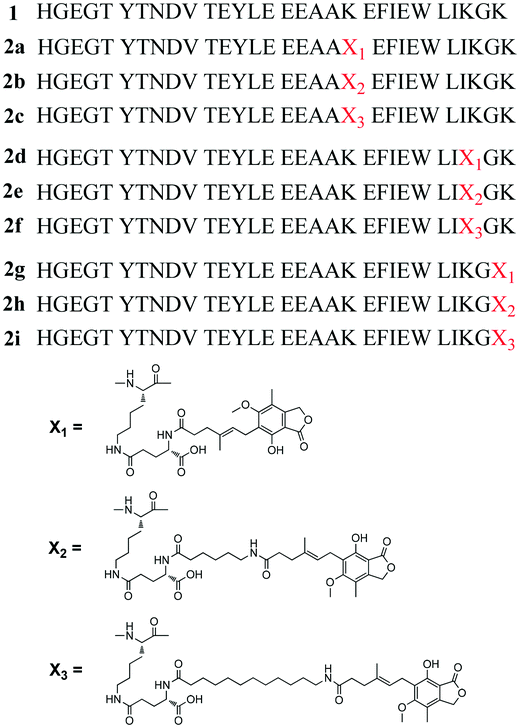 |
| | Fig. 1 Structures of 1 and MPA-modified Xenopus GLP-1 analogs (2a–2i). | |
Facilitating the physical interaction of peptide drugs with human serum albumin (HSA) is an effective way to improve the t½ of peptide drugs.11 When bound to HSA, proteolytic degradation and renal clearance is reduced because of steric effects.12 This approach was successfully used in the discovery of liraglutide and semaglutide.13 Previous research has also used this strategy to discover GLP-1 derivatives with prolonged t½ and hypoglycemic activities, such as fatty chain, dicoumarol and coumarin modified GLP-1s.14–16
Chemically modified peptides often accommodate the introduction of cysteine or a sulfur containing auxiliary. However, considering the potential immunogenicity caused by exogenous amino acid residues, such mutations are generally not suitable for therapeutic peptides. Alternatively, the amino group in the side chain of lysine (Lys) could serve as a proper group for the site specific modification of therapeutic peptides. Mycophenolic acid (MPA) is an immunosuppressive agent which extensively binds to HSA (97–98%).17 Considering the high HSA binding rate of MPA, it was hypothesized that the covalent coupling of MPA to 1 might offer a novel means to develop long-acting GLP-1 receptor agonists. The carboxyl group in MPA makes it easy to conjugate it with the Lys residue in peptide backbone of 1. However, to avoid serious reduction of the bioactivity of 1, the Lys conjugation site should be carefully investigated. In the present study, native MPA and two MPA derivatives with different lengths of fatty chain were designed and site specific conjugation through a γ-glutamyl linker was carried out on the three Lys residues of 1. A total of nine MPA-modified Xenopus GLP-1 analogs (2a–2i) were synthesized (Fig. 1). Compounds 2b and 2c which exhibited superior in vitro and in vivo bioactivities were selected for further modification to improve the stability and bioactivities, affording 3a–3d (Fig. 3). The influence of these modifications on glucose lowering, insulinotropic activity, stability, duration of action and chronic treatment effects of these compounds, was investigated.
Results and discussion
Design and synthesis of MPA-modified Xenopus GLP-1 analogs (2a–2i)
In previous research, alanine scanning was conducted on 1 to investigate the structure–activity relationship (SAR) of 1 (unpublished work). It was found that the replacement of Lys at position 20, 28, and 30 did not affect the in vitro receptor activation potency and in vivo glucose lowering activity of 1, suggesting that these positions were suitable sites for further modification. To compensate for the overall decreased water solubility caused by MPA conjugation, γ-glutamyl was used as a hydrophilic spacer between the MPA and the polypeptide. As the length of the fatty chain between MPA and the peptide backbone is important for albumin binding properties,11 two MPAs with a protracted fatty chain were designed. The peptide backbones in this study were synthesized using a fluorenylmethyloxycarbonyl (Fmoc)-based solid-phase peptide synthesis (SPPS) protocol [see ESI,† Scheme S1]. Rink Amide MBHA resin was used as a polymer support to obtain an amidated C-terminus. For site specific incorporation of MPA into peptides using solid-phase methods, the side chains of N-Fmoc-amino acids used for the peptide synthesis were protected with acid-labile groups (OtBu, tBu, Boc, and Trt), whereas the Lys to be modified was introduced using Fmoc-Lys(Dde)-OH. The N-terminal histidine (His) was replaced with Boc-His(Boc)-OH. The Dde groups were selectively removed using 2% hydrazine hydrate/dimethylformamide (DMF) (v/v), and Fmoc-Glu-OtBu was coupled with the amino group of Lys. For 2a, 2d and 2g, MPA was directly attached to the peptides through the amine of a glutamyl spacer. For 2b–2c, 2e–2f, and 2h–2i, Fmoc protected 6-aminocaproic acid or 12-aminolauric acid was used to obtain MPA analogues with an increased fatty chain length. Using this methodology, the peptide backbone was on the solid support during the whole reaction procedure, and the side reactions were minimized (Scheme 1). A total of nine MPA-modified Xenopus GLP-1 analogs (2a–2i, Fig. 1) were obtained with high yields. Crude products were purified using semi-preparative reversed phase high-performance liquid chromatography (RP-HPLC), and characterized using HPLC and micro time-of-flight mass spectrometry (MicroTOF MS; see ESI†).
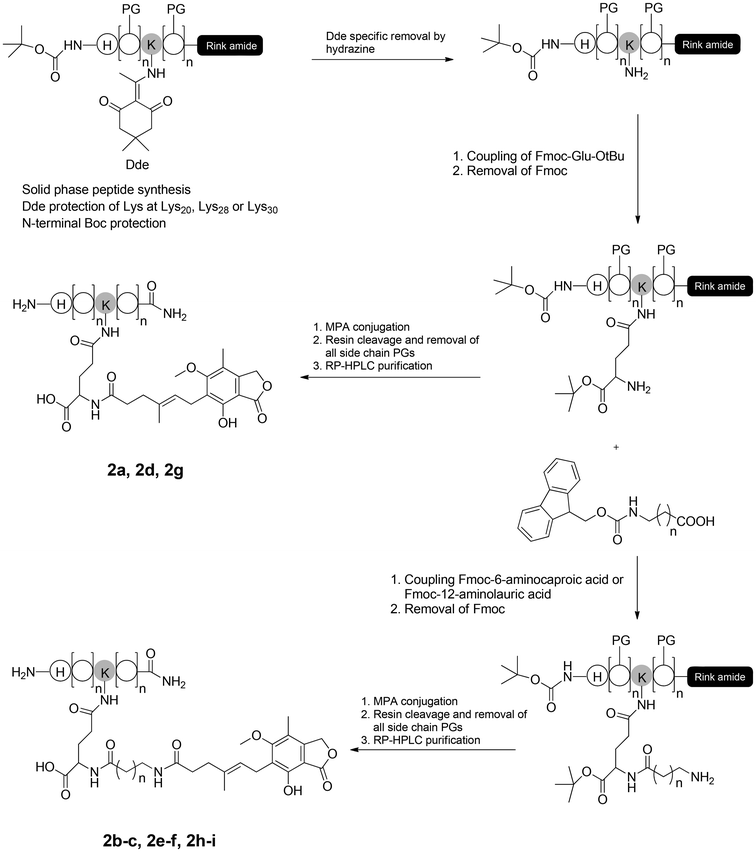 |
| | Scheme 1 Synthetic route of MPA-modified Xenopus GLP-1 analogs (2a–2i). PG: Acid labile protecting group. | |
Biological activities of the 2a–2i
Human embryonic kidney (HEK293) cells which reliably expressed the human GLP-1 receptor were used to assess the in vitro receptor activation potency of 2a–2i. As shown in Table 1, the chemical conjugation of MPAs to 1 had no significant negative effects on the receptor activation potency, and most of the analogs showed a high receptor activation potency. The MPA conjugation was well tolerated at position 20, regardless of the length of the alkyl chain of the MPAs, indicating that this position might not be involved in receptor activation. For Lys at position 28 and 30, an increase in the length of the alkyl chain of the MPAs led to decreased receptor activation potency. The greatest impact on receptor activation was observed by introducing MPAs at position 30, indicating that this position was not suitable for chemical modification. As compounds 2a–2i showed higher receptor activation potency in vitro, their in vivo antidiabetic activities were evaluated using intraperitoneal glucose tolerance testing (IPGTT) on Kunming mice, using liraglutide as the positive control. As shown in Fig. 2, the administration of liraglutide or 2a–2i (25 nmol kg−1, ip) significantly improved the glucose tolerance patterns. The blood glucose level in the control (saline) group rapidly increased to over 15 mmol L−1 at 15 min after a glucose challenge (2 g kg−1, ip), and then decreased slowly. Mice treated with liraglutide or 2a–2i demonstrated smaller glucose changes than that found in the controls. Consistent with the receptor activation results, 2a–2i with low EC50 (concentration of a drug that gives half-maximal response) values were also found to have better hypoglycemic activities in vivo. In particular, the antidiabetic effects of 2a–2cin vivo were better than those of liraglutide, as reflected by the calculated glucose area under the curve (AUC) values (Table 1). Because the glucose lowering activities of 2a–2c were similar, and a longer alkyl chain is usually more beneficial for HSA binding and in vivo half-life, 2b and 2c were finally selected for the following studies.
Table 1 The in vitro and in vivo bioactivities of 2a–2i
| Compounds |
EC50a (nM) |
AUCglucose 0–120minb |
|
Data are represented as EC50. Values are the means ± SD of three individual experiments and these were repeated three times (n = 3).
Glucose AUC values were calculated using IPGTT in Kunming mice. Means ± SD, n = 6.
|
| Liraglutide |
8.1 ± 2.4 |
584 ± 13 |
|
1
|
5.4 ± 1.9 |
461 ± 7 |
|
2a
|
6.2 ± 2.2 |
519 ± 25 |
|
2b
|
6.9 ± 1.7 |
522 ± 27 |
|
2c
|
7.3 ± 2.8 |
578 ± 28 |
|
2d
|
9.6 ± 3.2 |
668 ± 18 |
|
2e
|
11.8 ± 2.5 |
705 ± 28 |
|
2f
|
16.3 ± 2.7 |
758 ± 15 |
|
2g
|
12.1 ± 3.4 |
658 ± 20 |
|
2h
|
16.8 ± 5.1 |
801 ± 30 |
|
2i
|
23.5 ± 4.4 |
836 ± 18 |
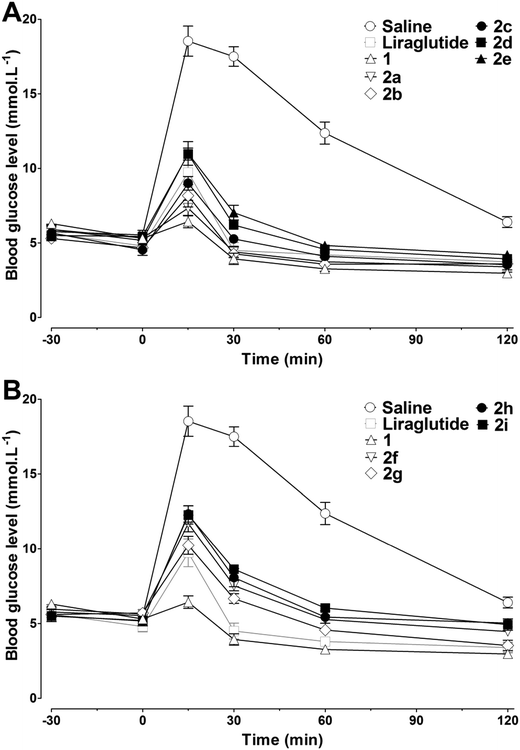 |
| | Fig. 2
In vivo bioactivities of liraglutide, 1, and 2a–2i tested using IPGTT in Kunming mice. Saline, liraglutide, 1, and 2a–2i (25 nmol kg−1) were injected ip at −30 min, followed by an ip glucose load (2 g kg−1) at 0 min. (A and B) Time–response curve of blood glucose (−30 to 120 min). Means ± SD, n = 6. | |
C-Terminal modification to increase the biological activities of 2b–2c
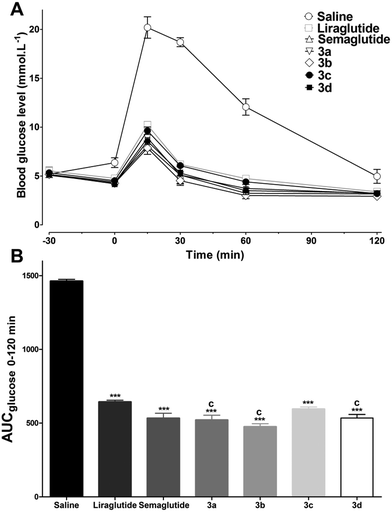
Previous research has confirmed that the bioactivity of exendin-4 was better than that of GLP-1, and the nine-AA sequence (PSSGA PPPS) at the C-terminal of exendin-4 plays an important role in the improved bioactivity.18 Furthermore, the C-terminal region of lixisenatide (PSSGA PPSKK KKKK) is similar to that of exendin-4, the main difference being the absence of a proline at position 38 and the addition of six Lys residues at position 39.19 These structural modifications significantly improved the GLP-1 receptor binding affinity as well as the stability of lixisenatide. Inspired by these studies, the C-terminal region of exendin-4 and lixisenatide was introduced into 2b or 2c, and four C-terminal modified 2b and 2c analogs (3a–3d, Fig. 3) were synthesized, purified and characterized using the same method as described previously (see ESI†). As shown in Table 2, compounds 3a–3d were found to have 1.4–2.5 fold higher receptor activation potency than their native forms (2b and 2c). Compounds 3b and 3d which have a lixisenatide C-terminal tail exhibited better receptor activation potency than 3a and 3c, indicating that the positively charged residues in C-terminal were more beneficial for the receptor activation. Furthermore, the in vivo hypoglycemic efficacies of 3a–3d were tested using IPGTT in Kunming mice. As shown in Fig. 4A, the administration of 3a–3d significantly enhanced glucose tolerance and exhibited comparable glucose lowering activities to that of liraglutide and semaglutide. Importantly, based on the calculated glucose AUC levels, no significant differences of antidiabetic effects were found between semaglutide and 3a–3d (Fig. 4B) at the dose of 25 nmol kg−1.
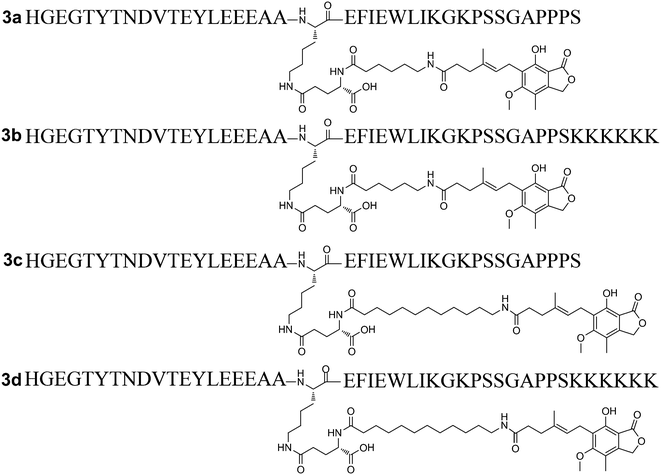 |
| | Fig. 3 Structures of C-terminal modified Xenopus GLP-1 analogs 3a–3d. | |
Table 2 The in vitro bioactivities of 3a–3d
| Compounds |
EC50a (nM) |
|
Data are represented as EC50. Values are the means ± SD of three individual experiments and these were repeated three times (n = 3).
|
| Liraglutide |
7.4 ± 1.6 |
|
3a
|
4.1 ± 1.1 |
|
3b
|
2.8 ± 0.7 |
|
3c
|
5.2 ± 2.1 |
|
3d
|
3.1 ± 0.9 |
 |
| | Fig. 4
In vivo bioactivities of liraglutide, semaglutide, and 3a–3d. (A) In vivo hypoglycemic activities of liraglutide, semaglutide, and 3a–3d tested using IPGTT in Kunming mice. Saline, liraglutide, semaglutide, and 3a–3d (25 nmol kg−1) were injected ip 30 min prior to ip glucose administration (2 g kg−1). Time–response curve of blood glucose (−30 to 120 min) are shown in panel A. (B) Calculated glucose AUC0–120min values. Means ± SD, n = 6. ***P < 0.001 versus saline, cP < 0.001 versus liraglutide. | |
Physicochemical characteristics and in vitro stabilities of 3a–3d
Improving the albumin binding affinity was an effective way to enhance the stability of peptide drugs. As MPA possesses a prominent HSA binding rate, it was predicted that the albumin binding affinity of 3a–3d could be significantly improved after the MPA conjugation. Thus, the albumin binding rates of 3a–3d were tested using an ultrafiltration method. As shown in Fig. 5A, the albumin binding rates of 3a and 3b were 64.9 ± 5.4% and 63.9 ± 2.9%, respectively, which were lower than those of liraglutide (81.8 ± 2.5%) and semaglutide (97.8 ± 1.4%). The albumin binding rates of 3c (84.8 ± 3.0%) and 3d (85.4 ± 2.7%) were similar to liraglutide and higher than those of 3a and 3b, indicating that a longer alkyl chain was more favorable for albumin binding. However, the albumin binding rates of 3c and 3d were still lower than those of semaglutide. The in vitro stabilities of 3a–3d were tested using incubation with rat plasma over 48 h, and then the results obtained were compared with those of liraglutide and semaglutide. As shown in Fig. 5B, liraglutide and semaglutide possessed a half-life of ∼31.5 h and >48 h at 37 °C, respectively, and the half-lives of 3a (∼19.3 h) and 3b (∼22.1 h) were shorter than those of liraglutide and semaglutide. Interestingly, the albumin binding rates of 3c and 3d were similar to those of liraglutide, whereas the in vitro stabilities of 3c (∼34.2 h) and 3d (∼39.8 h) were better than those of liraglutide. The improved stabilities of 3c and 3d may be attributed to the unique sequence of 1 and the C-terminal tail modification, which not only improved the bioactivities but also enhanced the stabilities of 3c and 3d. Semaglutide, which exhibited the highest albumin binding rate, also possessed the longest in vitro stability, revealing direct evidence of the albumin binding ability–stability relationship of GLP-1 analogues.
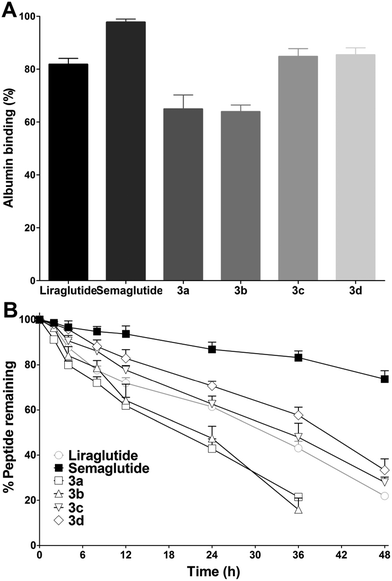 |
| | Fig. 5 Physicochemical and stability characterization of liraglutide, semaglutide, and 3a–3d. (A) The albumin binding capacities of liraglutide, semaglutide, and 3a–3d. (B) The degradation profiles of liraglutide, semaglutide, and 3a–3d incubated with rat plasma. Means ± SD, n = 3. | |
Glucose lowering and insulin secretion assay
As 3c and 3d exhibited superior bioactivities and stabilities, the hypoglycemic and insulinotropic activities of 3c and 3d in genetically diabetic mice (db/db) were evaluated further. As shown in Fig. 6A, blood glucose levels in the control (saline injected) group maintained a hyperglycemic state during 0–120 min, whereas liraglutide, semaglutide, 3c and 3d (25 nmol kg−1) administration potently reduced the blood glucose levels to ∼14.3, ∼11.0, ∼12.4 and ∼10.7 mmol L−1, respectively, at 15 min after glucose load (ip, 1 g kg−1). In particular, at the dose of 25 nmol kg−1, 3d exhibited a more potent hypoglycemic activity than that of liraglutide (P < 0.001, Fig. 6B) which was comparable to that of semaglutide. Furthermore, the plasma insulin levels were also determined during 0–120 min. As shown in Fig. 6C, the insulin levels in mice treated with liraglutide, semaglutide, 3c and 3d (25 nmol kg−1) were significantly enhanced to ∼15.8, ∼17.2, ∼16.5, ∼18.0 mIU L−1, respectively, at 15 min after the glucose load. The calculated insulin AUC values revealed that the insulinotropic activity of 3d was better than those of 3c (P < 0.05) and liraglutide (P < 0.01) at the dose of 25 nmol kg−1, and the insulinotropic activity of 3d was comparable to that of semaglutide (Fig. 6D). Importantly, the decreases in blood glucose levels in the 3c and 3d groups were accompanied by increases in plasma insulin levels, which was in accordance with a GLP-1-dependent mechanism.
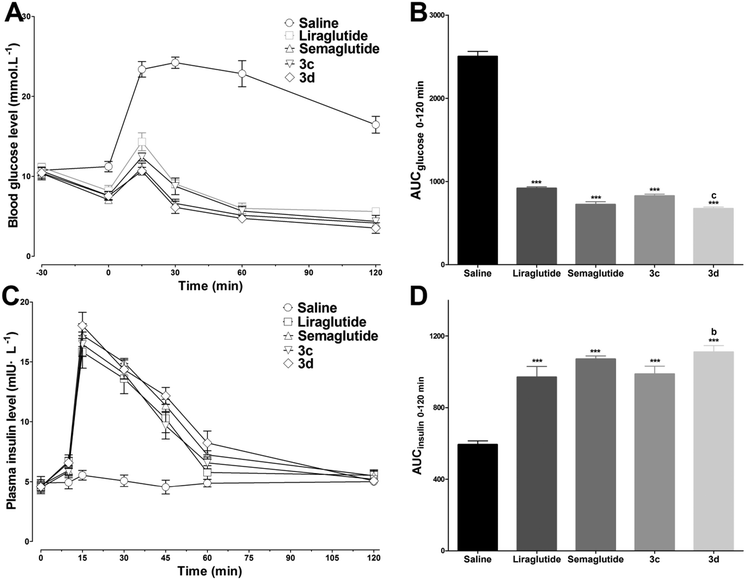 |
| | Fig. 6 Hypoglycemic and insulinotropic activities of 3c and 3d determined using IPGTT in db/db mice. Saline, liraglutide, semaglutide, 3c and 3d (25 nmol kg−1) were administered ip 30 min prior to the ip glucose load (1 g kg−1). (A) The time–response curve of blood glucose (−30 to 120 min). (B) Calculated glucose AUC0–120min values. (C) The time–response curve of plasma insulin (0 to 120 min). (D) Calculated insulin AUC0–120min values. Means ± SD, n = 6. ***P < 0.001 versus saline, bP < 0.01 versus liraglutide, cP < 0.001 versus liraglutide. | |
Antidiabetic duration tests
To test the long-acting hypoglycemic abilities of 3c and 3d further, two different antidiabetic duration tests were performed in db/db mice. Considering the in vitro stability results, liraglutide was selected as the positive control. First, to imitate the multiple meal-a-day pattern, a multiple oral glucose tolerance test (OGTT) was performed in fasting db/db mice. As illustrated in Fig. 7, the blood glucose levels increased rapidly in the control mice with saline and maintained a hyperglycemic state after each oral glucose load. The hypoglycemic effects of liraglutide were superior during 0–12 h, but were moderately reduced during 12–24 h. In accordance with the in vitro stability results, 3d, which exhibited the longest in vitro half-life was also found to possess the greatest in vivo antidiabetic duration, and the glucose lowering effect of 3d was relatively unchanged during the whole experimental period. Next, the antidiabetic duration for liraglutide, 3c and 3d were further assessed in non-fasting db/db mice. As shown in Fig. 8A, there was no notable difference in the lowest glucose level (∼12.0 mmol L−1) reached by liraglutide, 3c and 3d groups at a dose of 25 mmol kg−1. The rebound time from the lowest glucose level to a diabetic glucose level (>15 mmol L−1) of liraglutide was ∼13.0 h, whereas mice treated with 3c (∼18.9 h) and 3d (∼23.8 h) exhibited a significantly delayed rebound time. Furthermore, the dose dependency of the hypoglycemic effects of 3c and 3d were examined. At the highest dose (100 mmol kg−1), the rebound time from the lowest glucose level to a diabetic glucose level (>15 mmol L−1) of 3c (∼30.4 h) and 3d (∼38.4 h) were significantly enhanced. The calculated AUC0–48h values revealed that 3d possessed greater antidiabetic effects than 3c (P < 0.05), which was independent of the dose (Fig. 8B). In particular, the antidiabetic duration of 3d was nearly 40 h at the highest dose in db/db mice. It is well known that the drug clearance speed in mice is much faster than in humans. Thus, the hypoglycemic duration of 3d may be longer than the ∼40 h observed in the present study in humans.
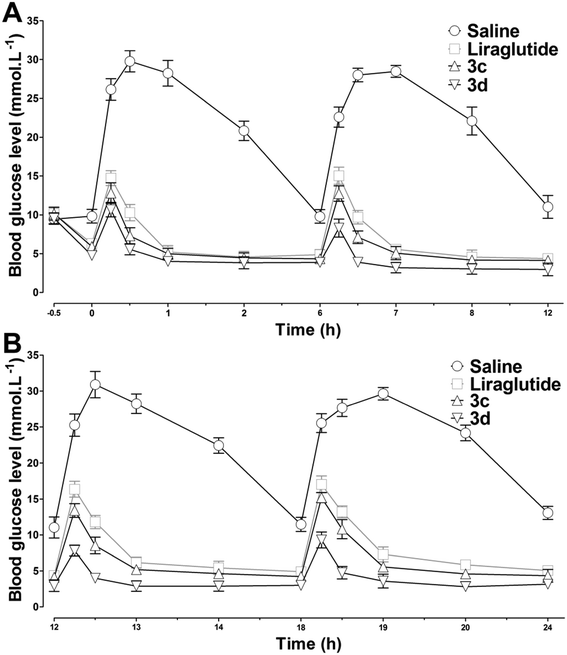 |
| | Fig. 7 Long-acting (24 h) hypoglycemic effects of 3c and 3d determined using the multiple OGTT in db/db mice. Mice were fasted for 18 h and saline, liraglutide, 3c or 3d (25 nmol kg−1) were administered ip, 30 min prior to the first oral glucose load (1.5 g kg−1), and the following OGTT were repeated at 6 h (A), 12 h and 18 h (B). The time–response curve of the blood glucose levels are shown in panels A and B. Means ± SD, n = 6. | |
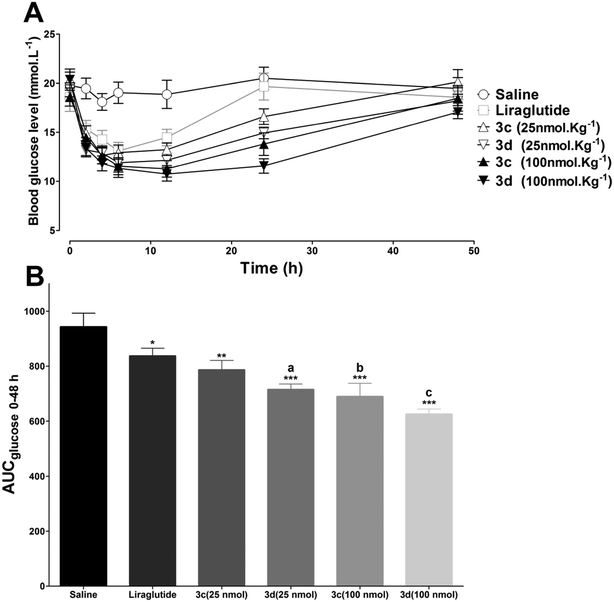 |
| | Fig. 8 Glucose lowering and stabilizing effects of liraglutide, 3c and 3d studied in non-fasting db/db mice. (A) Dose response for blood glucose lowering effects of liraglutide (25 nmol kg−1), 3c and 3d (25 or 100 nmol kg−1) in diabetic db/db mice after ip dosage. (B) Calculated glucose AUC0–48h values. *P < 0.05 versus saline, **P < 0.01 versus saline, ***P < 0.001 versus saline, aP < 0.05 versus liraglutide, bP < 0.01 versus liraglutide, cP < 0.001 versus liraglutide. Means ± SD, n = 6. | |
Chronic treatment effects of 3d
Long-term peripheral administration of GLP-1R agonists could achieve beneficial effects on T2DM, including glycated hemoglobin (HbA1c) and plasma lipids' reduction, food intake and weight gain suppression, increasing insulin sensitivity and β-cell neogenesis and/or differentiation induction.20 As 3d exhibited prominent long-acting hypoglycemic effects in vivo, effects of chronic ip administration of 3d were further studied in diabetic db/db mice. Based on the antidiabetic duration test results, liraglutide (25 mmol kg−1) was injected twice daily, and 3d (50 mmol kg−1) was injected once daily to achieve the same doses as liraglutide for five weeks. Mice were divided with matched HbA1c, a reliable index for long-term glycemic control. As shown in Fig. 9A, the HbA1c values stayed high in the control group after treatment, whereas the liraglutide and 3d treatments prevented the worsening of the HbA1c. Compared with day 0, the HbA1c in the 3d group was reduced by ∼6.7%, which was better than that in the liraglutide group (∼3.3%). The non-fasting blood glucose levels in saline treated mice stayed high during the treatment period, whereas the liraglutide and 3d treatments normalized the blood glucose levels (11.2–14.9 mmol L−1, Fig. 9B) throughout the five weeks. It is important to note, that the decreased non-fasting blood glucose levels were attributed to the insulinotropic activities of liraglutide and 3d, as reflected by the noticeably increased non-fasting plasma insulin concentrations in the liraglutide and 3d treated mice (Fig. 9C). It should be noted that the non-fasting plasma insulin concentrations in mice treated with 3d were slightly lower than those in the liraglutide group from day 22, indicating that the insulin sensitivity in the 3d treated mice was improved.
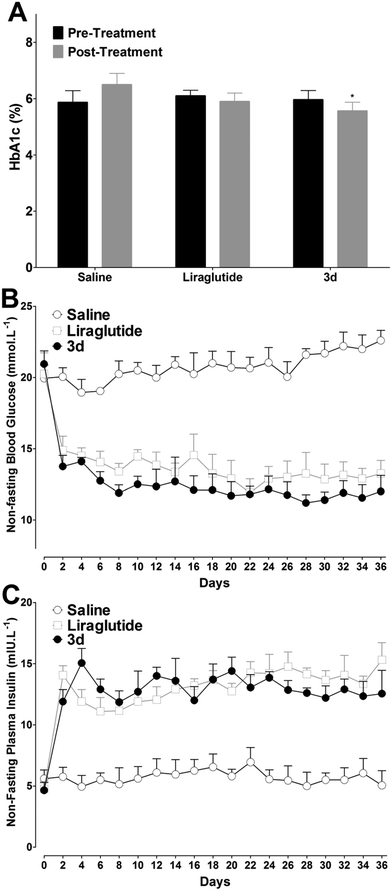 |
| | Fig. 9 The effect of chronically administered liraglutide and 3d treatment in db/db mice on HbA1c (A), non-fasting blood glucose levels (B) and non-fasting plasma insulin levels (C). Mean ± SD, n = 6. | |
Although continuous weight gains were observed in liraglutide and 3d treated mice, these weight gains were lower than those in the saline treated mice over the five week period. The relative body weight reductions compared with saline treated mice at day 36 were found to be ∼55% and ∼48% for 3d and liraglutide, respectively (Fig. 10A), and the food intake was also suppressed by both 3d and liraglutide treatments (Fig. 10B). At the end of the experiment, an IPGTT was performed to determine whether long-term 3d treatment improved the glucose tolerance. As shown in Fig. 10C, the glucose changes in saline treated mice were higher than those in the liraglutide and 3d groups, and this was independent of the measuring time. Furthermore, the glucose AUC0–120min was lower in 3d treated mice than that in liraglutide treated mice (Fig. 10C, inset). The enhanced glucose tolerance in 3d treated mice was presumably attributed to the protracted activity of β cell neogenesis and/or proliferation. Biochemical analysis revealed that the 3d treatment significantly decreased serum triglyceride (TG), total cholesterol (TC) and low-density lipoprotein (LDL) cholesterol, when compared to the saline group (P < 0.01, P < 0.01 and P < 0.05, respectively, Table 3). However, the high-density lipoprotein (HDL) cholesterol was only slightly reduced after 3d treatment. It is important to note, that the 3d treatment did not enhance the alanine aminotransferase (ALT) and aspartate aminotransferase (AST) values, indicating there is no hepatotoxicity after chronic treatment with 3d (Table 3).
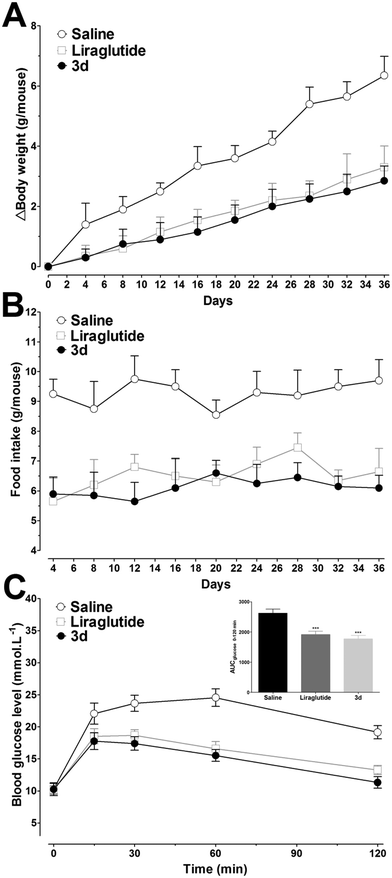 |
| | Fig. 10 The effects of chronic treatment using liraglutide and 3d in db/db mice. (A) Body weight gain. (B) Food intake. (C) Changes of blood glucose levels after ip administration of 1 g kg−1 glucose after five weeks of treatment. Inset: Calculated glucose AUC0–120min values. ***P < 0.001 versus saline. Means ± SD, n = 6. | |
Table 3 Biochemical analysis in db/db mice treated with liraglutide or 3d
|
|
Control |
Liraglutide |
3d
|
| TG: triglyceride, TC: total cholesterol, LDL-cholesterol: low-density lipoprotein cholesterol, HDL-cholesterol: high-density lipoprotein cholesterol, ALT: alanine aminotransferase, AST: aspartate aminotransferase. Means ± SD, n = 6. P < 0.05 versus control. P < 0.01 versus control. |
| TG (mmol L−1) |
2.3 ± 0.3 |
1.7 ± 0.2a |
1.4 ± 0.2b |
| TC (mmol L−1) |
6.5 ± 0.4 |
4.9 ± 0.4b |
4.5 ± 0.3b |
| LDL-cholesterol (mmol L−1) |
2.2 ± 0.3 |
1.4 ± 0.2a |
1.4 ± 0.1a |
| HDL-cholesterol (mmol L−1) |
1.7 ± 0.3 |
1.5 ± 0.2 |
1.6 ± 0.4 |
| ALT (IU L−1) |
61.4 ± 10.0 |
55.6 ± 6.1 |
56.9 ± 9.1 |
| AST (IU L−1) |
75.0 ± 7.4 |
65.7 ± 6.4 |
62.4 ± 7.5 |
Experimental
Materials and animals
Rink amide MBHA resin, N-Fmoc amino acids, diisopropylcarbodiimide (DIC), 1-hydroxybenzotriazole (HOBt), Fmoc-Lys(Dde)-OH, Fmoc-Glu-OtBu, Boc-His(Boc)-OH, liraglutide and semaglutide were obtained from GL Biochem (Shanghai) Ltd. (China). Kits for measuring the levels of cyclic adenosine monophosphate (cAMP) and insulin were purchased from Cisbio International (Bedford, MA, USA) and Nanjing Jiancheng Bioengineering Institute, (Jiangsu, China), respectively. Fmoc protected 6-aminocaproic acid and 12-aminolauric acid were purchased from Xi'an Ruixi Biological Technology Co., Ltd (Xi'an, China). HEK293 cell lines were obtained from Multispan, Inc (Hayward, CA, USA). The rat plasma was obtained from the Shanghai Jihe Biological Technology Co., Ltd (Shanghai, China). All other reagents were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) or Fisher Scientific (Pittsburgh, PA, USA) unless otherwise indicated. Male Kunming mice (20–25 g) and C57BL/6J-m+/+ Leprdb (db/db) mice (9 weeks old, 30–40 g) were obtained from the Comparative Medical Center of Yangzhou University (Yangzhou, China) and the Model Animal Research Center of Nanjing University (Nanjing, China), respectively. All the animals, six in a cage, were housed in an air-conditioned room (25 ± 2 °C) with a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light–dark cycle, and allowed free access to food (standard chow) and water. All animal studies were conducted in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (revised 2011), the Laboratory Animal Management Regulations in China, and approved by the institutional committee at Jiangsu Normal University.
:12 h light–dark cycle, and allowed free access to food (standard chow) and water. All animal studies were conducted in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (revised 2011), the Laboratory Animal Management Regulations in China, and approved by the institutional committee at Jiangsu Normal University.
General procedure for peptide synthesis
The peptide backbones of 1, 2a–2i, and 3a–3d were synthesized using standard Fmoc/tBu SPPS methodology using a semi-automated PSI-200 peptide synthesizer (Peptide Scientific Inc., USA).21 Rink amide MBHA amide resin was used with a loading of 0.382 mmol g−1. For 2a–2i and 3a–3d with an MPA side chain, the Lys to be modified was replaced with Fmoc-Lys(Dde)-OH and the N-terminal histidine was replaced with Boc-His(Trt)-OH. The Dde protection group was selectively removed by washing five times (10 min) with 2% hydrazine hydrate in DMF (v/v). Then, Fmoc-Glu-OtBu was added and coupled with the amino group of Lys using DIC/HOBT for 2.5 h, and the Fmoc protected group was removed using 20% piperidine/DMF (v/v). For 2a, 2d and 2g, MPA (4 equiv.) was dissolved with DIC (4 equiv.) and HOBt (4 equiv.) in DMF (5 ml), and coupled with the amine of glutamine (Glu) for 3 h. For 2b–2c, 2e–2f, and 2h–2i, Fmoc protected 6-aminocaproic acid or 12-aminolauric acid was firstly coupled with the amine of Glu, and the Fmoc was removed using 20% piperidine/DMF, then MPA (4 equiv.) was added and coupled using the same method as described previously. Finally, the crude products were cleaved from the resin using 1,2-ethanediol/phenol/water/thioanisole/trifluoroacetic acid (2.5:5:5:5:82.5) followed by precipitation in ether and then washing four times with ether. The crude analogues were purified using reverse-phase high performance liquid chromatography (RP-HPLC) (LC-20AP, Shimadzu) equipped with a C18 column (Hypersil GOLD, 250 × 20 mm, 12 μM, ThermoFisher Scientific). The purity of all the products was confirmed using analytical HPLC, and characterized using time-of-flight mass spectrometry (TOF-MS; micrOTOF QII Bruker) using an ESI method.
In vitro functional assay
HEK293 cells that expressed the hGLP-1 receptor were thawed and used for a functional assay, using a previously described method.15 Cells were suspended in assay buffer [Dulbecco's modified Eagle's medium (DMEM) growth medium, 20 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer, 1% penicillin–streptomycin, 2 mM L-Glu, and 0.5% fetal bovine serum]. Cells were plated out into 384-well microplates. Compounds to be tested were solubilized in dimethylsulfoxide and diluted in assay buffer and transferred to the microplate to reach assay concentrations of 1 × 10−13–1 × 10−6 M. Then, the plate was incubated in 5% carbon dioxide for 30 min at 37 °C. A Cisbio cAMP dynamic 2 kit was used to assay the cAMP concentrations using homogeneous time-resolved fluorescence technology. A multilabel reader (EnVision 2104 PerkinElmer, UK) was used (according to the manufacturer's instructions) to measure the fluorescence in each sample. The cAMP data was imported into GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA), and the EC50 values were determined using sigmoidal curve fitting.
IPGTT in Kunming mice
The acute hypoglycemic efficacies of liraglutide, semaglutide, 2a–2i and 3a–3d were determined in male Kunming mice using IPGTT.22 Briefly, mice (n = 6, 20–25 g, male) were fasted overnight, saline, liraglutide, semaglutide, 2a–2i and 3a–3d (25 nmol kg−1) were injected (ip) at t = −30 min, and then the mice were ip loaded with glucose (0 min, 25 nmol kg−1). The venous blood samples in each group were obtained from the tail vein at −30, 0, 15, 30, 60, and 120 min. Blood glucose levels were measured using a glucometer (GA-3, Sannuo, China).
Albumin binding assay
The albumin binding properties of liraglutide, semaglutide, and 3a–3d were determined using a modified ultra filtration method.23 Briefly, test compounds were dissolved in phosphate buffered saline (PBS; pH 7.4, 100 μg ml−1, 1 ml), and HSA (10 mg ml−1 in PBS, pH 7.4, 3 ml) was then added and the solution was then vortex mixed for 1 min. The mixture was incubated for 60 min at 37 °C. Then, 1 ml of the sample was loaded into a Centricon centrifugal filter device with a 30 kDa molecular weight cutoff (Millipore, Bedford, MA, USA). The samples were centrifuged for 30 min at 3500 rpm, and the content of the compound in the filtrate was determined using HPLC. In a similar way, the adsorption of each compound to the filter membrane was also investigated. The adsorption rate by the membrane was <4% in each case. The HSA binding testing using ultrafiltration was repeated three times for all the compounds studied.
In vitro stability
The in vitro stability of liraglutide, semaglutide, and 3a–3d were determined using a previously described method with some modifications.14 In brief, plasma was obtained from male Sprague Dawley rats (200–250 g) and stored at −20 °C until required. Liraglutide, semaglutide, and 3a–3d were dissolved in PBS (pH 7.4) and 0.5 ml of the tested compound was added into the plasma (1 ml) to achieve an initial concentration of 1000 ng ml−1 and then vortex mixed for 30 s. The mixture was incubated at 37 °C for 48 h. After 2, 4, 8, 12, 24, 36, and 48 h, a sample (100 μL) was removed from the incubation mixture and mixed with 200 μL acetonitrile containing 2% formic acid to precipitate the plasma protein. After centrifugation at 14000 rpm for 10 min, 50 μL of supernatant was injected into the LC-MS/MS system. The signals of the test compounds were identified using multiple reaction monitoring on a triple quadrupole MS (Sciex API 4000 mass spectrometer, Applied Biosystems, USA). The conditions for the RP-HPLC separation are described elsewhere.15 Degradation curves of all the tested compounds were determined in triplicate.
Glucoregulatory and insulinotropic assay
The effects of 3c and 3d on glucoregulation and insulinotropicity were determined using IPGTT on db/db mice.22 Briefly, male db/db mice (30–40 g, n = 6, 9 weeks) were fasted overnight for 18 h, and then saline (control), liraglutide, semaglutide, 3c or 3d (25 nmol kg−1) were injected ip at −30 min. Glucose (1 g kg−1) was administered ip at 0 min. Blood samples were obtained from the tail vein before glucose injection (−30 min) and at 0, 10, 15, 30, 45, 60, and 120 min. Blood glucose levels were tested immediately using a GA-3 one-touch glucometer. The remaining blood samples were centrifuged to obtain plasma samples, and plasma insulin levels were tested using a mouse insulin enzyme-linked immunosorbent assay (ELISA) kit (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China).
Long-acting glucoregulatory tests
The antidiabetic duration of 3c and 3d were determined using two different tests on fasting and non-fasting db/db mice.24,25 In a first experiment (multiple OGTT), db/db mice (n = 6, 30–40 g, 9 weeks) were fasted overnight for 18 h, and the mice were injected ip with saline (control), liraglutide, 3c and 3d (25 nmol kg−1) at −0.5 h. Glucose (1.5 g kg−1) was orally administered at 0 h, and blood samples in each group of mice were obtained at 0, 0.25, 0.5, 1, 2, and 3 h and measured using a GA-3 one-touch glucometer. In order to imitate the multiple meals a day pattern of mice and to test the long-acting antidiabetic effects of 3c and 3d, the glucose was loaded every 6 h in a 24 h period, and the testing intervals of the blood glucose levels were identical in each OGTT. The hypoglycemic efficacies of 3c and 3d were also evaluated in non-fasting db/db mice. Under non-fasting conditions, db/db mice (male, 30–40 g, 9 weeks, n = 6) were allowed free access to water and food. Each group of mice were injected ip with saline (control), liraglutide (25 nmol kg−1), 3c or 3d (25 or 100 nmol kg−1) at 0 h, and blood samples were collected at 0, 2, 4, 6, 12, 24 and 48 h and monitored using the same method as described previously.
Chronic treatment tests
Male db/db mice (n = 6, 30–40 g, 9 weeks) that were confirmed as being diabetic were divided into three groups with matched HbA1c (matched using a chemistry analyzer, Bayer Diagnostics, USA).21 The control group was injected ip with saline twice daily. Liraglutide (25 nmol kg−1) was used as positive control and administered ip twice daily, and 3d (50 nmol kg−1) was injected ip once daily in accordance with their antidiabetic duration profiles for 35 days. HbA1c was measured at day 0 and day 36. Non-fasting blood glucose levels and insulin concentrations were monitored at four day intervals. To account for the effects of the peptide induced reduction in food intake and body weight, food intake and body weight gain in each group of mice were determined every four days. A glucose tolerance test was performed following the 35 days of treatment. Each group of mice were fasted overnight for 18 h, and challenged with glucose (1 g kg−1, ip) followed by serial collection of blood for glucose analysis at 0, 15, 30, 60 and 120 min. The blood glucose levels determined using the GA-3 one-touch glucometer.
Data analysis
Results are expressed as means ± standard deviation (SD). Statistical significances were performed using a one-way analysis of variance (ANOVA), followed by post hoc Tukey's tests. AUC analyses for blood glucose and plasma insulin were made using GraphPad Prism version 5.0. P < 0.05 were considered to be significantly different.
Conclusions
Novel Xenopus GLP-1 analogs with covalently attached MPA as albumin binders were synthesized. Using reasonable site specific MPA conjugation and C-terminal modification, compounds 3a–3d were successfully constructed and these exhibited potent in vitro receptor activation potency and in vivo hypoglycemic activities. In vitro physicochemical characterization studies proved that MPA conjugation led to significantly enhanced albumin binding abilities and stabilities. Importantly, long-term 3d treatment achieved beneficial effects on HbA1c lowering and glucose tolerance normalization. The preclinical studies suggested that 3d has potential to be developed as an efficient GLP-1 receptor agonist for the treatment of T2DM. This study also demonstrates a novel means for half-life extension of peptide drugs, specifically, how MPA albumin binders can promote long-acting efficacy of GLP-1 analogs. Further customization of the MPA albumin binder in applications for other proteins or peptide pharmaceuticals is envisioned.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 81602964, 81602960 and 81703346), the Natural Science Foundation of Jiangsu Province (Grants No. BK20150243 and BK20161028), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, the Science and Technology Projects of Jiaxing (No. 2017AY33074) and the Key Project of Jiaxing University students' research training program (No. 85171739).
Notes and references
- J. J. Meier, Nat. Rev. Endocrinol., 2012, 8, 728–742 CrossRef PubMed.
- S. A. Sadry and D. J. Drucker, Nat. Rev. Endocrinol., 2013, 9, 425–433 CrossRef PubMed.
- G. Mingrone, S. Panunzi, A. De Gaetano, C. Guidone, A. Iaconelli, G. Nanni, M. Castagneto, S. Bornstein and F. Rubino, Lancet, 2015, 386, 964–973 CrossRef.
- A. Evers, T. Haack, M. Lorenz, M. Bossart, R. Elvert, B. Henkel, S. Stengelin, M. Kurz, M. Glien, A. Dudda, K. Lorenz, D. Kadereit and M. Wagner, J. Med. Chem., 2017, 60, 4293–4303 CrossRef PubMed.
- E. M. Bech, M. C. Martos-Maldonado, P. Wismann, K. K. Sørensen, S. B. van Witteloostuijn, M. B. Thygesen, N. Vrang, J. Jelsing, S. L. Pedersen and K. J. Jensen, J. Med. Chem., 2017, 60, 7434–7446 CrossRef PubMed.
- L. L. Baggio and D. J. Drucker, Gastroenterology, 2007, 132, 2131–2157 CrossRef PubMed.
- B. Manandhar and J.-M. Ahn, J. Med. Chem., 2015, 58, 1020–1037 CrossRef PubMed.
- C. Mapelli, S. I. Natarajan, J.-P. Meyer, M. M. Bastos, M. S. Bernatowicz, V. G. Lee, J. Pluscec, D. J. Riexinger, E. S. Sieber-McMaster and K. L. Constantine, J. Med. Chem., 2009, 52, 7788–7799 CrossRef PubMed.
- D. M. Irwin, M. Satkunarajah, Y. Wen, P. L. Brubaker, R. A. Pederson and M. B. Wheeler, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7915–7920 CrossRef.
- J. Han, X. Chen, Y. Wang, Y. Fei, F. Zhou, Y. Zhang, L. Liu, P. Si and J. Fu, Biochem. Pharmacol., 2017, 142, 155–167 CrossRef PubMed.
- K. Madsen, L. B. Knudsen, H. Agersoe, P. F. Nielsen, H. Thøgersen, M. Wilken and N. L. Johansen, J. Med. Chem., 2007, 50, 6126–6132 CrossRef PubMed.
- S. Son, S. Y. Chae, C. W. Kim, Y. G. Choi, S. Y. Jung, S. Lee and K. C. Lee, J. Med. Chem., 2009, 52, 6889–6896 CrossRef PubMed.
- J. Lau, P. Bloch, L. Schäffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz–Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef PubMed.
- J. Han, X. Huang, L. Sun, Z. Li, H. Qian and W. Huang, Biochem. Pharmacol., 2013, 86, 297–308 CrossRef PubMed.
- J. Han, L. Sun, Y. Chu, Z. Li, D. Huang, X. Zhu, H. Qian and W. Huang, J. Med. Chem., 2013, 56, 9955–9968 CrossRef PubMed.
- J. Han, L. Sun, X. Huang, Z. Li, C. Zhang, H. Qian and W. Huang, Br. J. Pharmacol., 2014, 171, 5252–5264 CrossRef PubMed.
- I. Nowak and L. M. Shaw, Clin. Chem., 1995, 41, 1011–1017 Search PubMed.
- L. Simonsen, J. J. Holst and C. F. Deacon, Diabetologia, 2006, 49, 706–712 CrossRef PubMed.
- J. Rosenstock, D. Raccah, L. Korányi, L. Maffei, G. Boka, P. Miossec and J. E. Gerich, Diabetes Care, 2013, 36, 2945–2951 CrossRef PubMed.
- D. Chen, J. Liao, N. Li, C. Zhou, Q. Liu, G. Wang, R. Zhang, S. Zhang, L. Lin and K. Chen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 943–948 CrossRef PubMed.
- B. Yang, X. Li, C. Zhang, S. Yan, W. Wei, X. Wang, X. Deng, H. Qian, H. Lin and W. Huang, Org. Biomol. Chem., 2015, 13, 4551–4561 Search PubMed.
- J. Han, Y. Wang, Q. Meng, G. Li, F. Huang, S. Wu, Y. Fei, F. Zhou and J. Fu, Eur. J. Med. Chem., 2017, 132, 81–89 CrossRef PubMed.
- H. Liao, J. Wu, E. Kuhn, W. Chin, B. Chang, M. D. Jones, S. O'Neil, K. R. Clauser, J. Karl, F. Hasler, R. Roubenoff, W. Zolg and B. C. Guild, Arthritis Rheum., 2004, 50, 3792–3803 CrossRef PubMed.
- T. H. Kim, H. H. Jiang, S. Lee, Y. S. Youn, C. W. Park, Y. Byun, X. Chen and K. C. Lee, Bioconjugate Chem., 2011, 22, 625–632 CrossRef PubMed.
- X. Cui, Q. Meng, Y. Chu, X. Gu, Y. Tang, F. Zhou, Y. Fei, J. Fu and J. Han, RSC Adv., 2016, 6, 94408–94416 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00471k |
|
| This journal is © The Royal Society of Chemistry 2018 |

 *a,
Junjie
Fu
b,
Lidan
Sun
c,
Yue
Han
a,
Qiuyi
Mao
a,
Fang
Liao
a,
Xinshi
Zheng
a and
Ke
Zhu
a
*a,
Junjie
Fu
b,
Lidan
Sun
c,
Yue
Han
a,
Qiuyi
Mao
a,
Fang
Liao
a,
Xinshi
Zheng
a and
Ke
Zhu
a










