
Synthesis and in vitro evaluation of substituted 3-cinnamoyl-4-hydroxy-pyran-2-one (CHP) in pursuit of new potential antituberculosis agents†
Zubair Shanib
Bhat‡
 ab,
Hafiz
Ul Lah‡
c,
Muzafar Ahmad
Rather
a,
Mubashir
Maqbool
a,
Tabassum
Ara
d,
Zahoor
Ahmad
*ab and
Syed Khalid
Yousuf
*bc
ab,
Hafiz
Ul Lah‡
c,
Muzafar Ahmad
Rather
a,
Mubashir
Maqbool
a,
Tabassum
Ara
d,
Zahoor
Ahmad
*ab and
Syed Khalid
Yousuf
*bc
aClinical Microbiology and PK/PD Division, CSIR-Indian Institute of Integrative Medicine, Sanatnagar, Srinagar, 190005, India. E-mail: zahoorap@iiim.ac.in; Fax: +91 1942441331; Tel: +91 194 2431253/55 Tel: +91 9906593222
bAcademy of Scientific and Innovative Research, Indian Institute of Integrative Medicine (CSIR), Sanatnagar, Srinagar, Jammu and Kashmir 190005, India. E-mail: khalidiiim@gmail.com
cMedicinal Chemistry Division, CSIR-Indian Institute of Integrative Medicine, Sanatnagar, Srinagar, 190005, India
dNational Institute of Technology-Srinagar, Jammu, Jammu & Kashmir 190006, India
First published on 6th December 2017
Abstract
Tuberculosis is an ever-evolving infectious disease that urgently needs new drugs. In the search for new antituberculosis agents, a library of 3-cinnamoyl-4-hydroxy-6-methyl-2H-pyran-2-ones (CHPs) (2a–2y) was synthesized and evaluated against a standard virulent laboratory strain of Mycobacterium tuberculosis H37Rv. Out of 25 compounds, 11, 5, 7 and 2 (2a and 2u) showed least, moderate, good and appreciable activities, respectively, based on minimum inhibitory concentrations (MICs). Both 2a and 2u exhibited an MIC value of 4 μg ml−1, which was close to those of standard antituberculosis drugs ethambutol, streptomycin and levofloxacin. Neither 2a nor 2u showed any activity against Gram-positive or Gram-negative bacteria and even against non-tuberculous mycobacterium, i.e. Mycobacterium smegmatis. Thus, like the antituberculosis drugs rifampicin, isoniazid and pretomanid, they are highly TB specific. All the pyrone-based chalcones showed no recognizable level of cytotoxicity against normal human kidney cell line (HEK-293) up to 80 μM concentration and 11 exhibited an IC50 ≤ 100 μM (highest tested concentration). On further investigation, both 2a and 2u proved to be nontoxic against four human cell lines but 2a proved to be a better choice as it did not reach IC50 even at 100 μM (highest tested concentration) while the IC50 of 2u was around 80 μM. In conclusion, our results demonstrate that 2a is specific against M. tuberculosis with no appreciable toxicity; its activity matches that of some clinically approved antituberculosis drugs and it therefore merits further evaluation.
Tuberculosis (TB) is an old but ever-evolving infectious disease that still poses a huge threat to the global human population.1 About a third of the world's population is harbouring the causative agent Mycobacterium tuberculosis (M. tuberculosis) as an asymptomatic latent TB infection (LTI).2 The chances of reactivation to active TB disease are higher in HIV-infected and other immune-compromised patients.3 HIV–TB co-infection is a deadly setback as a multitude of complications make the concurrent treatment of HIV–TB more complicated. These include drug–drug interactions,4 overlapping toxicity,5 non-adherence6 and TB-associated immune reconstitution inflammatory syndrome (IRIS).5 Another major challenge is drug resistance. Although the current first-line anti-TB drug regimen can achieve more than 95% efficacy, this is often reduced owing to the emergence of drug-resistant strains of M. tuberculosis7 so there is an urgent need for new drugs.8 Considering the failure rates during the discovery phase and clinical trials, the search for new compounds with low cytotoxicity and better efficacy is a daunting scientific challenge.9 Therefore it seems intellectually wise to take forward the core scaffolds that have already achieved some clinical relevance in humans.
In this direction, pyran-2-one (2P) represents the core structural subunit of a number of biologically significant natural and synthetic compounds.10 On one hand, pyran-2-one-based scaffolds have received FDA approval against HIV and, on the other hand, have displayed an appreciable level of anti-TB activity. (+)-Calanolide A isolated from Calophyllum lanigerum11 is the first pyran-2-one-based anti-HIV agent that significantly inhibited the growth of M. tuberculosis H37Rv.12 In particular, 4-hydroxy-pyran-2-one (4-HP) represents the active component of naturally isolated anti-TB agents like Pseudopyronine B13 and Myxopyronin14 and clinically approved anti-HIV agent like Tipranavir15 (Fig. 1).
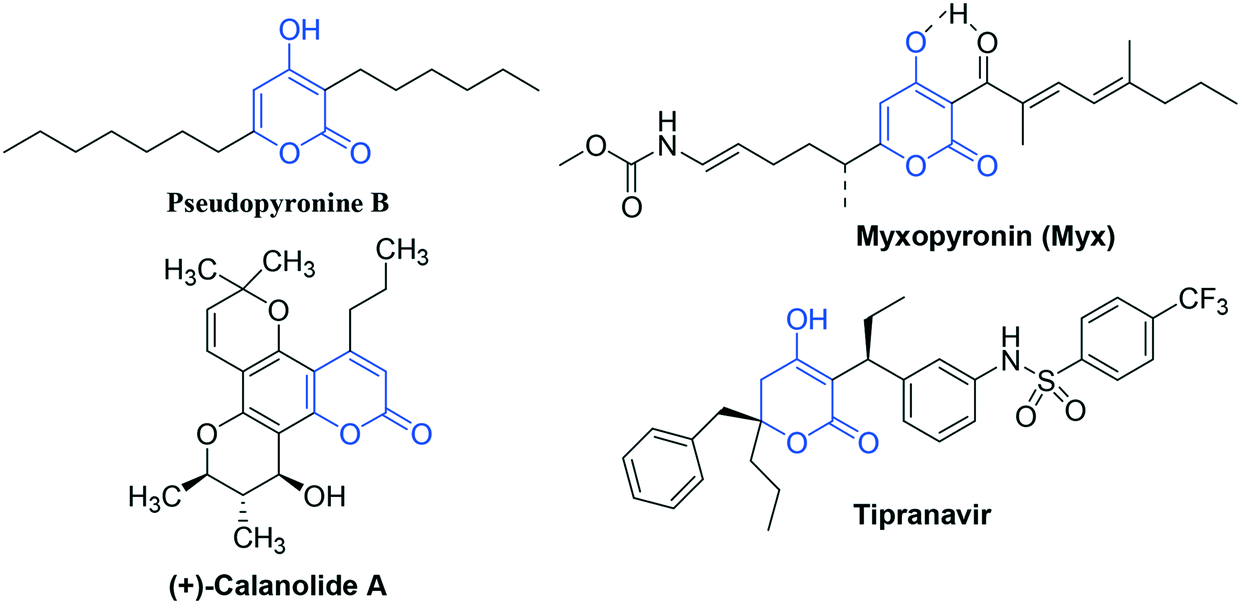 | ||
| Fig. 1 2-Pyrone-based antituberculosis and anti-HIV agents. | ||
Another class of compounds that is of great interest to medicinal chemists is chalcones (1,3-diaryl-2-propene-1-ones) owing to their simple chemistry, ease of synthesis and broad spectrum of pharmacological activities against major human diseases like cancer,16 inflammation,17 malaria,18 HIV19–21 and TB.22–28 The anti-TB activity of chalcones is attributed to their lipophilic nature, which allows them to easily penetrate the mycobacterial cell wall.29,30
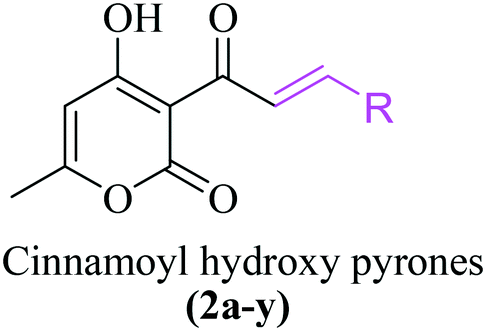
Interestingly, a class of cinnamoyl pyrone (CP)-based compounds exists that contains both a pyrone moiety (4-HP scaffold) as aromatic ring A and a cinnamoyl moiety as ring B, referred to as 3-cinnamoyl-4-hydroxy-6-methyl-2H-pyran-2-ones (CHPs). To date, the anti-HIV31 and anticancer32 potential of CHPs have been documented but no reports about their antituberculosis potential exist so far. Considering the fact that both pyrones and chalcones have shown some anti-TB potential and have attained some clinical relevance on one hand and the need for new antituberculosis drugs on the other hand, this study aimed to synthesize CHPs and exploit them for TB drug discovery. To the best of our knowledge, the results of this study show for the first time the promising antituberculosis potential of CHPs against M. tuberculosis.
A general synthesis of 3-cinnamoyl-4-hydroxy-6-methyl-2H-pyran-2-one derivatives 2a–2y is exhibited in Scheme 1.
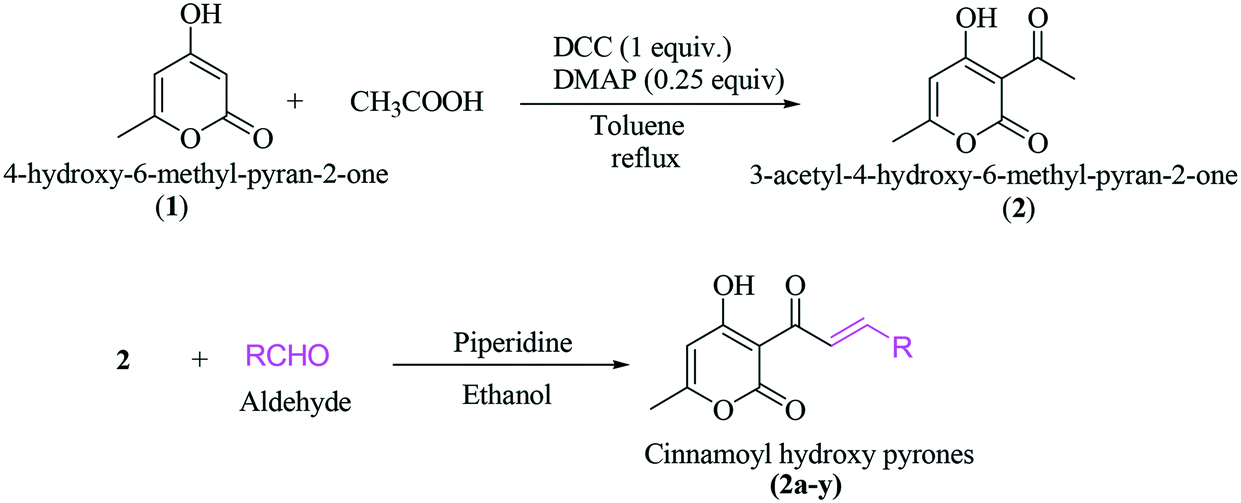 | ||
| Scheme 1 Synthesis of 3-cinnamoyl-4-hydroxy-6-methyl-2H-pyran-2-one derivatives. | ||
3-Acetyl-4-hydroxy-6-methyl-pyran-2-one (2) was synthesized from 4-hydroxy-6-methyl-pyran-2-one (1) using acetic acid, DCC and DMAP in toluene under reflux in an almost quantitative yield. The targeted cinnamoyl pyrones (2a–2y) were synthesized by reacting 3-acetyl-4-hydroxy-6-methyl-pyran-2-one (2) with a specific benzaldehyde in ethanol using catalytic piperidine under reflux in good to excellent yield (Table 1). The structures of all the synthesized compounds were characterized using 1H and 13C NMR. For example, the 1H NMR spectrum of 2a exhibited two doublet signals at 8.30 ppm (d, J = 15.7 Hz, 1H) and 7.94 ppm (d, J = 15.8 Hz, 1H), corresponding to the β- and α-H of the enone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, respectively. The peaks at 7.67 ppm (dd, J = 6.5, 2.8 Hz, 2H) and 7.39 ppm (dd, J = 11.3, 7.7 Hz, 3H) are attributed to five aromatic benzene C–H. The proton of C5 of the pyrone ring appeared as a singlet at 5.94 ppm. Similarly, the C6-methyl group showed a broad singlet at 2.26 ppm.
C, respectively. The peaks at 7.67 ppm (dd, J = 6.5, 2.8 Hz, 2H) and 7.39 ppm (dd, J = 11.3, 7.7 Hz, 3H) are attributed to five aromatic benzene C–H. The proton of C5 of the pyrone ring appeared as a singlet at 5.94 ppm. Similarly, the C6-methyl group showed a broad singlet at 2.26 ppm.
|
|
|||||
|---|---|---|---|---|---|
| S. no | Code | % yield | Structure (R) | M. tuberculosis MIC (μg ml−1) | M. smegmatis MIC (μg ml−1) |
| a Antituberculosis drugs RIF, INH, EMB, STR and LVX showed MICs values of 0.078, 0.312, 2.5, 1.25 and 2.5 μg ml−1, respectively, against M. tuberculosis. b LVX served as the positive control against M. smegmatis as RIF and INH have been reported to show 500- and 1000-fold higher MIC values against M. smegmatis (100 and 25 mg ml−1) compared to those of M. tuberculosis (0.2 and 0.02 μg ml−1, respectively) and therefore cannot act as controls. LVX exhibited an MIC of 2.5 μg ml−1 against M. smegmatis. | |||||
| 1 | 2a | 85 |
|
4 | >128 |
| 2 | 2b | 81 |
|
8 | >128 |
| 3 | 2c | 79 |

|
256 | >128 |
| 4 | 2d | 80 |
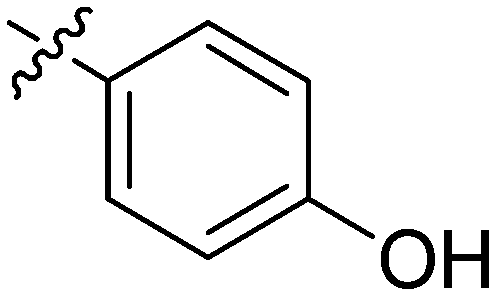
|
64 | >128 |
| 5 | 2e | 78 |

|
16–32 | >128 |
| 6 | 2f | 82 |
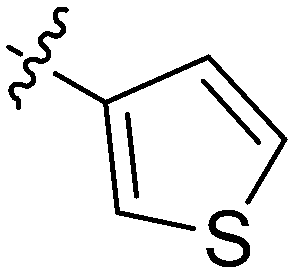
|
32–64 | >128 |
| 7 | 2g | 77 |

|
32–64 | >128 |
| 8 | 2h | 80 |

|
8 | >128 |
| 9 | 2i | 81 |

|
256 | >128 |
| 10 | 2j | 83 |
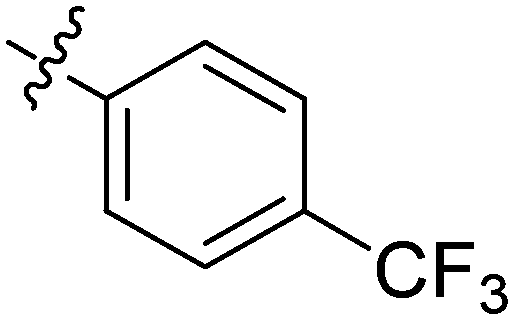
|
256 | >128 |
| 11 | 2k | 79 |

|
256 | >128 |
| 12 | 2l | 78 |
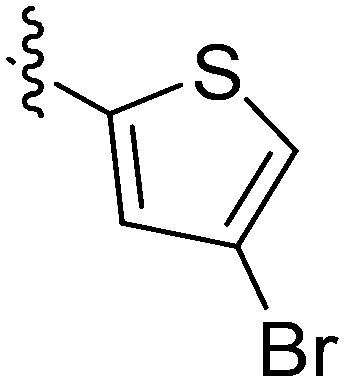
|
4–8 | >128 |
| 13 | 2m | 80 |
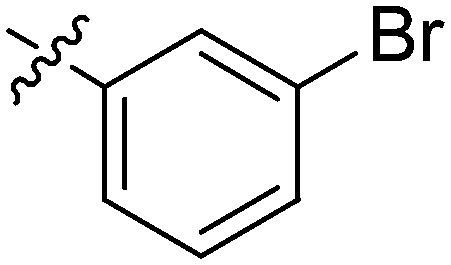
|
4–8 | >128 |
| 14 | 2n | 77 |
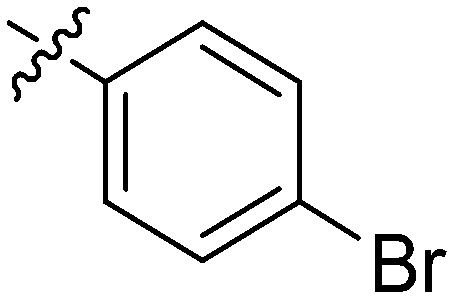
|
8–16 | >128 |
| 15 | 2o | 81 |
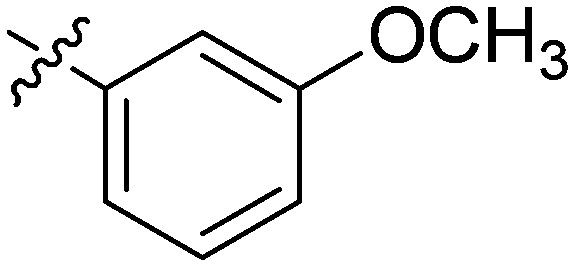
|
8–16 | >128 |
| 16 | 2p | 82 |

|
32 | >128 |
| 17 | 2q | 79 |
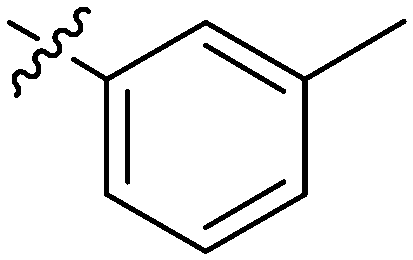
|
32 | >128 |
| 18 | 2r | 82 |

|
16 | >128 |
| 19 | 2s | 82 |
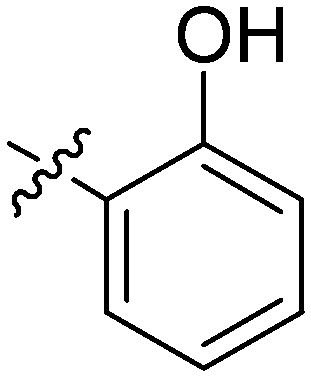
|
8–16 | >128 |
| 20 | 2t | 78 |
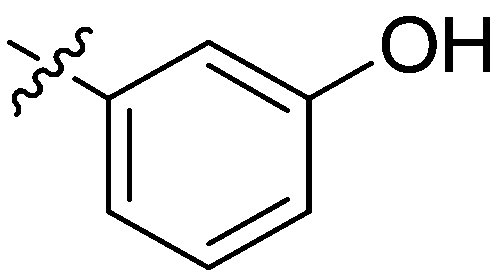
|
8–16 | >128 |
| 21 | 2u | 83 |

|
4 | >128 |
| 22 | 2v | 80 |

|
4–8 | >128 |
| 23 | 2w | 81 |
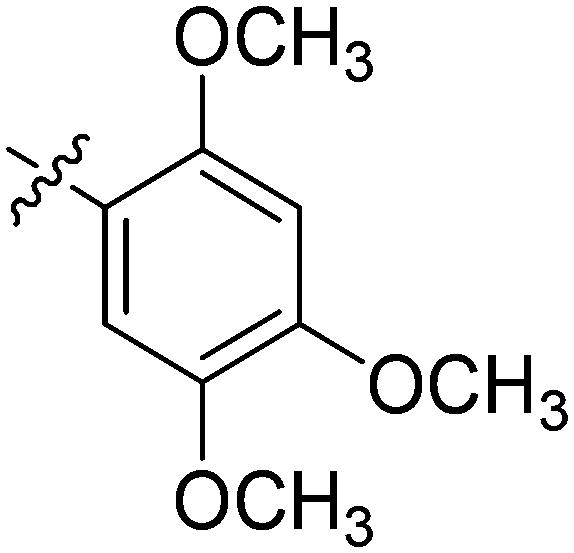
|
8 | >128 |
| 24 | 2x | 83 |
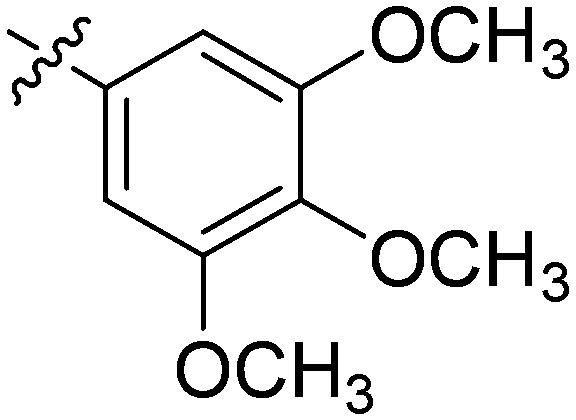
|
8 | >128 |
| 25 | 2y | 80 |
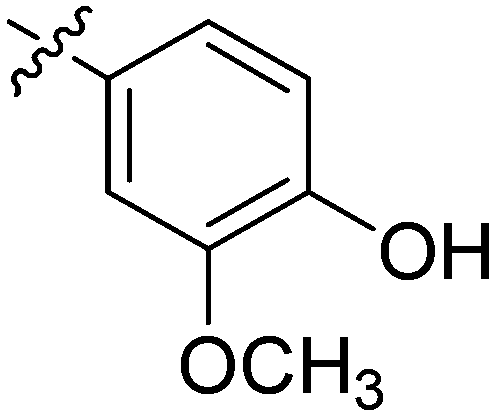
|
32 | >128 |
Antimicrobial susceptibility testing (AST) was done by determining the MIC of antimicrobial agents and test compounds by 7H9 broth microdilution assay, as discussed previously with slight modifications.33 Briefly, Middlebrook 7H9 broth with 10% ADC (albumin, dextrose and catalase) containing the CHP-based test compounds in the concentration range 0.25–128.0 μg ml−1 were added in duplicate in 96-well titer plates (Nest Biotech, China). Each well was inoculated with 50 μl of appropriately diluted mid-log phase M. tuberculosis H37Rv culture corresponding to ∼1 × 105 CFU ml−1. Isoniazid (INH), rifampicin (RIF), ethambutol (EMB), streptomycin (STR) and levofloxacin (LVX) were used as positive controls and drug-free broth served as the negative control.
Original CFU were verified by plating serial 10-fold dilutions of the inoculum onto Middlebrook 7H11 agar plates that were incubated at 37 °C and then read after four weeks. The MIC was taken as the lowest concentration (μg ml−1) of an antibiotic that inhibits the visible growth after two weeks of incubation. MIC against M. smegmatis was determined as above but the reading was taken after only 72 h owing to its fast-growing nature.
The MICs of standard antituberculosis drugs RIF, INH, EMB, STR and LVX were found to be 0.078, 0.312, 2.5, 1.25 and 2.5 μg ml−1, respectively, against M. tuberculosis H37Rv. MIC results of all the CHP derivatives (2a–2y) against M. tuberculosis are presented in Table 1. Among the 25 derivatives, 11 did not show any attractive activity i.e. their MIC values were ≥32 μg ml−1. Five compounds (2n, 2o, 2r, 2s and 2t) showed moderate antituberculosis activity with MIC values ranging from 8 to 16 μg ml−1 (Table 1). Seven compounds (2b, 2h, 2l, 2m, 2v, 2w and 2x) showed good anti-TB potential reflected by their MIC values ranging from 4 to 8 μg ml−1 (Table 1). Two compounds (2a and 2u) displayed excellent antituberculosis activity against M. tuberculosis with MIC values of 4 μg ml−1 (Table 1), which is close to those of standard antituberculosis drugs EMB, STR and LVX, thereby reflecting the promising antituberculosis potential of these two compounds that strongly merits further evaluation. This significant antituberculosis potential could be due to 2-pyrone-based polyketides, which represent a diverse class of secondary metabolites with crucial roles in M. tuberculosis. Indeed a putative chalcone synthase (PKS11) and a tri-ketide, as well as tetra-ketide pyrone synthase (PKS18) in M. tuberculosis, have been found to synthesize a unique cyclic 5-methyl-6-alkyl-4-hydroxy-2-pyrone (MAHP) via various intermediates. These small polyketide molecules are finally incorporated in the M. tuberculosis cell wall, where they are believed to regulate permeability.34,35 This is one of the ways by which pyrone-based molecules attack M. tuberculosis. Additionally, pyrones have been shown to inhibit M. tuberculosis transcription, such as rifampicin, but even in rifampicin-resistant strains. Thus, there are experimental indications as to how these molecules might work against M. tuberculosis. Further reasons for this anti-TB activity include the chalcones side, which owing to its lipophilic nature easily penetrates the mycobacterial cell wall and hence causes damage.29,30 It is to be noted that none of the molecules exhibit appreciable activity against M. smegmatis (Table 1).
Based on our experimental results, the structure–activity relationship (SAR) of this class of anti-TB agents with respect to benzene ring can be summarised as follows: 2a is the most potent molecule so it can be concluded that substitution of any sort in the benzene ring leads to a decrease in activity. Though the –OC2H5 group is well tolerated, its position is very important since its presence at the para position proved to be detrimental to the activity. Though the presence of halogens and hydroxyl groups is tolerated, electron-withdrawing groups like –CF3 proved to be unfavourable for activity. In general, the presence of alkyl and alkoxy groups also decreases the potency of the molecules. Additionally, replacement of the phenyl ring with heterocyclic rings like furan and thiophene also decreased the antituberculosis activity.
Since first-line antituberculosis drugs are specifically active against M. tuberculosis only and exhibit very poor activity against other microbes, we therefore evaluated the two most potent CHPs (2a and 2u) against four Gram-positive bacteria [Staphylococcus aureus (ATCC 25923), Staphylococcus epidermidis (ATCC 12228), Micrococcus luteus (ATCC 10240), and Bacillus subtilis (ATCC 11774)] and four Gram-negative bacteria [Escherichia coli (ATCC 10536), Enterococcus faecalis (ATCC 51299), Pseudomonas aeruginosa (ATCC 10145), and Klebsilla pneumonia (ATCC BAA-2146)]. Strains were revived according to written instructions, plated onto Muller Hinton Agar (MHA) and incubated at 37 °C for 24 h. The MIC was determined by a previously described method with minor modification using MH broth and a reading was taken after 24 h of incubation at 37 °C.36 The second-line anti-TB drug levofloxacin (LVX) served as the positive control owing to its broad spectrum of activity. LVX exhibited MICs values of 0.078–0.156, 0.078, 0.039–0.078, 0.39, 0.078, 0.312–0.625, 0.156 and 0.625–1.25 μg ml−1 against S. aureus, B. subtilis, M. luteus, S. epidermidis, E. coli, P. aeruginosa, E. faecalis and K. Pneumonia, respectively; these MIC values of LVX are well in agreement with reported ones (Table 2). The MIC values of 2a and 2u observed in this study are presented in Table 2. None of them exhibited any significant activity against Gram-positive bacteria: S. aureus, B. subtilis, S. epidermidis and M. luteus, or Gram-negative bacteria: E. coli, E. faecalis, K. pneumonia and P. aeruginosa (Table 2). Full visual turbidity in the 96-well plate was observed even up to the highest tested concentration of 128 μg ml−1 for all compounds against each bacteria. Our results clearly show that 2a and 2u exhibit unique activity specifically against M. tuberculosis. This observation is of great significance for developing CHP-based antituberculosis drugs as they will be less likely to interfere with normal human flora.
| Type of bacteria | Bacterial strain | MIC (μg ml−1) | ||
|---|---|---|---|---|
| 2a | 2u | LVXa | ||
| a LVX is a standard broad-spectrum antibacterial and anti-TB drug that served as the positive control. | ||||
| Gram-positive | S. aureus | >128 | >128 | 0.078–0.156 |
| S. epidermidis | >128 | >128 | 0.039–0.078 | |
| M. luteus | >128 | >128 | 0.078 | |
| B. subtilis | >128 | >128 | 0.39 | |
| Gram-negative | E. coli | >128 | >128 | 0.078 |
| E. faecalis | >128 | >128 | 0.156 | |
| P. aeruginosa | >128 | >128 | 0.312–0.625 | |
| K. pneumonia | 128 | 128 | 0.625–1.25 | |
Among the critical factors that allow any molecule to proceed successfully during the drug discovery process is selective toxicity towards pathogenic microbes and none/less toxicity on mammalian cells as it is through this characteristic that activity and toxicity get separated and the therapeutic index becomes acceptable. We therefore evaluated the cytotoxicity of CHP-based derivatives in concentrations ranging from 1 to 80 μM (1 μM, 20 μM, 40 μM and 80 μM) against a normal human kidney (HEK-293) cell line, which was purchased from the National Centre for Cell Science (NCCS), Pune, India. This was done on the basis of percentage growth inhibition assessed by MTT assay. Overall, the compounds showed lesser growth inhibition of HEK-293 cells up to 80 μM concentration and thus CHPs were found to be nontoxic at concentrations that are usually high-end concentrations for such studies. We therefore evaluated all compounds at a much higher concentration of 100 μM. 11 compounds (2x, 2w, 2v, 2u, 2q, 2p, 2n, 2j, 2k, 2l, and 2m) inhibited HEK cell growth by ≥50% at this highest tested concentration and thus exhibited an IC50 value of 100 μM. Five compounds (2b, 2i, 2r, 2t and 2y) showed 45 to 50% inhibition at 100 μM, thus their IC50 is >100 μM but will be close to this value. Nine compounds did not even inhibit 45% of growth at the highest experimental concentration and thus their IC50 is clearly >100 μM, demonstrating them to be safe for human cells.
The toxicity of the two most potent compounds with respect to antituberculosis activity, 2a and 2u (MIC = 4 μg ml−1), was further evaluated in the concentration range 0.0–100 μM against four human cell lines, namely HEK-293, breast cancer (MCF-7), colon cancer (HCT-116) and prostate cancer (PC-3), by double dilution method using the MTT cell viability assay. MCF-7, HCT-116 and PC3 cells were obtained from the National Cancer Institute (NCI), USA. The results of this study as the mean ± standard deviation of percentage growth inhibition are presented in Fig. 2i–iv. Both compounds 2a and 2u showed some dose-dependent growth inhibition response against all cell lines (Fig. 2i–iv). At the highest experimental concentration, 2a inhibited <50% (38–44%) of the growth of three cell lines HEK-293, MCF-7 and PC3, and inhibited 50% of the growth of the HCT-116 cell line at the same concentration (Fig. 2i–iv). In contrast, 2u inhibited 67–70% of the growth of all the cell lines tested at the same concentration and its IC50 appeared to be 80 μM against all cell lines (Fig. 2i–iv). Therefore, 2a displayed a clear advantage over 2u with respect to the toxicity profile. Furthermore, for 2a the MIC is 4 μg ml−1 against M. tuberculosis and only 40–50% inhibition was observed at 100 μM (25.6 μg ml−1); this demonstrates its broad therapeutic window. From antituberculosis activity to cytotoxicity studies, it is clear that compound 2a is the choice for future studies.
 | ||
| Fig. 2 Cytotoxicity profile of 3-cinnamoyl-4-hydroxy-6-methyl-2H-pyran-2-ones (2a and 2u) expressed as percentage growth inhibition. i) Normal human embryonic kidney (HEK-293); ii) breast cancer (MFC-17); iii) colon cancer (HCT-116) and iv) prostate cancer (PC3) cell lines. Values are mean ± SD of three sets. | ||
Conclusion
Out of 25 compounds subjected to antimycobacterial susceptibility testing, two compounds displayed potent anti-TB activity (2a and 2u) that matches that of some known antituberculosis drugs. 2a and 2u were found to be ineffective against a panel of Gram-positive and Gram-negative bacteria, and even against M. smegmatis, thereby reflecting their specific antituberculosis activity, as reported for some antituberculosis drugs. Further cytotoxicity data significantly favoured 2a over 2u. The structure–activity relationship reveals that substitution of any sort in the benzene ring leads to a decrease in activity. Undoubtedly, therefore, 2a merits further evaluation with respect to its anti-TB potential.Ethical approval
This article does not contain any study involving animals.Conflicts of interest
All authors declare that there is no conflict of interest.Acknowledgements
The authors are highly thankful to Director IIIM, Dr. Ram A Vishwakarma for unconditional support. DST-India is acknowledged for funding under the INSPIRE faculty programme (project GAP-1179). Thanks are due to UGC-GOI for the Fellowship and the Academy of Scientific and Innovative Research (AcSIR) for doctoral programme registration of Zubair Shanib Bhat. This work was supported by funding from Council of Scientific and Industrial Research Govt. of India (MLP-6010 and BSC-203) and by Ramalingaswami Fellowship grant from Department of Biotechnology Govt. of India (GAP-1160) to ZA. IIIM publication No: IIIM/2160/2017.References
- A. Zumla, M. Raviglione, R. Hafner and C. F. von Reyn, N. Engl. J. Med., 2013, 368, 745–755 CrossRef CAS PubMed.
- E. W. Campion, H. Getahun, A. Matteelli, R. E. Chaisson and M. Raviglione, N. Engl. J. Med., 2015, 372, 2127–2135 CrossRef PubMed.
- J. Bruchfeld, M. Correia-Neves and G. Källenius, Cold Spring Harbor Perspect. Med., 2015, 5, a017871 CrossRef PubMed.
- J. Mannu, P. Jenardhanan and P. P. Mathur, Med. Chem. Res., 2014, 23, 905–917 CrossRef CAS.
- E. M. Shankar, R. Vignesh, R. Ellegård, M. Barathan, Y. K. Chong, M. K. Bador, D. V. Rukumani, N. S. Sabet, A. Kamarulzaman, V. Velu and M. Larsson, Pathog. Dis., 2014, 70, 110–118 CrossRef CAS PubMed.
- P. Naidoo, K. Peltzer, J. Louw, G. Matseke, G. McHunu and B. Tutshana, BMC Public Health, 2013, 13, 396 CrossRef PubMed.
- R. J. O'Brien and P. P. Nunn, Am. J. Respir. Crit. Care Med., 2001, 163, 1055–1058 CrossRef PubMed.
- World Health Organisation, WHO | Multidrug-resistant tuberculosis (MDR-TB), http://www.who.int/tb/challenges/mdr/en/.
- A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, Nature, 2011, 469, 483–490 CrossRef CAS PubMed.
- Z. S. Bhat, M. A. Rather, M. Maqbool, H. U. Lah, S. K. Yousuf and Z. Ahmad, Biomed. Pharmacother., 2017, 91, 265–277 CrossRef CAS PubMed.
- Y. Kashman, K. R. Gustafson, R. W. Fuller, J. H. Cardellina, J. B. McMahon, M. J. Currens, R. W. Buckheit, S. H. Hughes, G. M. Cragg and M. R. Boyd, J. Med. Chem., 1992, 35, 2735–2743 CrossRef CAS PubMed.
- Z. Q. Xu, W. W. Barrow, W. J. Suling, L. Westbrook, E. Barrow, Y. M. Lin and M. T. Flavin, Bioorg. Med. Chem., 2004, 12, 1199–1207 CrossRef CAS PubMed.
- A. C. Giddens, L. Nielsen, H. I. Boshoff, D. Tasdemir, R. Perozzo, M. Kaiser, F. Wang, J. C. Sacchettini and B. R. Copp, Tetrahedron, 2008, 64, 1242–1249 CrossRef CAS.
- H. Irschik, K. Gerth, G. Höfle, W. Kohl and H. Reichenbach, J. Antibiot., 1983, 36, 1651–1658 CrossRef CAS PubMed.
- C. Flexner, G. Bate and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2005, 4, 955–956 CrossRef CAS PubMed.
- D. K. ar Mahapatra, S. K. umar Bharti and V. Asati, Eur. J. Med. Chem., 2015, 98 Search PubMed.
- S. Bano, K. Javed, S. Ahmad, I. G. Rathish, S. Singh, M. Chaitanya, K. M. Arunasree and M. S. Alam, Eur. J. Med. Chem., 2013, 65, 51–59 CrossRef CAS PubMed.
- B. Insuasty, J. Ramírez, D. Becerra, C. Echeverry, J. Quiroga, R. Abonia, S. M. Robledo, I. D. Vélez, Y. Upegui, J. A. Muñoz, V. Ospina, M. Nogueras and J. Cobo, Eur. J. Med. Chem., 2015, 93, 401–413 CrossRef CAS PubMed.
- H. Sharma, S. Patil, T. W. Sanchez, N. Neamati, R. F. Schinazi and J. K. Buolamwini, Bioorg. Med. Chem., 2011, 19, 2030–2045 CrossRef CAS PubMed.
- A. Hameed, M. I. Abdullah, E. Ahmed, A. Sharif, A. Irfan and S. Masood, Bioorg. Chem., 2016, 65, 175–182 CrossRef CAS PubMed.
- A. L. Cole, S. Hossain, A. M. Cole and O. Phanstiel, Bioorg. Med. Chem., 2016, 24, 2768–2776 CrossRef CAS PubMed.
- F. Macaev, Med. Chem., 2014, 4, 487–493 CAS.
- T. L. B. Ventura, S. D. Calixto, B. De Azevedo Abrahim-Vieira, A. M. T. De Souza, M. V. P. Mello, C. R. Rodrigues, L. S. De Mariz E Miranda, R. O. M. A. De Souza, I. C. R. Leal, E. B. Lasunskaia and M. F. Muzitano, Molecules, 2015, 20, 8072–8093 CrossRef CAS PubMed.
- B. R. Copp, Nat. Prod. Rep., 2003, 20, 535–557 RSC.
- A. Suksamrarn, A. Chotipong, T. Suavansri, S. Boongird, P. Timsuksai, S. Vimuttipong and A. Chuaynugul, Arch. Pharmacal Res., 2004, 27, 507–511 CrossRef CAS.
- P. M. Sivakumar, S. P. Seenivasan, V. Kumar and M. Doble, Bioorg. Med. Chem. Lett., 2007, 17, 1695–1700 CrossRef CAS PubMed.
- D. Patel, P. Kumari and N. B. Patel, Med. Chem. Res., 2013, 22, 726–744 CrossRef CAS.
- P. M. Sivakumarl, V. Kumar, S. P. Seenivasan, J. Mohanapriya and M. Doble, Adv. Biomed. Res., Proc. 7th WSEAS Int. Conf. Math. Biol. Ecol. (MABE ‘10), Proc. Int. Conf. Med. Physiol. (PHYSIOL. ‘10), Proc. Int. Conf. Biochem. Med. Chem. (BIOMEDCH ‘10), 2010, pp. 168–172 Search PubMed.
- P. J. Brennan and H. Nikaido, Annu. Rev. Biochem., 1995, 64, 29–63 CrossRef CAS PubMed.
- J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2012, 7, 863–875 CrossRef CAS PubMed.
- K. Ramkumar, K. V. Tambov, R. Gundla, A. V. Manaev, V. Yarovenko, V. F. Traven and N. Neamati, Bioorg. Med. Chem., 2008, 16, 8988–8998 CrossRef CAS PubMed.
- Q. Y. Lan, Q. L. Liu, J. Cai and A. W. Liu, Int. J. Clin. Exp. Pathol., 2015, 8, 155–163 CAS.
- Z. Ahmad, S. Tyagi and A. Minkowski, et al. , Indian J. Med. Res., 2012, 136, 808–814 CAS.
- P. Saxena, G. Yadav, D. Mohanty and R. S. Gokhale, J. Biol. Chem., 2003, 278, 44780–44790 CrossRef CAS PubMed.
- K. Gokulan, S. E. O'Leary and W. K. Russell, et al. , J. Biol. Chem., 2013, 288, 16484–16494 CrossRef CAS PubMed.
- M. A. Rather, A. M. Lone, B. Teli, Z. S. Bhat, P. Singh, M. Maqbool, B. A. Shairgojray, J. M. Dar, S. Amin, K. Y. Syed and Z. Ahmad, MedChemComm, 2017, 8, 2133–2141 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00366h |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2018 |