
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic bead encapsulation above 20 kHz with triggered drop formation†
Iain C.
Clark
a and
Adam R.
Abate
 *b
*b
aDepartment of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, CA, USA
bDepartment of Bioengineering and Therapeutic Sciences, California Institute for Quantitative Biosciences (QB3), Chan Zuckerberg Biohub, University of California, San Francisco, San Francisco, CA, USA. E-mail: adam@abatelab.org
First published on 26th October 2018
Abstract
Microsphere beads are functionalized with oligonucleotides, antibodies, and other moieties to enable specific detection of analytes. Droplet microfluidics leverages this for single-molecule or -cell analysis by pairing beads and targets in water-in-oil droplets. Pairing is achieved with devices operating in the dripping regime, limiting throughput. Here, we describe a pairing method that uses beads to trigger the breakup of a jet into monodispersed droplets. We use the method to pair 105 Human T cells with polyacrylamide beads ten times faster than methods operating in the dripping regime. Our method improves the throughput of bead-based droplet workflows, enabling analysis of large populations and the detection of rare events.
1. Introduction
Molecular biology utilizes multiple reaction steps that require reagent addition, incubation, and purification. Solid supports, like functionalized beads, are widely used because they can be tailored with useful physical and chemical properties. Beads are easily functionalized to capture small molecules, proteins, or nucleic acids and they can be purified by centrifugation, magnetic separation, or fluorescent activated sorting. The benefits of beads extend to microfluidic applications;1–4 single beads are discrete and biocompatible analytical sensors that are employed for single-cell or single-molecule analysis. However, for this function, beads must be paired with their analytes and compartmentalized so that subsequent reactions occur independently. Water-in-oil emulsions are ideally suited for this purpose and have been routinely utilized in bead-based workflows.Numerous examples of bead-based droplet workflows are reported in the literature, and commercial products are expanding these capabilities to non-microfluidic labs (10× Genomics, Mission Bio, 1CellBio). Beads functionalized with oligonucleotides are used to barcode and RNA sequence single cells,1,5 to prime RT-PCR or PCR detection of specific RNA and DNA sequences,4,6 to copy single molecules via PCR (BEAMing),2,7 and to barcode DNA molecules for phasing studies.8 Beads also encode DNA libraries and bind protein products for in vitro protein engineering.3 Antibody coated beads are used for high-throughput immunoassays9 and detection of cytokines.10,11 Lysing and trapping cellular DNA and RNA within hydrogel beads is used for digital droplet PCR and RT-PCR,6 for overlap PCR to link functional and phylogenetic markers,12 and for massively parallel genome sequencing.13
A critical part of bead-based droplet workflows involves pairing beads with the analyte of interest (cells, nucleic acids, proteins) in droplets. Drop making is typically performed by co-flowing beads with targets in a dripping regime to produce a monodispersed emulsion. To ensure single bead-target encapsulations, beads and targets are diluted such that one in approximately every hundred drops contains a bead and target. This limits the throughput of applications that pair two discrete particles (e.g. cells and beads) in drops.
To increase efficiency and throughput, elastic packed beads that flow uniformly into the drop maker can be used, such that nearly every drop contains a single bead.14 However, these techniques operate in the low flow rate dripping regime, limiting throughput and the number of targets analyzed. For higher throughput, bulk drop generation like vortexing, stirring, or pipetting is used. This produces polydispersed emulsions that yield uncontrolled assays and are difficult to process with important microfluidic steps like merging and sorting. To enable high throughput bead-based droplet assays, a method is needed that rapidly encapsulates beads in monodispersed drops.
In this paper, we demonstrate bead encapsulation ten times faster than previously described.14 Our method uses beads to internally trigger the breakup of an otherwise stable jet, generating drops of controlled size. When applied to unpacked rigid beads, the resultant emulsion consists of a population of large polydispersed empty drops, and another of small, uniform, bead-containing drops. When applied to packed elastic beads that flow regularly into the jet, the jet breaks into uniform droplets of a size determined by the bead frequency and flow rates. We demonstrate the utility of this approach by encapsulating single T cells at over 20 kHz drop generation. Our approach increases the throughput of bead and target co-encapsulation in microfluidics, enabling the analysis of larger populations and the detection of rare cells.
2. Results and discussion
Water and oil co-flowed in a hydrophobic microfluidic channel produces water-in-oil droplets in a capillary number (Ca) dependent manner. Such devices can form droplets in different regimes. At low Ca, drops form through a quasistatic plugging and squeezing mechanism, while at moderate Ca, shearing of the continuous phase against the dispersed phase becomes important (Fig. 1A). At higher Ca, the dispersed phase flows as a stable jet without breaking up (Fig. 1B). If cells,15 particles,16 or beads are introduced into the jet, they can seed Rayleigh–Plateau instabilities, breaking it into drops (Fig. 1C); however, since these discrete entities are dispersed randomly in the droplet phase, the breakup is irregular, yielding a polydispersed emulsion. To produce a monodispersed emulsion, uniformly periodic perturbations must be applied to the jet, which can be achieved by introducing air bubbles.17 Here, we extend this concept to packed elastic beads which, like air bubbles, flow with regular periodicity, and thus break the jet into monodispersed drops containing single beads (Fig. 1D). Additional aqueous can be introduced via side channels to independently adjust droplet volume in the triggering regime (Fig. 1E).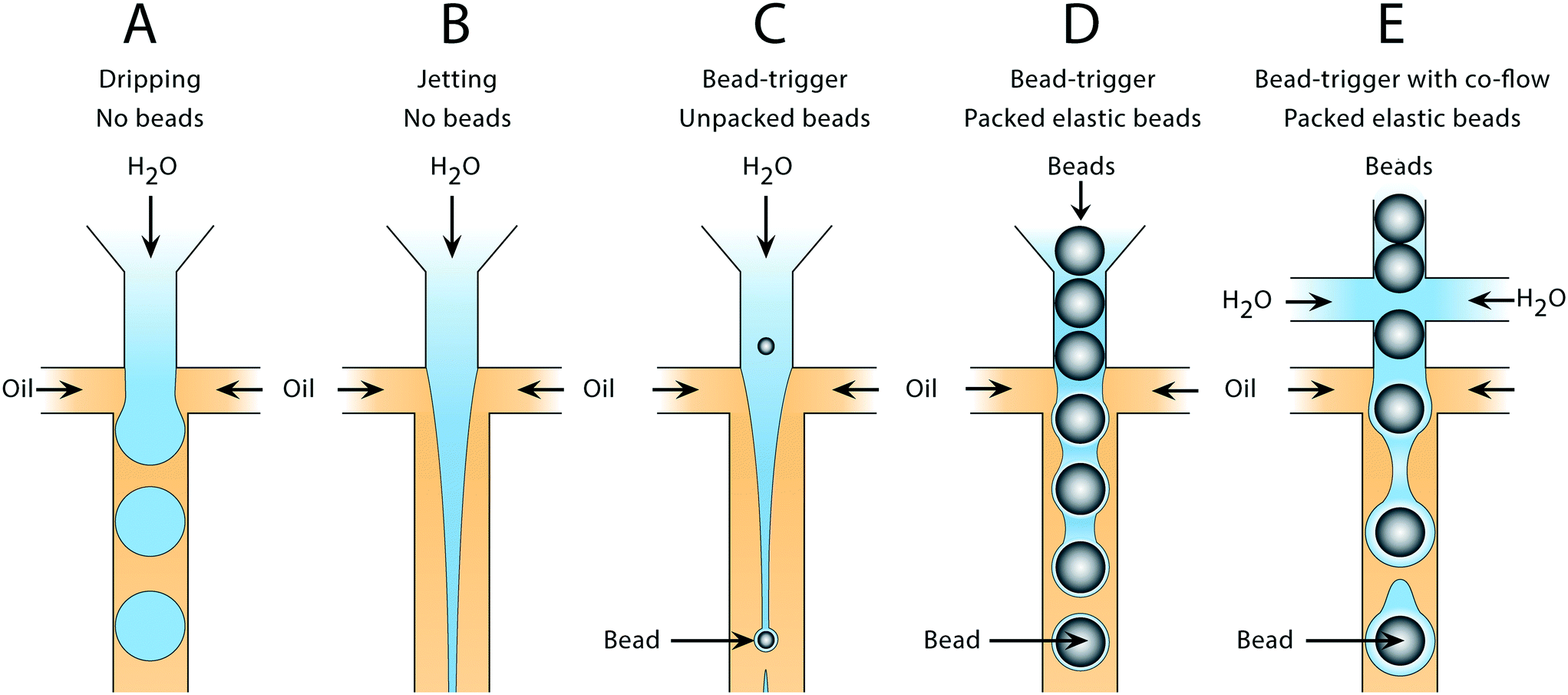 | ||
| Fig. 1 Droplet generation in the dripping and jetting flow regimes using bead-triggering. In the absence of a perturbation, the jet remains stable and drops are not formed in the main channel. Introduction of a bead causes an internal perturbation that triggers jet breakup and drop formation. A) Drop formation in the dripping regime without beads. B) Stable jet formation at high capillary number without beads. C) Drop formation using unpacked rigid beads at limiting dilution to trigger breakup of the dispersed phase. D) Drop formation using packed beads to trigger breakup without additional co-flow. E) Drop formation using packed beads to trigger breakup of a co-flowed dispersed phase. | ||
2.1 Formation of monodispersed bead-containing droplets in a polydispersed emulsion
For rigid beads that cannot be packed due to clogging, the sample is diluted, yielding less than 1 in 10 drops containing a bead. When paired with diluted cells, less than 1 in 100 drops contain a bead-cell pair, making the process wasteful and limiting overall throughput since all the empty drops must still be produced. Bead triggering runs the device under jetting conditions, increasing throughput by ten fold. To demonstrate that unordered beads can be used to trigger drop formation and increase device speed, we form a stable jet by co-flowing 2% agarose and 1× PBS containing rigid beads (Toyopearl HW-65S). The device design and beads are identical to a recently published single cell RNA-sequencing workflow (Drop-seq). The jet, unperturbed, does not breakup in the channel (Fig. 2A). The rigid bead disrupts the streamlines within the jet, perturbing the oil–water interface, and seeding a Rayleigh-Plateau instability that breaks the jet (Fig. 2A). For jets of similar state, the breakup process is reproducible, yielding drops that are substantially larger than the particles, and uniform. The resultant emulsion consists of large polydispersed empty drops, and a second population of small monodispersed bead-containing drops.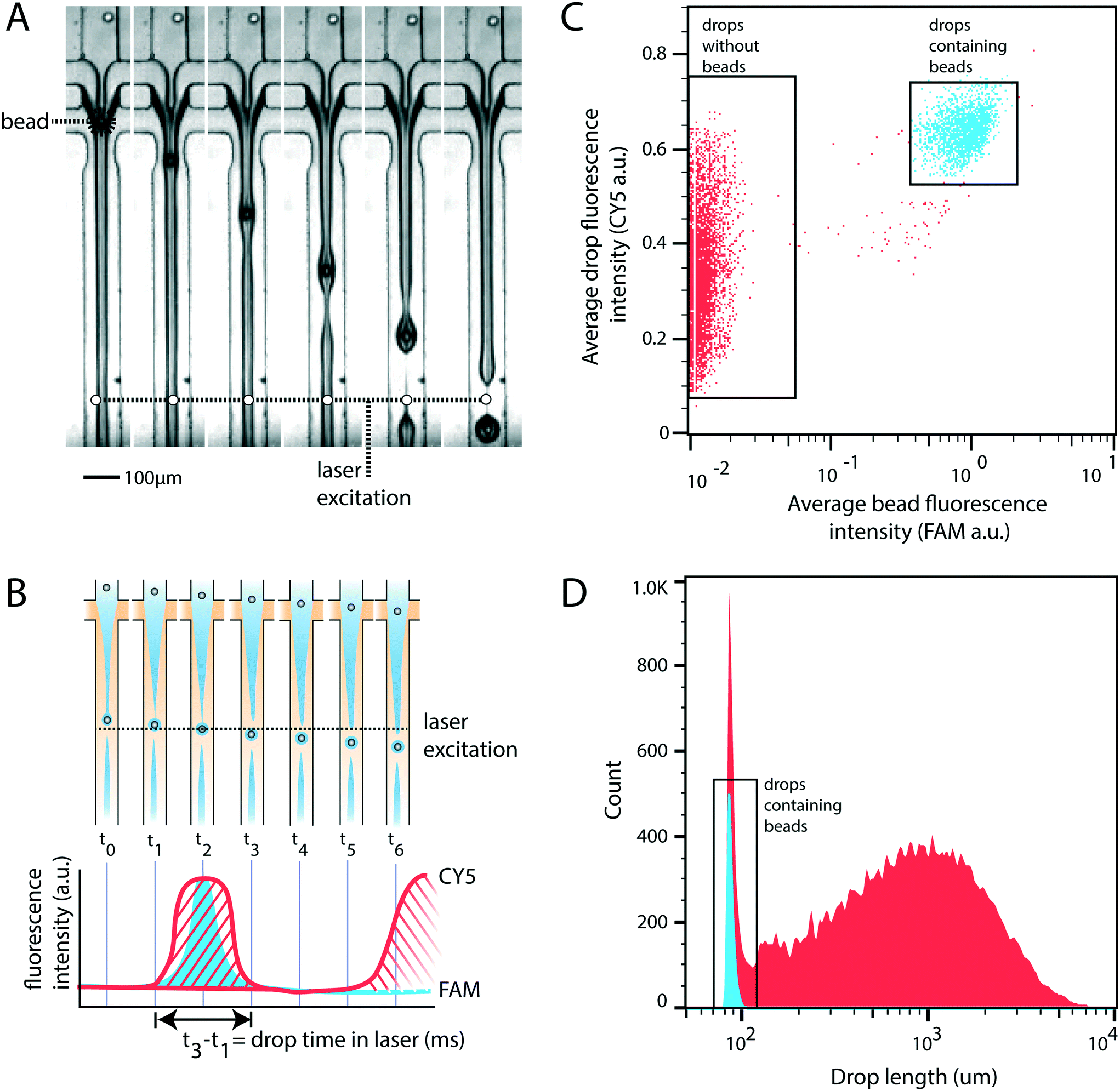 | ||
| Fig. 2 Stable jet breakup using unpacked rigid beads. A) Frames from a video of device operation showing stable jet formation and bead-induced jet breakup. B) Cartoon representation of the experimental setup used to measure drop size and the presence of bead containing drops. Drop size is calculated as the time a drop spends in the excitation window (t3–t1). C) Droplet cytometry analysis of drop formation identifies empty drops and drops containing beads. D) Histogram of drop size. Blue represents bead-containing drops identified in panel C. Red represents non-bead containing drops identified in panel C. | ||
To characterize the efficiency and uniformity of bead-triggered droplets, we use laser induced fluorescence to accumulate statistics on thousands of bead-encapsulation events. Hydroxylated methacrylic polymer beads (Toyopearl HW-65S) stained with Eva Green and visible on the FAM channel are co-flowed with an aqueous solution containing BSA conjugated CY5. Drops travel through a laser to excite the dyes (Fig. 2B). Emitted light passes through a series of filters and is collected using photomultiplier tubes.9 Timetraces of fluorescent intensity are collected, drops are detected by the presence of a peak, and the average peak fluorescence is determined for each channel. This allows us to determine which drops contain a bead (Fig. 2C). The time that a drop spends flowing through the laser is proportional to its length, providing a measure drop size distributions during bead triggering. Bead containing drops show a tight size distribution and are smaller than drops resulting from random un-triggered breakup of the jet (Fig. 2D). This demonstrates that while the overall emulsion is polydispersed, a monodispersed population of droplets containing beads is generated. These droplets could potentially be recovered by filtration or flow fractionation. For single-device workflows that do not require additional microfluidic steps, the resulting emulsion is functionally equivalent to a monodispersed emulsion. All drops containing beads are uniform in size; biochemical reactions involving beads therefore remain uniform with respect to reagents and products. For example, if the droplet captures mRNA using beads (Drop-seq) or performs a PCR off bead primers (BEAMing), those reactions will occur in droplets of uniform size. An additional benefit of jet triggering is that it creates drop sizes that are smaller than the nozzle, and therefore smaller than could be produced in a normal dripping regime. This prevents clogging of the device, which is especially important when using rigid beads that easily clog channels.
2.2 High throughput encapsulation of packed elastic beads
To generate a monodispersed emulsion of bead-containing drops, elastic beads are packed. This allows beads to be injected at regular intervals, preventing uneven spacing between beads, which would result in empty drops.14 During packed bead loading in a dripping regime, droplet generation occurs independently of the presence of a bead (Fig. 3A). Single bead-per-drop loading is achieved by tuning flow rates to synchronize bead injection with drop formation (Fig. 3A). As a result, the loading of elastic microspheres is limited by the ability to operate in a dripping regime, which constrains the throughput of bead-based workflows. Combining bead packing with jet triggering exploits periodic bead injection to trigger regular jet breakup and monodispersed droplet formation.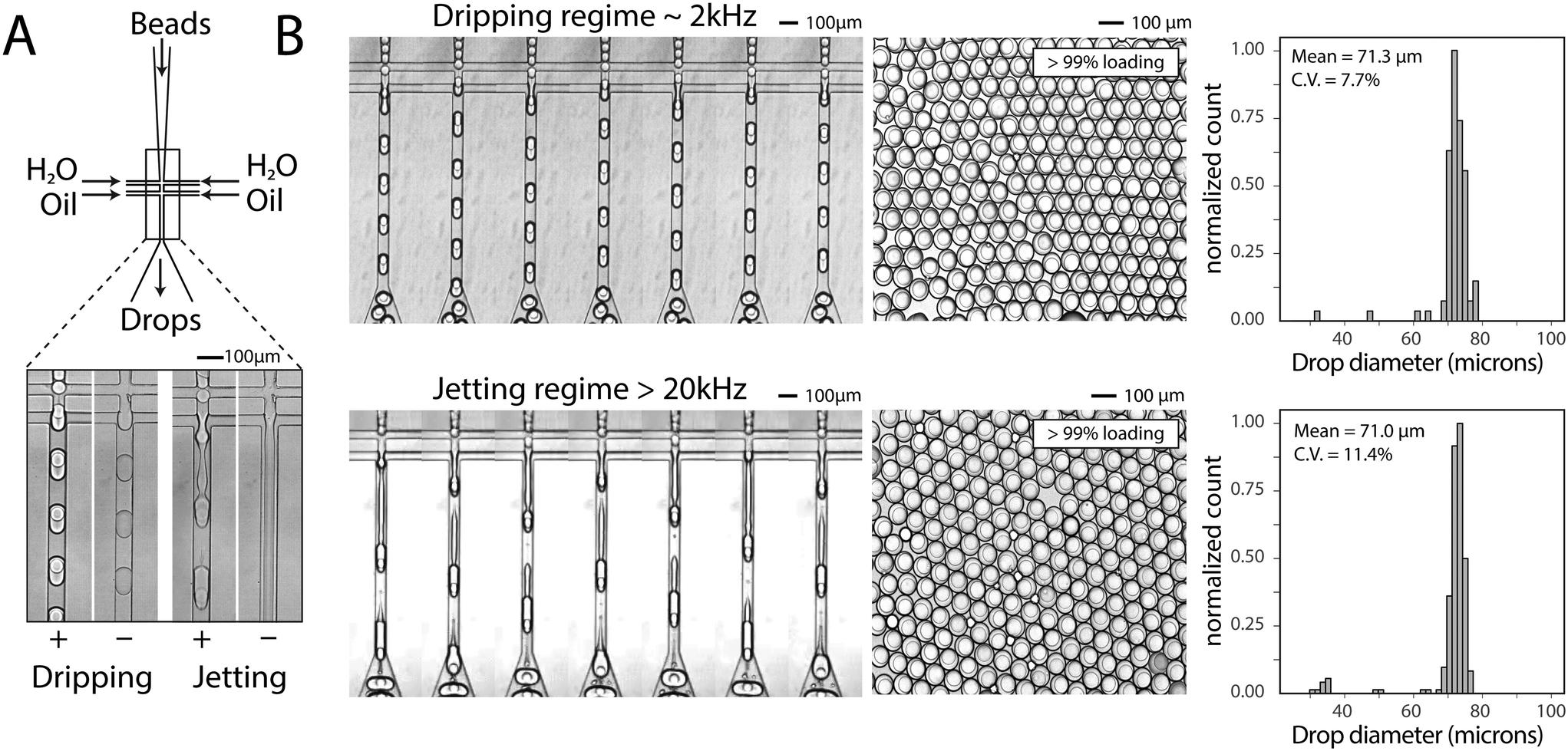 | ||
| Fig. 3 Bead-dependent drop formation in the jetting regime with an aqueous co-flow. A) Device schematic with fluid inlets and outlets. Device operation in the dripping and jetting regimes with and without 50 μm beads. B) Frames from a video of device operation in the dripping (top) and jetting (bottom) regimes. The resultant emulsions and drop diameter histograms are shown to the right of time lapse images. | ||
Co-flowing an additional aqueous allows for reagent addition and independent control of drop size (Fig. 3A). Co-flowing is especially important in cases where bulk addition of reagents is not experimentally feasible. For example, cells and lysis buffer, enzymes and their substrates, and components of chemical reactions often must be co-flowed on-chip to prevent mixing outside of droplets. In the absence of a bead, the aqueous co-flow forms a stable jet that does not break in the microfluidic channel. Introduction of the bead induces drop formation (Fig. 3A). High-speed video frames capture the elongation of the fluid jet and subsequent bead-dependent drop formation (Fig. 3B, bottom). Operation in this regime produces bead-containing drops of similar percent loading (>99%) and size uniformity to a typical dripping regime (Fig. 3B).
If the beads are hydrogels, it is often possible to soak reagents into pores and an aqueous co-flow is not necessary. In this case, bead-triggering occurs even without co-flow, but is difficult to visualize because packing removes excess aqueous phase and no jet forms. The dispersed phase is made entirely of beads with interstitial fluid. In this scenario, packed beads trigger drop formation and the flow rate can be increased well into a typical aqueous jetting regime. We compare the point at which a dispersed water phase transitions from dripping into jetting to the point at which tightly packed beads transition from single into multi-core drops (Fig. 4A). Compared to non-bead droplet generation, bead triggering operates efficiently at higher capillary numbers and high dispersed phase flow rates (6000 μl h−1) (Fig. 4B).
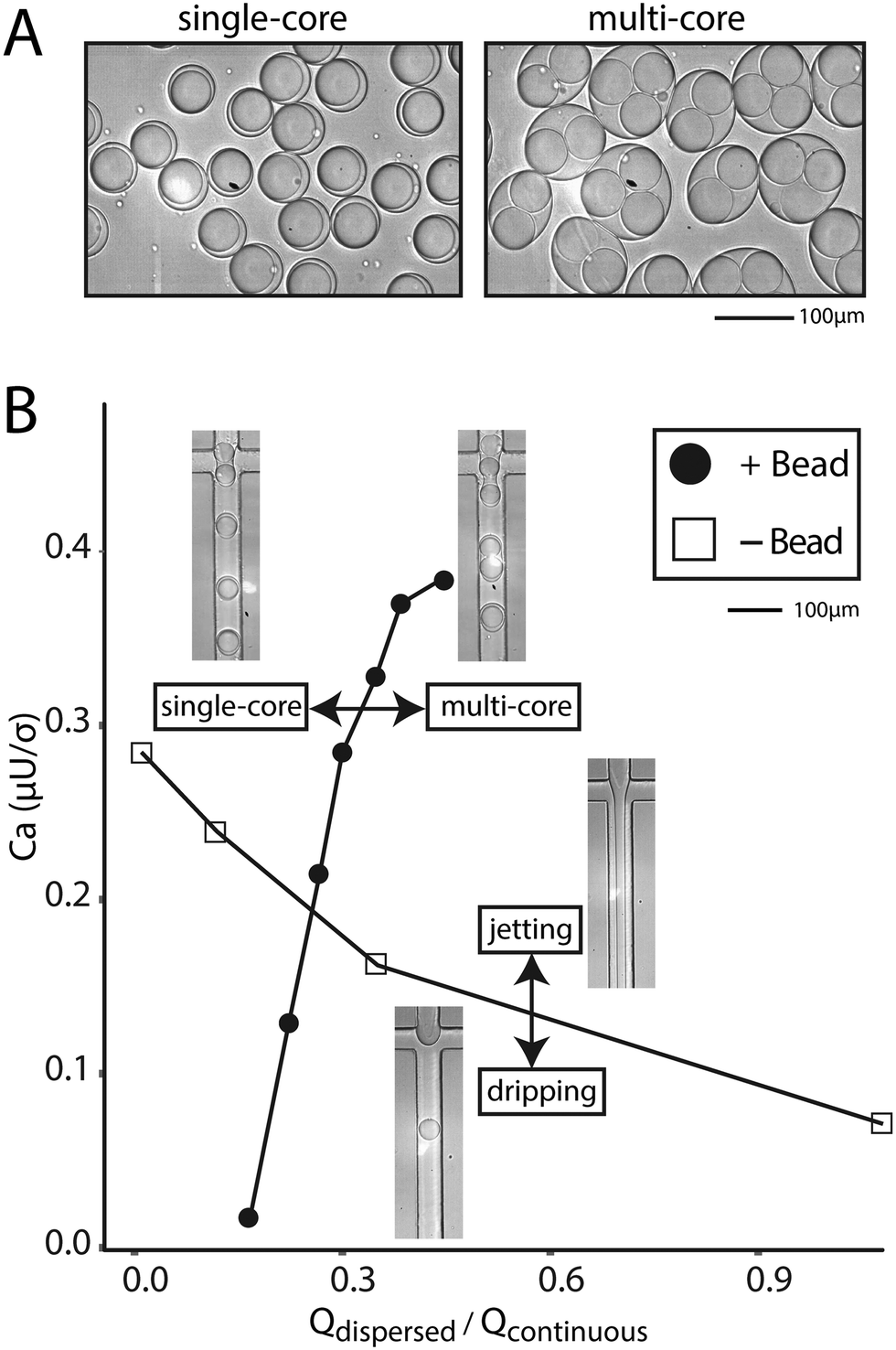 | ||
| Fig. 4 Bead-dependent drop formation in the jetting regime without an aqueous co-flow. A) Microscope images of single-bead and multi-bead containing drops. B) Phase diagram of the transition from single- to multi-bead drops as a function of capillary number and flow rate ratio. Images of devices depict operation in particular regions. | ||
2.3 Bead-cell pairing above 20 kHz
The principle utility of bead-triggered drop formation is that it enables an increase in throughput with minimal reduction in emulsion quality. We demonstrate this by encapsulating 50 μm polyacrylamide beads in monodispersed droplets at high speeds. The drop maker is run with the two aqueous streams at 4000 μl h−1 each and oil at 8000 μl h−1 (Fig. 5A), corresponding to a predicted encapsulation frequency of approximately 23 kHz. To measure the frequency of drop making, we record fluorescent time trace data of flowing droplets (Fig. 5B, inset) and compute the power spectrum of the intensity (Fig. 5B). The peak of this spectrum agrees with the estimates of drop-making frequency based on flow rate and demonstrates an order of magnitude increase in throughput compared to running in the dripping regime.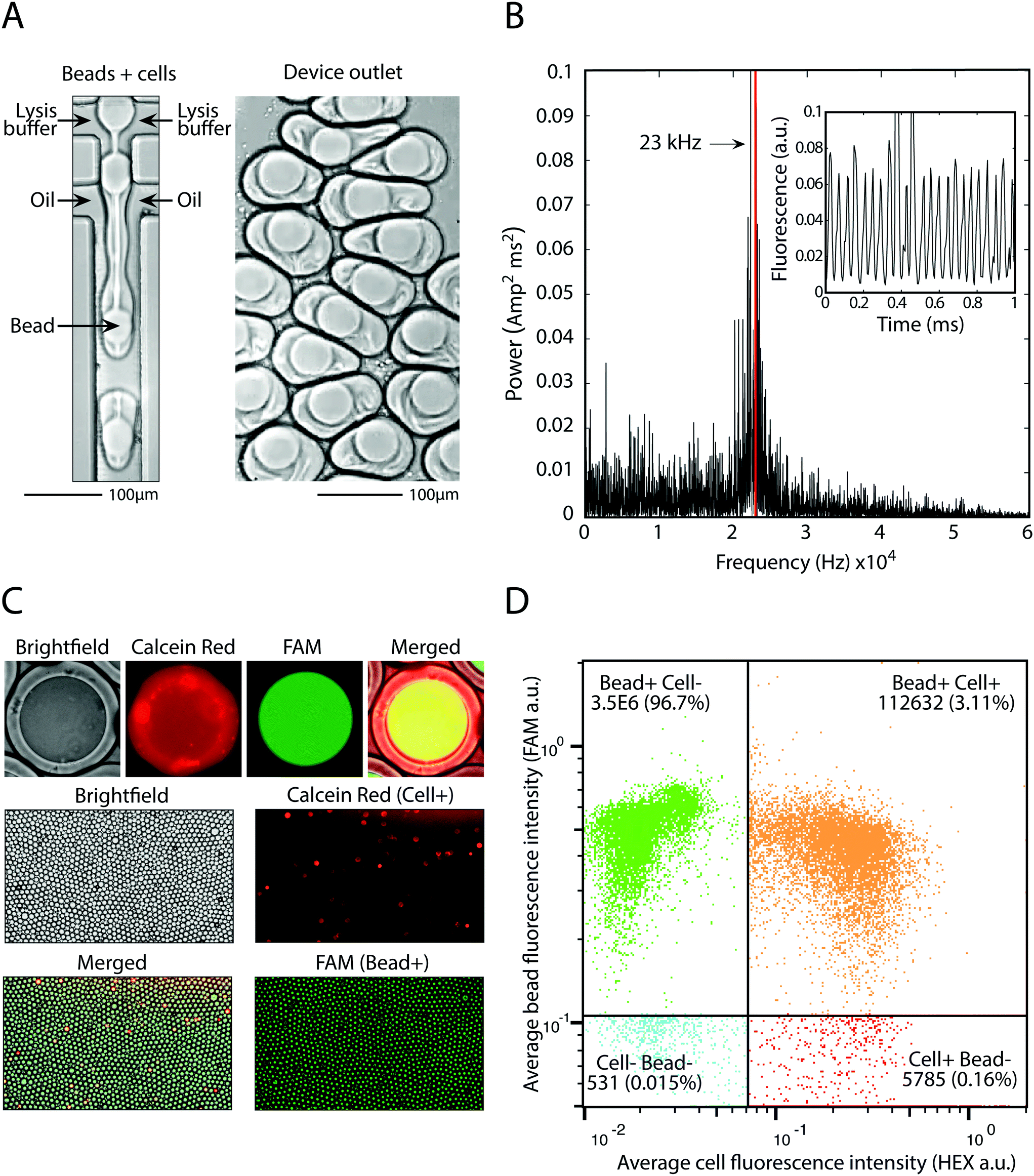 | ||
| Fig. 5 Pairing cells with beads at 23 kHz. A) Device operation and the resultant droplets in the outlet. B) Power spectrum of fluorescent time trace data (inset) used to calculate drop making frequency. C) Fluorescence microscopy of FAM-stained beads and calcein red cells demonstrates bead loading and cell-bead pairing. D) High throughput analysis of millions of bead-containing droplets and hundreds of thousands of bead-cell pairings using a droplet cytometer. | ||
Using this approach we pair cells with beads at high speed. We dilute cells to achieve one cell per 20 drops and pair these cells with beads at 23 kHz, easily achieving 1 kHz pairing. This allows pairing of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells with single beads in minutes, or 10 million cells in several hours. Cells are stained with calcein red and beads are polymerized with a fluorescent oligonucleotide so that each can be detected on the FAM and HEX channels, respectively (Fig. 5C). Upon on-chip mixing with lysis buffer, cells release the calcein red dye and the entire drop becomes fluorescent. Analysis of drop fluorescence using microscopy demonstrates the precise loading of one-bead per drop, as well as pairing of cells with beads (Fig. 5C). To accurately measure the throughput of this approach, we analyze the drops with our droplet cytometer. In the 3.5 million drops analyzed, 99.8% contain a fluorescent bead, and 3.1% contain a bead-cell pair, close to the 5% cell loading predicted based on the cell concentration used. This demonstrates that bead-triggered encapsulation and pairing functions with a speed and accuracy capable of processing millions of cells.
000 cells with single beads in minutes, or 10 million cells in several hours. Cells are stained with calcein red and beads are polymerized with a fluorescent oligonucleotide so that each can be detected on the FAM and HEX channels, respectively (Fig. 5C). Upon on-chip mixing with lysis buffer, cells release the calcein red dye and the entire drop becomes fluorescent. Analysis of drop fluorescence using microscopy demonstrates the precise loading of one-bead per drop, as well as pairing of cells with beads (Fig. 5C). To accurately measure the throughput of this approach, we analyze the drops with our droplet cytometer. In the 3.5 million drops analyzed, 99.8% contain a fluorescent bead, and 3.1% contain a bead-cell pair, close to the 5% cell loading predicted based on the cell concentration used. This demonstrates that bead-triggered encapsulation and pairing functions with a speed and accuracy capable of processing millions of cells.
3. Conclusion
Beads are widely used in both commercial and academic droplet workflows because useful mechanical and chemical properties can be produced or purchased, enabling the chemical and enzymatic coupling of oligonucleotides, proteins, and antibodies. However, such workflows are subject to an upper limit on throughput, governed by the Capillary number dependent transition from dripping to jetting. This paper presents a mechanism of drop formation that utilizes internal perturbation of a stable jet by beads, enabling bead-based workflows to operate above 20 kHz. This mechanism drives drop generation of viscous jets, co-flowed reagents, and packed beads with no additional aqueous phase, making it widely applicable to most bead-based applications. Bead-triggering is particularly useful for analysis of large populations and the study of rare cells that require processing of millions of bead-containing drops.4. Methods
4.1 Device fabrication
SU-8 2025 photoresist (MicroChem, Westborough, MA, USA) is used to make master structures on a 3-inch silicon wafer using standard photolithography techniques. Curing agent and PDMS prepolymer (Momentive, Waterford, NY, USA; RTV 615) are mixed 1:10, degassed in a vacuum chamber, poured onto the master mold in a petri dish, further degassed until no bubbles are present, and baked at 65 °C for 4 hours. PDMS replicas are removed from the master, punched with a 0.75 mm biopsy punch (TedPella, Inc., Redding, CA, USA; Harris Uni-Core 0.75), bonded to glass slides (75 × 50 × 1.0 mm, 12-550C, Fisher Scientific) using a plasma bonder (Technics Plasma etcher), and placed at 150 °C for ten minutes to strengthen bonds. Devices are treated with Aquapel with a five-minute contact time and purged with air, rendering them hydrophobic. Devices are baked for at least 30 minutes at 65 °C to evaporate any remaining Aquapel.
4.2 Rigid bead encapsulation
Rigid 20–40 μm beads made from hydroxylated methacrylic polymer (Toyopearl HW-65S, Tosoh Bioscience) are used to demonstrate the formation of monodispersed bead-containing droplets at limiting dilution. Beads are suspended in 68% OptiPrep DensityGradient Medium (Sigma-Aldrich), and co-flowed with 2% agarose (Ultra-low Gelling Temperature Agarose, Type IX-A, Sigma-Aldrich) in 1× PBS. The device, identical to one previously described,1 is fabricated at a height of 100 μm. Flow rates are 2000 μl h−1 bead-containing solution, 2000 μl h−1 agarose, and 40000 μl h−1 2% ionic Krytox, prepared as previously described.18
4.3 Droplet cytometry
The custom microfluidic cytometer is designed as previously described.9 The system is equipped with three lasers (473 nm, 532 nm, 638 nm). EVA green (1×) stained Toyopearl HW-65S beads are detected with the 473 nm laser (Fig. 2). BSA conjugated with Cy5 is used to measure drop size with the 638 nm laser (Fig. 2). Drop length is calculated using the time a drop spends in the laser, the total flow rate, and the cross sectional channel dimensions (125 μm × 125 μm). Calcein red-orange is used to detect cells with the 532 nm laser (Fig. 5). Acrydited primers containing FAM are polymerized during droplet polyacrylamide formation and detected with the 473 nm laser (Fig. 5). Drop fluorescence is calculated as the integral of peak intensity divided by the drop size. Results are exported and analyzed in FlowJo.4.4 Hydrogel synthesis
Elastic hydrogels for bead-packing experiments are made microfluidically using a bubble-trigger device.19 A solution containing 8% Acrylamide with crosslinker (40% acrylamide/bis solution, 19:1, Biorad), 200 mM TRIS pH 8.3, and 0.3% ammonium persulfate in water is used as the dispersed phase. The continuous phase consists of 2% ionic krytox with 1% N,N,N′,N′-tetramethylethylenediamine (ThermoFisher). Solidification occurs for 1 hour at room temperature. Hydrogel beads are recovered by adding an equal volume of 20% 1H,1H,2H,2H-perfluoro-1-octanol, mixing gently, centrifuging, and removing the oil phase with a pipet. Hydrogel beads are washed thrice with water containing 0.1% Tween-20. Hydrogel beads in water are 50 μm in diameter with a coefficient of variation of <10%.
4.5 Packed bead encapsulation
Prior to use, solidified polyacrylamide beads are filtered using a 70 μm cell strainer (Corning Falcon Cell Strainer) to remove large beads and prevent device clogging. This step is necessary to ensure consistent operation of the encapsulation device. Beads are packed in a syringe by centrifugation (Sorvall ST40R) at 4700 rpm for 10 minutes using a custom 3D printed syringe adapter. Supernatant is removed and beads are injected onto microfluidic devices. We find that high speeds compress beads and result in higher levels of bead jamming and bursting than under dripping conditions. This is improved using PE/5 tubing instead of PE/2 tubing (Scientific Commodities).For co-flow experiments in the dripping regime, 200 μl h−1 1× PBS with 0.1% Tween, 200 μl h−1 packed polyacrylamide beads, and 600 μl h−1 oil are used. For co-flow experiments in the jetting regime, 4000 μl h−1 1× PBS with 0.1% Tween, 4000 μl h−1 packed polyacrylamide beads and 6000 μl h−1 oil are used. For experiments without a co-flow, we determine the phase transition as follows. Points represent the average of two measurements: the first measurement is the transition from dripping to jetting (or single-core to multi-core) the second measurement is the transition from jetting to dripping (or multi-core to single-core). Aqueous flow rate is held constant and oil flow rate is increased above and then decreased below the transition point. Aqueous flow rate is changed and the same procedure is completed to generate phase boundaries. Capillary number is calculated using the continuous phase flow rate, viscosity of the continuous phase (HFE-7500, 1.24 × 10−3 kg m−1 s−1) and interfacial tension (4 mN m−1) assuming a square channel cross-section of 55 μm × 55 μm.
4.6 Cell-bead pairing
T cells (Jurkat) are cultured in an incubator containing 5% CO2 at 37 °C using Advanced RPMI 1640 (Gibco/Invitrogen) with 10% heat inactivated FBS, 100 units per ml penicillin and 100 μg ml−1 streptomycin. Cells are stained using calcein (25 μM calcein red-orange, AM, C34851, ThermoFisher) in 1× PBS, and incubated on ice for 30 min. Cells are washed twice (HBSS, no calcium, no magnesium, 14170112, ThermoFisher) and re-suspended in 18% OptiPrep Density Gradient Medium (Sigma-Aldrich) in HBSS. Polyacrylamide beads are polymerized with 10 μM Acyridited oligonucleotides (IDT) containing a FAM labeled 3′ end. The flow rate is 4000 μl h−1 50 μm beads, 4000 μl h−1 lysis buffer (0.1% LiDS, 1 mM EDTA, 20 mM TRIS 8.3, 500 mM LiCl), and 8000 μl h−1 oil (Biorad Droplet Generation Oil for EvaGreen #1864005). To quickly find conditions to generate monodispersed bead-triggered drops, a jet is formed in the absence of beads. Drops are collected into a 10 mL syringe for later re-injection and detection. To calculate the speed of drop generation, timetrace data of fluorescence intensity was collected for flowing droplets and analyzed in Matlab using the fast fourier transform (fft) function. The power spectrum was calculated as the square of the absolute value of fft divided by the number of samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by amfAR, The Foundation for AIDS Research (109537-61-RGRL) and by the NIH: National Institute of Allergy and Infectious Diseases (AI129206).References
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed
.
- D. Dressman, H. Yan, G. Traverso, K. W. Kinzler and B. Vogelstein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8817–8822 CrossRef CAS PubMed
- A. Sepp, D. S. Tawfik and A. D. Griffiths, FEBS Lett., 2002, 532, 455–458 CrossRef CAS PubMed
- R. Novak, Y. Zeng, J. Shuga, G. Venugopalan, D. A. Fletcher, M. T. Smith and R. A. Mathies, Angew. Chem., 2010, 123, 410–415 CrossRef
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed
- H. Zhang, G. Jenkins, Y. Zou, Z. Zhu and C. J. Yang, Anal. Chem., 2012, 84, 3599–3606 CrossRef CAS PubMed
- F. Diehl, M. Li, Y. He, K. W. Kinzler, B. Vogelstein and D. Dressman, Nat. Methods, 2006, 3, 551–559 CrossRef CAS PubMed
- E. Borgström, D. Redin, S. Lundin, E. Berglund, A. F. Andersson and A. Ahmadian, Nat. Commun., 2015, 6, 7173 CrossRef PubMed
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed
- T. Konry, M. Dominguez-Villar, C. Baecher-Allan, D. A. Hafler and M. L. Yarmush, Biosens. Bioelectron., 2011, 26, 2707–2710 CrossRef CAS PubMed
- V. Chokkalingam, J. Tel, F. Wimmers, X. Liu, S. Semenov, J. Thiele, C. G. Figdor and W. T. S. Huck, Lab Chip, 2013, 13, 4740–4744 RSC
- S. J. Spencer, M. V. Tamminen, S. P. Preheim, M. T. Guo, A. W. Briggs, I. L. Brito, D. A. Weitz, L. K. Pitkänen, F. Vigneault, M. P. Juhani Virta and E. J. Alm, ISME J., 2016, 10, 427–436 CrossRef CAS PubMed
- F. Lan, J. R. Haliburton, A. Yuan and A. R. Abate, Nat. Commun., 2016, 7, 11784 CrossRef CAS PubMed
- A. R. Abate, C.-H. Chen, J. J. Agresti and D. A. Weitz, Lab Chip, 2009, 9, 2628–2631 RSC
- M. Chabert and J. L. Viovy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3191–3196 CrossRef CAS PubMed
- I. Cohen, H. Li, J. L. Hougland, M. Mrksich and S. R. Nagel, Science, 2001, 292, 265–267 CrossRef CAS PubMed
- A. R. Abate and D. A. Weitz, Lab Chip, 2011, 11, 1713–1716 RSC
- C. J. DeJournette, J. Kim, H. Medlen, X. Li, L. J. Vincent and C. J. Easley, Anal. Chem., 2013, 85, 10556–10564 CrossRef CAS PubMed
- Z. Yan, I. C. Clark and A. R. Abate, Macromol. Chem. Phys., 2016, 218, 1600297 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc00514a |
| This journal is © The Royal Society of Chemistry 2018 |