
Fully automated, on-site isolation of cfDNA from whole blood for cancer therapy monitoring†
 *ab
*ab
Abstract
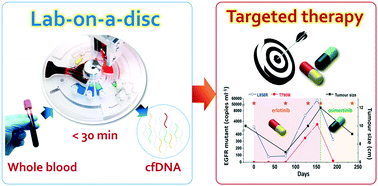
The potential utility of circulating tumour DNA (ctDNA) in patient blood for cancer diagnostics and real-time monitoring of disease progression is highly recognized. However, the lack of automated and efficient methods for cell-free DNA (cfDNA) isolation from peripheral blood has remained a challenge for broader acceptance of liquid biopsy in general clinical settings. Here, we demonstrate a lab-on-a-disc system equipped with newly developed, electromagnetically actuated, and reversible diaphragm valves that allows fully automated and rapid (<30 min) isolation of cfDNA from whole blood (>3 ml) to achieve high detection sensitivity by minimizing the degradation of fragile ctDNA as well as contamination of wild-type DNA from abundant blood cells. As a proof of concept study, we used the lab-on-a-disc to isolate cfDNA from patients with non-small cell lung cancer and successfully detected epidermal growth factor receptor gene mutations (L858R, T790M) during targeted drug therapy. The proposed lab-on-a-disc enables a fully automated, rapid, and point-of-care cfDNA enrichment starting from whole blood to facilitate the wide use of liquid biopsy in routine clinical practice.
- This article is part of the themed collections: Personalised Medicine: Liquid Biopsy and Celebrating Excellence in Research: 100 Women of Chemistry
 Please wait while we load your content...
Please wait while we load your content...

