
Rapid and label-free identification of single leukemia cells from blood in a high-density microfluidic trapping array by fluorescence lifetime imaging microscopy†

Abstract
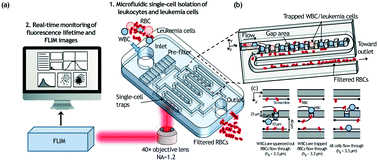
The rapid screening and isolation of single leukemia cells from blood has become critical for early leukemia detection and tumor heterogeneity interrogation. However, due to the size overlap between leukemia cells and the more abundant white blood cells (WBCs), the isolation and identification of leukemia cells individually from peripheral blood is extremely challenging and often requires immunolabeling or cytogenetic assays. Here we present a rapid and label-free single leukemia cell identification platform that combines: (1) high-throughput size-based separation of hemocytes via a single-cell trapping array, and (2) leukemia cell identification through phasor approach and fluorescence lifetime imaging microscopy (phasor-FLIM), to quantify changes between free/bound nicotinamide adenine dinucleotide (NADH) as an indirect measurement of metabolic alteration in living cells. The microfluidic trapping array designed with 1600 highly-packed addressable single-cell traps can simultaneously filter out red blood cells (RBCs) and trap WBCs/leukemia cells, and is compatible with low-magnification imaging and fast-speed fluorescence screening. The trapped single leukemia cells, e.g., THP-1, Jurkat and K562 cells, are distinguished from WBCs in the phasor-FLIM lifetime map, as they exhibit significant shift towards shorter fluorescence lifetime and a higher ratio of free/bound NADH compared to WBCs, because of their glycolysis-dominant metabolism for rapid proliferation. Based on a multiparametric scheme comparing the eight parameter-spectra of the phasor-FLIM signatures, spiked leukemia cells are quantitatively distinguished from normal WBCs with an area-under-the-curve (AUC) value of 1.00. Different leukemia cell lines are also quantitatively distinguished from each other with AUC values higher than 0.95, demonstrating high sensitivity and specificity for single cell analysis. The presented platform is the first to enable high-density size-based single-cell trapping simultaneously with RBC filtering and rapid label-free individual-leukemia-cell screening through non-invasive metabolic imaging. Compared to conventional biomolecular diagnostics techniques, phasor-FLIM based single-cell screening is label-free, cell-friendly, robust, and has the potential to screen blood in clinical volumes through parallelization.
- This article is part of the themed collection: RSC papers by NanoEngineering for Medicine and Biology 2018 Speakers
 Please wait while we load your content...
Please wait while we load your content...

