
A microfluidic chip capable of generating and trapping emulsion droplets for digital loop-mediated isothermal amplification analysis†
 *abc
*abc
Abstract
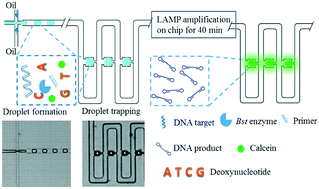
Loop-mediated isothermal amplification (LAMP) is a nucleic acid amplification technique that rapidly amplifies specific DNA molecules at high yield. In this study, a microfluidic droplet array chip was designed to execute the digital LAMP process. The novel device was capable of 1) creating emulsion droplets, 2) sorting them into a 30 × 8 droplet array, and 3) executing LAMP across the 240 trapped and separated droplets (with a volume of 0.22 nL) after only 40 min of reaction at 56 °C. Nucleic acids were accurately quantified across a dynamic range of 50 to 2.5 × 103 DNA copies per μL, and the limit of detection was a single DNA molecule. This is the first time that an arrayed emulsion droplet microfluidic device has been used for digital LAMP analysis. When compared to microwell digital nucleic acid amplification assays, this droplet array-based digital LAMP assay eliminates the constraint on the size of the digitized target, which was determined by the dimension of the microwells for its counterparts. Moreover, the capacity for hydrodynamic droplet trapping allows the chip to operate in a one-droplet-to-one-trap manner. This microfluidic chip may therefore become a promising device for digital LAMP-based diagnostics in the near future.
 Please wait while we load your content...
Please wait while we load your content...

