
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResonant laser–SNMS for spatially resolved and element selective ultra-trace analysis of radionuclides
Michael
Franzmann
 *ab,
Hauke
Bosco
a,
Linda
Hamann
a,
Clemens
Walther
a and
Klaus
Wendt
b
*ab,
Hauke
Bosco
a,
Linda
Hamann
a,
Clemens
Walther
a and
Klaus
Wendt
b
aInstitute for Radioecology and Radiation Protection (IRS), Leibniz University of Hannover, Germany. E-mail: franzmann@irs.uni-hannover.de; Tel: +49 511 762 3069
bInstitute of Physics, University of Mainz, Staudingerweg 7, 55128 Mainz, Germany
First published on 21st March 2018
Abstract
The newly developed resonant laser–SNMS system at the IRS Hannover combines the high spatial and decent mass resolution of a TOF-SIMS instrument with the element-selective process of resonant laser ionization. This setup was characterized by use of synthetic uranium, plutonium and technetium samples to prepare and demonstrate the performance for measurements on environmental samples. The laser–SNMS system will be applied for the detection, visualization and ultra-trace analysis of radionuclide containing nanoparticles in environmental samples with strongly reduced or even completely omitted chemical preparation. The necessary suppression of isobaric contamination was demonstrated as well as the first detection of uranium containing particles in a completely unaltered soil sample.
1 Introduction
Ultra-trace analysis of uranium, plutonium and technetium is of major interest for the investigation of radionuclide mobility, transport behavior and distribution in radioactively contaminated environments as well as for nuclear safeguard and forensics applications. To understand the underlying processes, commercially available technology is often either not sufficiently sensitive or selective. Correspondingly, dedicated instruments are developed for specific needs, often tailored accordingly by combination of different experimental methods and analytical steps as so-called hyphenated techniques. Secondary ion mass spectrometry (SIMS) is a standard technique for direct and spatially resolved analysis of hot particles,1,2 but its application already on uranium as most dominant contribution is limited by isobaric interferences. Previous chemical separation to suppress interfering species always implies dissolution of the sample material and would consequently destroy any kind of structural information, which should be visualized. A technology which is extensively free of isobaric interferences is resonance ionization mass spectrometry (RIMS). It constitutes another ultra-trace analytic technique, which is routinely applied for analysis of radionuclides.3,4 RIMS uses the characteristic atomic transitions for optical multi-step excitation and ionization and therefore intrinsically possesses the required element selectivity. Resonant laser–SNMS (Secondary Neutral Mass Spectrometry) is based on the combination of the direct and spatially resolving ablation of sample constituents by sputtering (adopted from SIMS) with subsequent laser resonance ionization of the sputtered neutral atomic fraction. The successful application of resonant laser–SNMS for ultra-trace analysis of radionuclide-containing nanoparticles was demonstrated at the Institute for Nuclear Chemistry of the Johannes Gutenberg-Universität Mainz by Erdmann et al.5 already in 2009 using a prototype setup. On the basis of this development, a technically mature Laser–SNMS system was developed and put into operation at the Institute for Radioecology and Radiation Protection (IRS) of the Leibniz-Universität Hannover. The analytic device is based upon a commercial state-of-the-art TOF-SIMS instrument and implies a dedicated high power Ti:sapphire laser system jointly forming one of the key components for the research project SIRIUS (Secondary laser Ionization of Radioactive Isotopes for Ultra-trace analysis with Spatial resolution) funded by the German BMBF. It was set up with special emphasis on the aspect to localize, analyze and visualize ultra-trace amounts and distributions of any kind of radionuclides in environmental samples. This paper describes the initial analytic characterization of the new system using synthetic and well quantified samples containing different trace and ultra-trace amounts of uranium, plutonium and technetium radioisotopes. Additional systematic investigations focused on the suppression of isobaric contaminations and the detection of radionuclide containing so-called hot particles in environmental samples demonstrating the performance and the capabilities of this new analytical tool for radioecology.2 Experimental
2.1 TOF-SIMS instrument
The TOF.SIMS 5 of the manufacturer IONTOF is a sophisticated analytical device, which comprises a selection of different ion guns and several sample preparation accessories. The primary ion gun is a bismuth cluster source providing a 30 keV pulsed ion beam for static SIMS. With an optimized focus for surface imaging the gun allows lateral resolution of down to 80 nm. The TOF-SIMS instrument is equipped with two additional sputter guns. The first one uses cesium or oxygen ions for low-energy depth profiling, while the second sputter gun provides a 20 keV argon cluster ion beam with adjustable cluster size of up to 10.000. The resulting low energy per atom enables depth profiling of structures containing large organic molecules without significant dissociation.Mass spectrometry of the sputtered secondary ions is performed by a time-of-flight analyzer involving a gridless reflectron and a high sensitive single ion detection, leading to a mass resolution of up to 12.000. The foreseen application of direct ultra-trace analysis of radionuclides in a sample implies the complete suppression of any kind of organic background or atomic isobaric interferences stemming from the stable constituents, which limit conventional SIMS. The attempt requires additional improvement of sensitivity and selectivity, which can only be met by enclosing a dedicated ionization step on top of the sputtering process as found in multi-step resonance ionization by well tuned laser light.
2.2 Ti:sapphire laser system for resonance ionization
Resonant laser ionization provides high elemental selectivity by stepwise excitation and ionization using strong optical dipole transitions along a multi-step excitation ladder, which is characteristic for each chemical element based upon its unique atomic structure (shown in Fig. 1).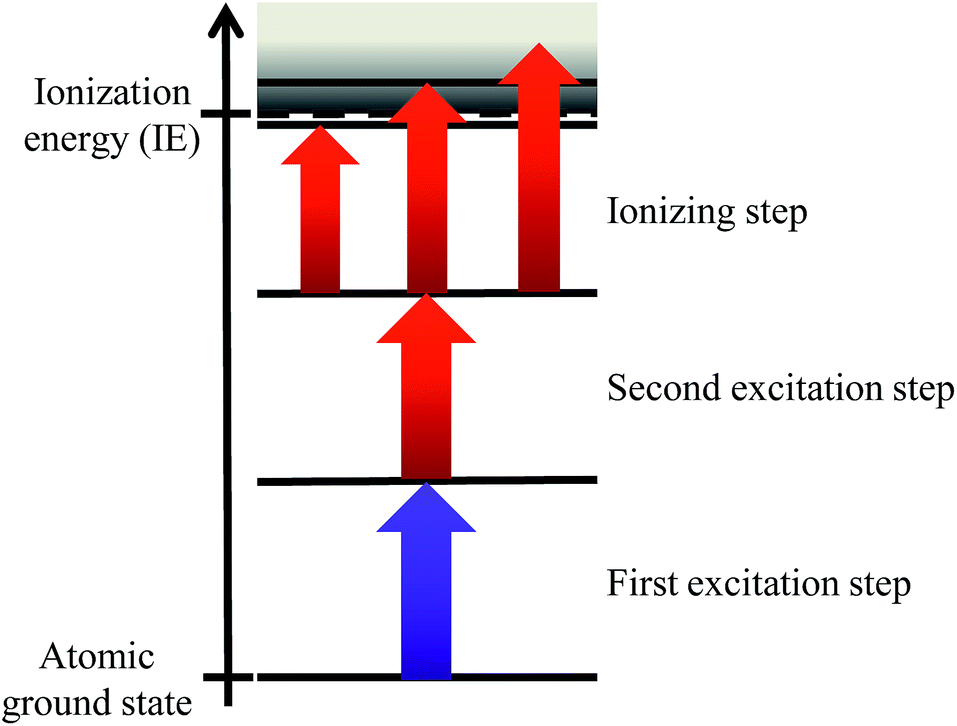 | ||
| Fig. 1 Three-step excitation scheme for resonant laser ionization including three methods for the final ionizing step: the ionization can be achieved through bound Rydberg levels slightly below the IE, by excitation to autoionizing levels above the IE or by using a non-resonant process. | ||
The element of interest is efficiently ionized while any kind of isobaric background stemming from atomic or molecular constituents is primarily unaffected and consequently suppressed. The applied high power Ti:sapphire laser system for resonance ionization at IRS was initially designed and adapted to the special needs of this project by the LARISSA group at the Institute of Physics of the Johannes Gutenberg-Universität Mainz. The layout of the laser cavities and the overall setup including shaping, transport, overlap and spectral, spatial and temporal control of the laser beams is based on experiences from a number of widely similar laser systems utilized for atomic spectroscopy, for production of isotopically pure ion beams at radioactive ion beam facilities and for different applications of trace analyses.6 The setup consists of three tunable Ti:sapphire lasers, delivering up to 4 W laser power each at a repetition rate of 10 kHz with 5–9 GHz spectral bandwidth. The Ti:sapphire lasers are pumped independently by three frequency doubled, commercial Nd:YAG lasers. This arrangement simplifies the precise control of the timing overlap of the three laser pulses (pulse duration 20–50 ns) with the pulsed sputtering and extraction of the SIMS instrument and with each other. The broad wavelength range of 690–960 nm, which can be covered by Ti:sapphire lasers combined with optional frequency doubling offers the possibility to use just three lasers to match a multitude of ionization schemes of many different radionuclides. Adjustment times from one element to another amount to just several minutes in favorable cases and do not exceed a few hours, thus even allowing for a limited sequential multi element analysis.
2.3 Combined laser–SNMS system
The combination of the commercial SIMS instrument with a dedicated laser system for resonance ionization is sketched in Fig. 2 in a simplified way.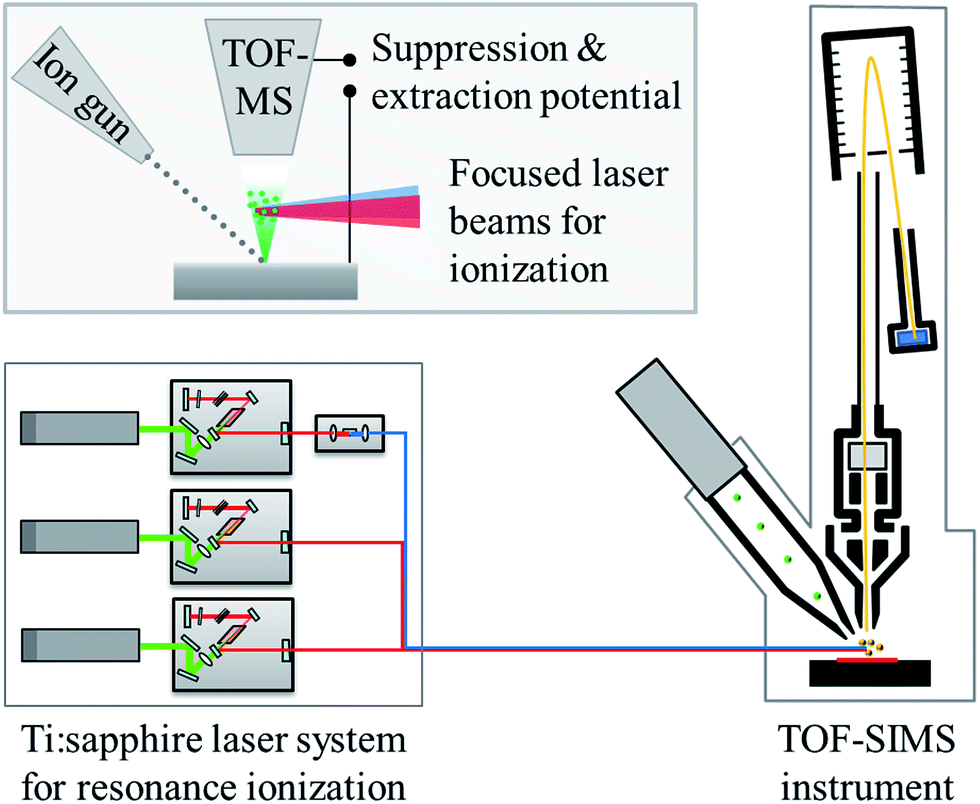 | ||
| Fig. 2 Schematic representation of the two main parts of the laser–SNMS system. The upper inset shows a magnification of the sputter and ionization region. | ||
In contrast to SIMS, SNMS utilizes the neutral fraction of the sputtered particles instead of the sputtered secondary ions. The ions produced directly by the sputtering process are suppressed by an electrical field, while the sputtered neutrals expand as a cloud into the vacuum above the sample surface. The spatially and temporally overlapped laser beams are focused into this cloud to initiate the stepwise excitation and ionization of the atoms of a chosen element. As a suppression potential is turned off right before the laser pulse and an attractive potential is switched on immediately afterwards, the laser ions are extracted into the TOF mass analyzer. Due to this efficient background suppression the signal-to-noise ratio in laser–SNMS spectra is much higher than in comparable SIMS spectra, often by orders of magnitude. Additionally, resonant laser ionization allows to clearly identify the ion signal of the selected element as a direct quantification of background can be carried out by simply detuning the wavelength of one of the lasers. Resonant laser ionization is applicable only to ground state atoms, which leads to the high selectivity for atomic mass spectrometry but also prevents the analysis of molecular species. Therefore, the laser–SNMS system at IRS Hannover can be switched from SNMS to SIMS analysis within seconds to offer the advantages of both techniques.
It is well known, that the sputtering process generates approximately up to a thousand times more neutral than positively or negatively charged ionic species. Intuitively a significant gain in the efficiency of laser–SNMS as compared to SIMS would thus be expected. However, in the experiment the laser ion signal of SNMS ranges in most cases in the same level as the sputter ion signal from SIMS. This fact is ascribed to the limited ionization efficiency for laser–SNMS, which is influenced by several parameters. Firstly, the overlap between the laser foci and the expanding particle cloud is limited to a small volume and must match the acceptance region of the mass analyzer. The very small distance of only 1.5 mm between the sample surface and the extraction electrode requires a tight focusing of the laser beams and very careful overlapping. During the delay of typically 200 ns, which is needed after the initial sputter pulse to suppress the sputter ions, the sputtered neutral particle cloud expands significantly and partly drifts out of this small laser interaction region. Additional losses are caused by the limited ionization efficiency of some optical excitation and ionization schemes, which are seemingly even affected by the conditions in the expanding atom cloud. The latter effect is still under investigation.
3 Results and discussion
3.1 Characterizing the laser–SNMS system
Individual excitation schemes for resonant laser ionization of a specific element could vary in ionization efficiency according to different cross sections and lifetimes of the utilized energy levels and optical transitions. Furthermore, the available power for each laser step and the contribution of non-resonant ionization of the specific element or an isobaric species has a major influence on the resulting laser–SNMS spectrum. Therefore it was necessary to characterize the new laser–SNMS system with synthetic standard samples of every element of interest as preparation for any kind of analytical measurements on environmental samples. The first characterization measurements were performed on the elements uranium, plutonium and technetium, which are of primary interest within the SIRIUS project. The available standard material was dissolved in nitric acid (0.1–0.3 M). 200 nl to 1 μl of this solution was pipetted onto a sample holder and vaporized for reliable deposition of the sample material. The influence of topology and conductivity of the sample was strongly reduced for characterization by application of a thin layer of sample material on a polished and electrically grounded aluminum sample holder.The excitation schemes of U, Pu and Tc as listed in Table 1, were developed in former spectroscopic experiments of the LARISSA group at the Institute of Physics of the Johannes Gutenberg-Universität Mainz or with its participation in collaborating institutions, respectively.4,7,8
| Isotope | First excitation step [nm] | Second excitation step [nm] | Ionizing step [nm] |
|---|---|---|---|
| 238U | 415.514 | 767.461 | 749.239 |
| 242Pu | 420.770 | 847.261 | 750.239 |
| 99Tc | 429.824 | 395.151 | 838.244 |
 | ||
| Fig. 3 Comparison of laser–SNMS spectra of uranium, plutonium and technetium. (a) Laser–SNMS spectrum of uranium including non-resonant ionized UO, UO2 and UC2. (b) Laser–SNMS spectrum of plutonium including non-resonant ionized PuO. (c) Laser–SNMS spectrum of technetium without a non-resonant ionized signal. | ||
These additional oxide and dioxide ion signals, which could even surpass the atomic ion signal could strongly increase the sensitivity for localization of smallest samples content, if combined with the resonant laser ion signal. Furthermore, they provided additional information about matrix effects and the chemical composition of the samples, while not interfering with the ion signal of the element of interest. A non-resonant ion signal that would interfere with the resonant ion signal could be avoided by changing the ionization scheme or by a reduced output power of the related laser. A formation of oxides after the sputtering process was excluded by spatial resolved analysis of the sample surface. The areas of origin of the atomic and molecular ion species deviate from each other, revealing strongly reduced or oxidized regions on the sample surface most likely due to interaction with the aluminum sample holder, which was verified by SIMS analysis.
| Isotope | Saturation power [mW] | ||
|---|---|---|---|
| FES | SES | AI | |
| 238U | 2.0(7) | 32(10) | >2300, not saturated |
| 242Pu | 10(5) | 25(18) | >1800, not saturated |
| 99Tc | 0.3(1) | 3.5(8) | >2400, not saturated |
The first and second excitation steps (FES & SES) were saturated with only a fraction of the available laser power, whereas for the ionizing step no saturation was obtained. The latter effect is ascribed to the fact, that all three schemes use spectrally broad autoionizing levels (AI) situated above the ionization energy, which are rapidly depopulated by ionization preventing saturation.
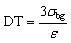 | (1) |
The challenge of efficiency measurements with a system based on static SIMS is the low sputter rate. The complete consumption of sample material for an efficiency measurement necessitates a very small and still well defined amount of sample material. Such a synthetically produced and quantified standard sample was only available for uranium. The suitable uranium sample material was provided by the Research Center Jülich in the form of synthetic uranium particles with a diameter of 1.3–1.4 μm consisting of a well defined number of 1.37(4) × 1010 atoms of the IRMM-183 standard material.11 These particles were deposited separately on a silicon waver for optimum analytical conditions considering conductivity and topology of the sample.
The efficiency determined in this way amounted to 2.63(7) × 10−5 for consideration of the atomic uranium ion signal alone, which matches the efficiency range of comparable systems.12 By accepting also the non-resonantly ionized uranium oxide and dioxide signal counts the efficiency is raised by a factor of 7 to 2.05(5) × 10−4, while the significance in the element assignment is still preserved. The lower efficiency of the purely atomic uranium is primarily limited by the high prevalence of oxidized uranium in the sample. Correspondingly, the efficient resonant laser ionization process is only affecting the small atomic fraction, while non-resonant ionization of the uranium oxides is much less efficient but affects a much larger share of the sample material. The resulting calculated DT is 2.12(8) × 106 atoms for atomic uranium and 3.58(9) × 105 atoms for acceptance of the total uranium signal. Unfortunately, this calculated DT represents the best achievable value for complete consumption of the sample material, which is no feasible limit for ultra-trace analysis on environmental samples. The synthetic particles for efficiency measurements were made of pure triuranium octoxide (U3O8) without any matrix material. Achieving similar DTs in environmental samples requires sputtering of all enclosing organic and inorganic material, which is much too time consuming. Therefore a more meaningful DT must be determined under realistic experimental conditions involving suitable sample compositions. This step was achieved by analysis of dilution series made up from standard materials for all three elements of interest. The measured DT is determined by the smallest trace amount delivering a detectable resonant ion signal in a reasonable time of up to 1 hour. Resulting DTs are shown in Table 3; for the aforementioned case of uranium they differ from the idealistically calculated DT by two to three orders of magnitude.
| Isotopes | |||
|---|---|---|---|
| 238U | 242Pu | 99Tc | |
| Efficiency | 2.63(7) × 10−5 | ||
| Calculated DT [atoms] | 2.12(8) × 106 | ||
| Sample concentration | 1 ppb | 10 ppb | 2 ppb |
| Measured DT [atoms] | 5.2(16) × 108 | 5.1(15) × 109 | 2.2(16) × 109 |
3.2 Influence of argon cluster sputtering
In typical environmental samples radionuclide containing particles are often covered by layers of organic material. Additionally, the exposed surfaces of the particles itself could have undergone much stronger oxidation processes than the particle core. A higher degree of oxidation implies a significant reduction of the resonantly ionizable atomic fraction of the radionuclide after the sputtering process. Therefore, the supplementary argon cluster sputter gun, which is primarily attached for organic depth profiling,13 was used for in situ sample preparation. Organic contaminations on the surface of environmental samples were reduced through argon cluster sputtering by 1–3 orders of magnitude strongly dependent on the sample material. This beneficial removal of significantly oxidized surface layers by application of an atomic argon ion gun was demonstrated for uranium on a U3O8 particle in former experiments.14 The results were validated in this work with argon cluster sputtering for test samples made of plutonium and uranium. A gain in the resonantly ionized ion signal of approximately one order of magnitude was achieved, as shown in Fig. 4.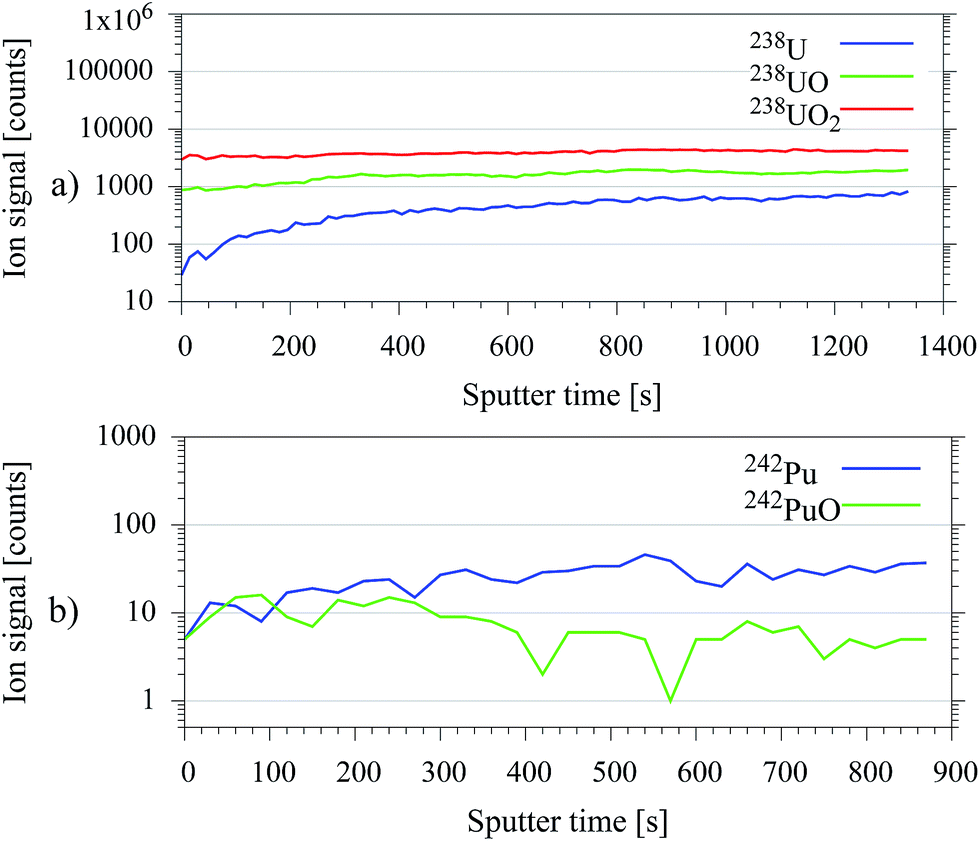 | ||
| Fig. 4 Ion signal development during continuous argon cluster sputtering of (a) resonantly ionized U in comparison to non-resonantly ionized UO, UO2 (b) for resonantly ionized Pu and non-resonantly ionized PuO. | ||
However, this result relies on a lower degree of oxidation in the particle core. Therefore, it is not universal and could fail e.g. on particles, which were exposed to environmental influences for longer periods.
3.3 Analytical measurements
Resonant laser–SNMS offers the unique advantage of direct, in situ and spatially resolved detection, imaging and analysis of hot particles in chemically unprepared environmental samples with suppression of all isobaric interferences. These in general are ubiquitous, stemming either from organic constituents or, in particular regarding radionuclide determination from the nuclear fuel cycle, are caused by the highly predominant 238U and 238UH for measurements of 238Pu and 239Pu. Therefore, the first analytical applications of the IRS laser–SNMS system focused on particle detection and analysis involving uranium suppression.| Mass (isotopes) | ||||||
|---|---|---|---|---|---|---|
| 235 | 238 | 239 | 240 | 241 | 242 | |
| Uranium | 0.23% | 89.98% | ||||
| Plutonium | 0.24% | 5.39% | 2.54% | 0.70% | 0.74% | |
| Americium | 0.19% | |||||
For demonstration of the capability and strength of uranium suppression in laser–SNMS the MOX sample was measured initially with the standard SIMS method to provide a rough overview and quantification of the overall content. The outstanding selectivity of resonant laser–SNMS was demonstrated by application of laser–SNMS on the same sample immediately afterwards, once applying a laser ionization scheme for uranium and alternatively one for plutonium. The mass spectra of these measurements are presented in Fig. 5.
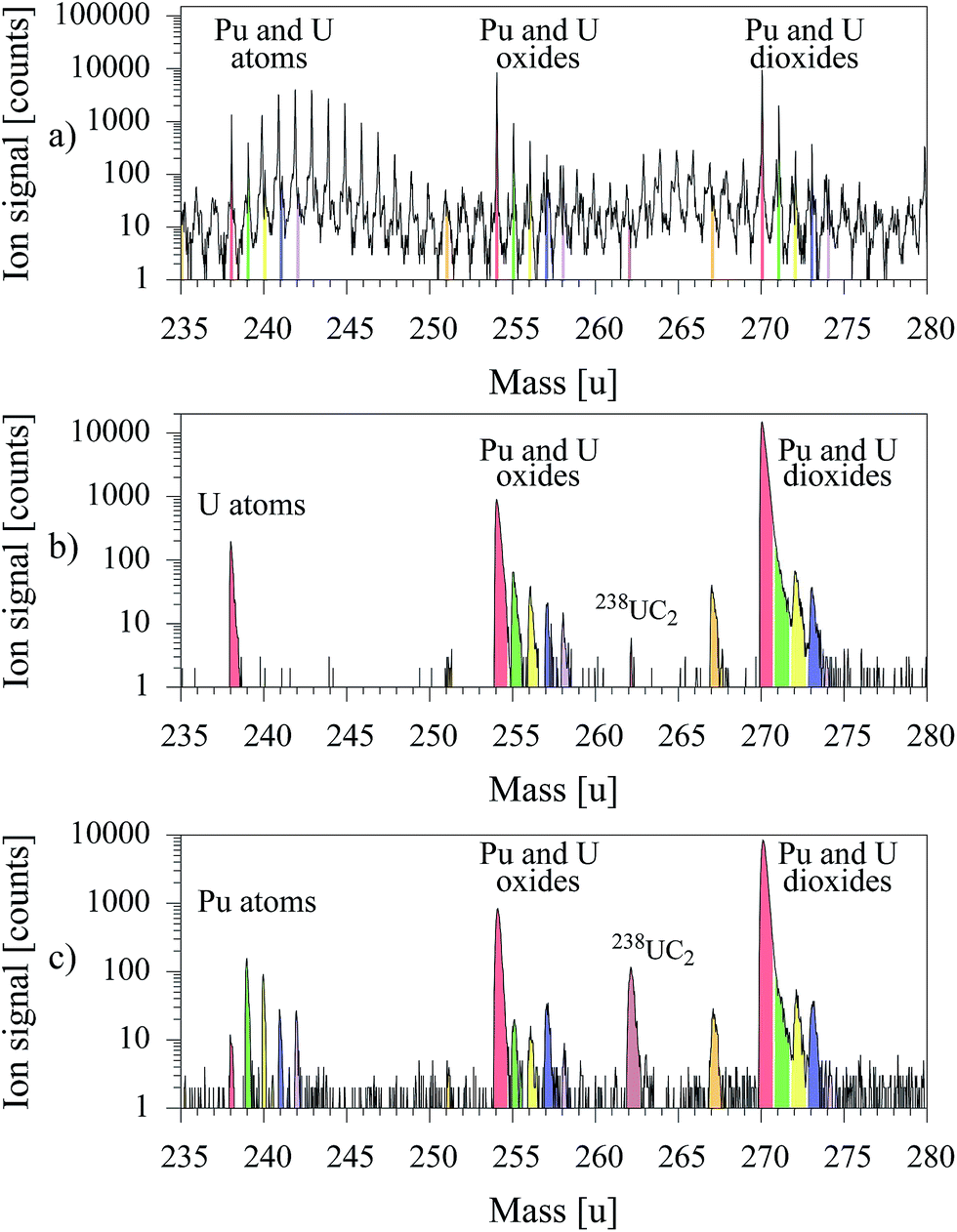 | ||
| Fig. 5 Mass spectra of a MOX sample taken by: (a) conventional SIMS, (b) laser–SNMS with resonant ionization of U, (c) laser–SNMS with resonant ionization of Pu. | ||
In the conventional SIMS spectrum depicted in Fig. 5a, signals are observed on each individual mass value. The peaks of 238U, 238UO and 238UO2 can be identified, but are covered or at least strongly superimposed by contributions from other ionized atomic or molecular species with similar or even higher intensity. Even the leading mass peaks of plutonium as atom, oxide or dioxide, expected at mass 239, 255 or 271 can not be positively identified. All this unspecific background is almost entirely suppressed in the mass spectra of laser–SNMS for both settings of laser wavelengths, addressing either uranium (Fig. 5b) or plutonium (Fig. 5c). Both spectra show clean and undisturbed atomic ion signals of the resonantly ionized element and rather identical, combined ion signals of non-resonantly ionized oxides and dioxides of both elements. A rather pronounced peak at mass number 262, identified as uranium carbide, is unexpectedly observed for lasers tuned resonantly to the plutonium transitions in comparison to the small peak for lasers tuned to uranium while in both spectra any other contributions are completely suppressed.
For uranium the ratio of resonant ionization of the atom to non-resonant ionization of oxide and dioxide in this specific sample amounts to about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:100, while for Pu the resonant atomic signal is strongest by about a factor of 2 in comparison to the non-resonant signal of the molecular species. This discrepancy in the behavior of the two actinide elements once again confirms the extremely high reactivity of uranium, which e.g. leads to fast and strong oxidation processes and prevents any treatment or analysis of U atoms with reasonable efficiency as e.g. attempted by laser isotope enrichment. If the laser wavelengths are set to the plutonium ionization scheme, the plutonium isotope ratio can be analyzed avoiding any disturbing interference from 238U or 241Am. Isotope ratios, as extracted from the integrated individual counts of the peaks perfectly reproduce the specified values for all isotopes as shown in Table 5.
:6:100, while for Pu the resonant atomic signal is strongest by about a factor of 2 in comparison to the non-resonant signal of the molecular species. This discrepancy in the behavior of the two actinide elements once again confirms the extremely high reactivity of uranium, which e.g. leads to fast and strong oxidation processes and prevents any treatment or analysis of U atoms with reasonable efficiency as e.g. attempted by laser isotope enrichment. If the laser wavelengths are set to the plutonium ionization scheme, the plutonium isotope ratio can be analyzed avoiding any disturbing interference from 238U or 241Am. Isotope ratios, as extracted from the integrated individual counts of the peaks perfectly reproduce the specified values for all isotopes as shown in Table 5.
| Isotopes | |||||
|---|---|---|---|---|---|
| 238Pu | 239Pu | 240Pu | 241Pu | 242Pu | |
| Specified | 2.52% | 56.14% | 26.42% | 7.25% | 7.67% |
| Measured | 2.8(8)% | 58(3)% | 25(2)% | 6.7(10)% | 7.1(10)% |
The minor trend in the measurement of too high results for lighter and too low values for higher masses, well within the error limit, is ascribed to the influence of the isotope shift. In general, the laser wavelengths were adjusted to the maximum of the resonant transitions of 240Pu, the bandwidth of the Ti:sapphire lasers of 5–9 GHz, as given above, does not cover the full range of isotope shifts for all measured plutonium isotopes in the individual excitation steps. Addressing higher precision in isotope ratio determination, e.g. for determination of sample origin, this effect can be accounted for in future applications by successive laser adjustment to the precise resonant transitions of each isotope. Alternatively a quantification of this effect on a calibrated standard sample can be used. The isotope shift in the optical transitions of uranium somewhat exceeds that of plutonium with values of up to 2 GHz per mass unit. With the lasers optimized to 238U correspondingly a resonant laser ionization signal for atomic 235U on mass 235 was neither expected nor observed. However, a rough estimate of this isotope ratios for uranium can be approximated from the non-resonantly ionized oxide and dioxide species, which were measured simultaneously to the resonant signal of plutonium. The resulting isotope ratios, shown in Table 6, rather well match the specified isotope content.
| Isotopes | ||
|---|---|---|
| 235U | 238U | |
| Specified | 0.25% | 99.75% |
| Approximated via oxides | 0.22(7)% | 99.8(17)% |
| Approximated via dioxides | 0.27(2)% | 99.7(5)% |
After identification of suspected regions, these alpha particle emitting areas were transferred onto the sample holder of the TOF-SIMS 5 apparatus on the indium foil and were analyzed with resonant laser–SNMS tuned to uranium. A corresponding mass spectrum of a selected region including a single uranium particle is giving in Fig. 6. Ion signals for atomic uranium as well as oxides and dioxides were observed for both isotopes 235 and 238 together with a peak at mass number 248 ascribed to ThO and a weak unidentified signal on mass number 256. The signal ratio of the resonantly ionized uranium atoms to non-resonantly ionized uranium oxide and dioxide is different to that for synthetic standards due to the higher degree of oxidation. Besides the non-resonant ion signals of UO and UO2 an additional significant non-resonant signal of 232ThO was measured on mass number 248.
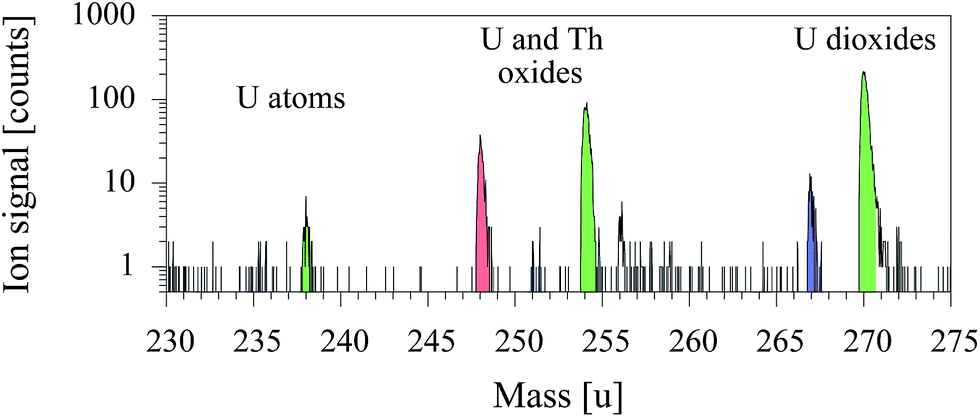 | ||
| Fig. 6 Laser–SNMS spectrum of a selected region (21 × 21 μm) of a soil sample from Kabul. | ||
The spatially resolved visualization of the ion signal is depicted in the four two dimensional images of Fig. 7.
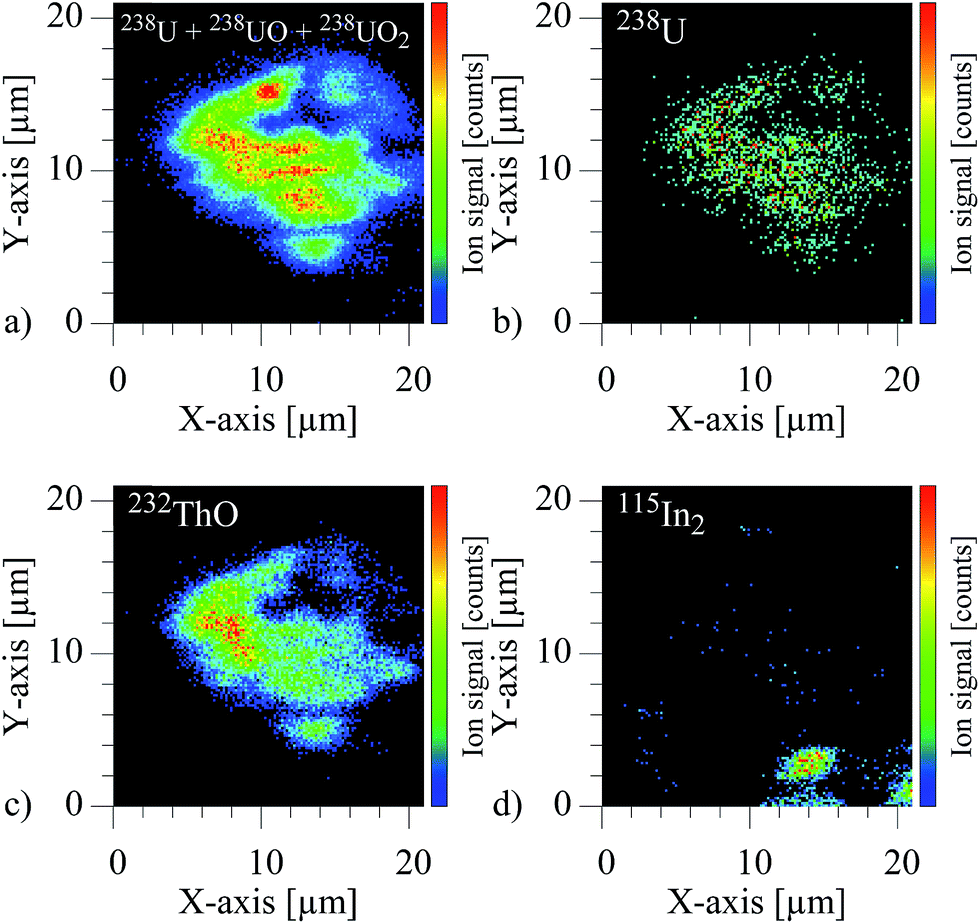 | ||
| Fig. 7 (a) Combined ion signal image of 238U, 238UO and 238UO2 with inhomogeneous distributed maxima. (b) Resonant ion signal image of 238U. (c) Non-resonant ion signal image of 232ThO with differently located maximum compared to the uranium oxide and dioxide. (d) Non-resonant ion signal image of 115In2, which was used for sample fixation. | ||
These show the individual ion images of the combined uranium species, pure atomic uranium, ThO and the carrier foil In. For the actinide elements the contours clearly identifies a particle of approximately 16 × 14 μm containing both, uranium and thorium, while only an insignificant signal of In is observed at one edge of the imaged area. The intensity patterns in addition show variation of uranium to oxide or uranium to thorium distributions, which can be related to inhomogeneities and surface structures of the particle. A clear, unquestionable indication for an uranium content in the particle was found by using the element selectivity of the resonant ion signal on mass number 238, despite the small fraction of atomic uranium in the laser–SNMS signals. Additional information like the shape of the particle and the specific inhomogeneous distribution of uranium and thorium inside the particles was correspondingly extracted from the much stronger non-resonant ion signals of 238UO2 and 232ThO.
4 Conclusions
We have shown, that the new laser–SNMS system at the Institute for Radioecology and Radiation Protection in Hannover meets all requirements for spatially resolved ultra-trace analysis of radionuclides. Low saturation laser powers of several mW confirmed an ideal overlap between the three laser foci and the sputtered neutral particle cloud. In the present status of development the apparatus delivers a decision threshold (DT) of 5 × 108 to 5 × 109 atoms. On top of that, further potential for optimization of the technique was indicated by the two orders of magnitude lower estimated DT, which were derived from efficiency measurements on uranium. Furthermore, the overall efficiency might still be enhanced by a possible reduction of the oxidized fraction through suitable reduction agents or carriers or by modification of the ion extraction, which are under investigation presently. The first analytical measurements of the laser–SNMS system presented here demonstrated that isobaric contaminations, whether from organic background or other interfering elemental or molecular species, can be efficiently suppressed, which enables the measurement of reliable isotope ratios and element selective visualization for radioactively contaminant particles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been financially supported by the German Federal Ministry of Education and Research (BMBF) in the joint project SIRIUS. We also thank M. Tanha for taking and providing of the soil samples from Kabul. Additionally, M. Franzmann thanks the Siebold-Sasse foundation for financial support.References
- Y. Ranebo, P. Hedberg, M. Whitehouse, K. Ingeneri and S. Littmann, J. Anal. At. Spectrom., 2009, 24, 277–287 RSC.
- F. Esaka, K. Esaka, C. Lee, M. Magara, S. Sakurai, S. Usuda and K. Watanabe, Talanta, 2007, 71, 1011–1015 CrossRef CAS PubMed.
- N. Trautmann, G. Passler and K. Wendt, Anal. Bioanal. Chem., 2004, 378, 348–355 CrossRef CAS PubMed.
- K. Wendt, C. Geppert, C. Mattolat, G. Passler, S. Raeder, F. Schwellnus, K. Wies and N. Trautmann, Anal. Bioanal. Chem., 2012, 404, 2173–2176 CrossRef CAS PubMed.
- N. Erdmann, J.-V. Kratz, N. Trautmann and G. Passler, Anal. Bioanal. Chem., 2009, 395, 1911–1918 CrossRef CAS PubMed.
- T. Kron, Y. Liu, S. Richter, F. Schneider and K. Wendt, J. Phys. B: At., Mol. Opt. Phys., 2016, 49, 185003 CrossRef.
- S. Raeder, A. Hakimi, N. Stöbener, N. Trautmann and K. Wendt, Anal. Bioanal. Chem., 2012, 404, 2163–2172 CrossRef CAS PubMed.
- B. A. Bushaw, S. Raeder, S. L. Ziegler and K. Wendt, Spectrochim. Acta, Part B, 2007, 62, 485–491 CrossRef.
- N. Erdmann, M. Betti, F. Kollmer, A. Benninghoven, C. Grüning, V. Philipsen, P. Lievens, R. E. Silverans and E. Vandeweert, Anal. Chem., 2003, 75, 3175–3181 CrossRef CAS PubMed.
- D. Willingham, M. R. Savina, K. B. Knight, M. J. Pellin and I. D. Hutcheon, J. Radioanal. Nucl. Chem., 2013, 296, 407–412 CrossRef CAS.
- R. Middendorp, M. Dürr, A. Knott, F. Pointurier, D. Ferreira Sanchez, V. Samson and D. Grolimund, Anal. Chem., 2017, 89, 4721–4728 CrossRef CAS PubMed.
- K. Knight, M. Savina, B. Isselhardt, I. Hutcheon, S. Prussin and M. Pellin, Proc. Radiochim. Acta, 2011, 1, 37 Search PubMed.
- E. Niehuis, R. Möllers, D. Rading, H.-G. Cramer and R. Kersting, Surf. Interface Anal., 2013, 45, 158–162 CrossRef CAS.
- N. Erdmann, J.-V. Kratz, N. Trautmann, G. Passler, M. Betti, P. Lievens, R. E. Silverans and E. Vandeweert, Spec. Publ. - R. Soc. Chem., 2006, 305, 122–124 CAS.
- M. R. Tanha, PhD. thesis, Gottfried Wilhelm Leibniz University Hannover, 2017.
- M. R. Tanha, S. Bister, E. Mühr-Ebert, F. Tawussi, A. Hölzer, B. Riebe, S. Schneider, L. Hamann, A. Ikeda-Ohno, M. Schulze, F. R. Khalid, M. A. Storai and C. Walther, 2018, submitted.
| This journal is © The Royal Society of Chemistry 2018 |