
SI-traceable quantification of sulphur in copper metal and its alloys by ICP-IDMS
Pranee
Phukphatthanachai
 abc,
Ulrich
Panne
ac,
Norbert
Jakubowski
a and
Jochen
Vogl
*a
abc,
Ulrich
Panne
ac,
Norbert
Jakubowski
a and
Jochen
Vogl
*a
aBundesanstalt für Materialforschung und -prüfung (BAM), Unter den Eichen 87, 12205 Berlin, Germany. E-mail: jochen.vogl@bam.de
bNational Institute of Metrology (Thailand), 3/4-5 Moo 3, Klong 5, Pathum Thani, Thailand 12120. E-mail: pranee@nimt.or.th
cHumboldt University, Department of Chemistry, Brook-Taylor 2, 12489 Berlin, Germany
First published on 15th November 2017
Abstract
Previously applied methods for the quantification of sulphur in copper and other pure metals revealed a lack of SI-traceability and additionally showed inconsistent results, when different methods were compared. Therefore, a reference procedure is required which allows SI-traceable values accompanied by a sound uncertainty budget. In this study a procedure was developed for the quantification of total sulphur in copper at low concentration levels using inductively coupled plasma-isotope dilution mass spectrometry (ICP-IDMS). The major part of the copper matrix was separated by adding ammonia which forms a complex with the copper while releasing the sulphur followed by chromatographic separation using a weak cation resin. After that the sulphur fraction was further purified by chromatographic means using first an anion exchange method and second a chelating resin. The developed procedure shows high performance, especially concerning high efficiency in matrix removal (>99.999%) while keeping the recovery of sulphur above 80%. Procedure blanks are in the order of 3–53 ng resulting in LOD and LOQ values of 0.2 μg g−1 and 0.54 μg g−1, respectively. The procedure is sufficient to facilitate value assignment of the total sulphur mass fraction in reference materials. Additionally, relative measurement uncertainties were calculated to be below 1% and the measurement results were traceable to the SI. The procedure reported in this study is a new reference procedure for sulphur measurement in copper, being fit for two major purposes, certification of reference materials and assignment of reference values for inter-laboratory comparison.
1. Introduction
Copper is an essential metal for different organisms and it has been one of the most useful metals known to man. It is the third most important metal in industrial applications, and it is widely used in unalloyed and alloyed conditions. The copper industry in Europe has an estimated turnover of about €45 billion, and copper prices have increased by a factor of 4 in the last decade. Mainly copper is used in electricity and energy, building construction, engineering and transportation.1 The world's copper supply comes from two main sources: mining/refining and recycling. Copper ores are normally associated with sulphur, and copper can be extracted from chalcocite (Cu2S, copper sulphide), chalcopyrite (CuFeS2, copper iron sulphide) and cuprite (Cu2O, copper oxide). Although there are more than 400 copper alloys in use, the applications mainly focus on copper–zinc alloys (brass), copper–tin alloys (bronze), copper–nickel alloys, copper–beryllium alloys and pure copper (unalloyed), which is mainly used in electrical engineering.Keeping up the quality of copper and copper alloys in technology requires specific reference materials. In general, copper reference materials can be divided into two major classes, materials which are characterized by their copper mass fraction or purity (main element, kg kg−1) and materials which are characterized by impurities (other elements). Sulphur (S) is one of the major impurities in copper which directly influences its chemical, physical, and mechanical properties such as the colour, hardness, tensile strength, and heat treatment of copper.2,3 Therefore, the determination of the sulphur content in copper is necessary for many technological applications.
Available copper reference materials (RMs) providing data for the sulphur mass fraction are compiled in Table 1. Roughly half of the listed materials are certified for their sulphur mass fractions; the other half of the reference materials only provide additional/information values for the sulphur mass fraction. A large part of the materials being certified for sulphur show relative measurement uncertainties of 7–30%, whereas all other materials only provide relative standard deviations of the inter-laboratory comparison or no uncertainty data at all. The reviewed information emphasizes the lack of reference procedures, which can provide sufficiently small measurement uncertainties and which are suitable as reference procedures, especially for the certification of reference materials.
| Material no. | Description | Copper mass fraction in, kg kg−1 | Sulphur mass fraction | CV/AIa | U rel inb, % | Comment | Producer, country | Year of issue | Available |
|---|---|---|---|---|---|---|---|---|---|
| a CV = certified value, AI = additional information for sulphur. b Relative measurement uncertainty. | |||||||||
| BAM-Y001 | Pure copper | 99.9970 ± 0.001 | 5.4 ± 1.6 μg g−1 | CV | 29.6 | Certified by spectrophotometry | BAM, DE | 2004 | ✓ |
| BAM-M376a | Pure copper | — | 133 ± 19 μg g−1 | AI | 14.3 | The uncertainty larger than expected or in case of possible inhomogeneities | BAM, DE | 2016 | ✓ |
| BAM-M385 | Pure copper | — | 31.2 ± 1.5 μg g−1 | CV | 4.8 | Certified by inter-laboratory comparison | BAM, DE | 2003 | ✗ |
| BAM-227 | Copper chips | 85.57 ± 0.03 | 0.122% ± 0.010% (2SD) | — | 8.2 | Certified by photometry | BAM, DE | 1979 | ✗ |
| BAM-228 | Copper chips | 85.34 ± 0.03 | 0.036% ± 0.004% (2SD) | — | 11.2 | Certified by coulometry and photometry | BAM, DE | 1979 | ✓ |
| NIST SRM399 | Unalloyed copper – Cu VI | 99.79 ± 0.01 | (10) μg g−1 | AI | — | — | NIST, US | 1993 | ✓ |
| NIST SRM400 | Unalloyed copper – Cu VII | 99.70 ± 0.02 | (9) μg g−1 | AI | — | — | NIST, US | 1986 | ✓ |
| NIST SRM457 | Unalloyed copper IV (solid) | 99.97 ± 0.18 | (4 ± 1) μg g−1 | AI | 25.0 | Certified by inter-laboratory comparison including NIST | NIST, US | 2013 | ✓ |
| NIST SRM494 | Unalloyed copper – Cu I | 99.91 ± 0.01 | (15 ± 3) μg g−1 | CV | 20.0 | Certified by inter-laboratory comparison | NIST, US | 1986 | ✓ |
| NIST SRM495 | Unalloyed copper – Cu II | 99.94 ± 0.01 | (13 ± 2) μg g−1 | CV | 15.4 | Certified by inter-laboratory comparison | NIST, US | 1987 | ✓ |
| NIST SRM498 | Unalloyed copper V (solid) | 99.98 ± 0.01 | (11) μg g−1 | AI | — | — | NIST, US | 1993 | ✓ |
| NIST SRM885 | Refined copper | — | (18 ± 3) μg g−1 | CV | 16.7 | — | NIST, US | 1991 | ✓ |
| NIST SRM1034 | Unalloyed copper | 99.96 not certified | (2.8 ± 0.2) μg g−1 | CV | 7.1 | Certified by ID-TIMS technique | NIST, US | 1982 | ✓ |
| BCR-017-B | Pure copper | — | (10.4 ± 0.6) μg g−1 | CV | 5.8 | Certified by inter-laboratory comparison | IRMM, BE | 1989 | ✓ |
| IARM 278A | Tellurium copper | 99.5 | (20 ± 3) μg g−1 | CV | 15 | Certified by inter-laboratory comparison | ARMI, UK | 2007 | ✓ |
| 39X 17866 | Residuals in copper (CHILL-CAST) | — | (520 ± 30) μg g−1 | AI | 5.8 | — | MBH Analytical Ltd, UK | 2014 | ✓ |
| 39X 17871 | Residuals in copper (CHILL-CAST) | — | (52 ± 4) μg g−1 | AI | 7.4 | — | MBH Analytical Ltd, UK | 2013 | ✓ |
Sayi et al.4 reviewed the different techniques available to determine the total sulphur mass fraction in inorganic compounds such as gravimetry, titrimetry, spectrophotometry, combustion, and mass spectrometry. The techniques were presented depending on the measurement range for the sulphur mass fraction and the corresponding precision in various sample types. The obtained precision of those techniques is in the range of 0.2–10% relative standard deviation. One of the most powerful techniques for the determination of sulphur in metal samples is glow discharge mass spectrometry (GDMS). Major advantages of this technique are a short analysis time and a simple sample preparation process, both making it fit for routine analysis. The disadvantages of GDMS, however, are relatively high measurement uncertainties of 20% and more and the requirement of matrix matched standards for obtaining reliable results. Classical methods such as carrier gas hot extraction/combustion analysis are used for sulphur analysis by converting the sulphur in the sample into sulphur dioxide in a high purity oxygen atmosphere, followed by the measurement of the sulphur dioxide by using thermal conductivity or infrared absorption spectrometry. This technique is quite fast but the precision is in the range of 2–10% (expressed in RSD) which is not fit for our purposes.4
In recent years, inductively coupled plasma-mass spectrometry (ICP-MS) has been investigated for the quantification of sulphur in low level materials. Martinez-Sierra et al. clearly reviewed the technical problems of sulphur analysis by ICP-MS such as required mass resolution and potential interference on the basis of various publications. Most of the applications, however, are focused on organic samples such as fuel, protein, and pharmaceuticals.6
A major challenge for the quantification of sulphur in copper (alloyed/unalloyed) by ICP-MS is the copper matrix itself, causing matrix effects and making extensive cleaning of cones and extraction lenses necessary after measurements. Matschat et al. investigated the analysis of high-purity metals (including copper) by high resolution ICP-MS.7 They found that the copper matrix shows strong matrix effects on the sensitivity, resulting from Cu deposition on the cones. The relative decrease in the sensitivity amounts to about 70% when aspirating a 5000 mg L−1 copper solution.7 Moreover, the compared analytical methods for analysing impurities in pure copper, but the number of reported datasets were small while the standard deviations were large.8 Most commonly matrix effects in ICP-MS are being reduced by sufficient dilution, often with dilution factors of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and higher. In the case of sulphur analysis in copper such high dilution factors are ineffective, because as a consequence sulphur is diluted to the medium to low ng g−1 range making measurements with sufficiently low measurement uncertainties (<10% relative) impossible.
000 and higher. In the case of sulphur analysis in copper such high dilution factors are ineffective, because as a consequence sulphur is diluted to the medium to low ng g−1 range making measurements with sufficiently low measurement uncertainties (<10% relative) impossible.
The application of isotope dilution mass spectrometry (IDMS) can overcome some of these limitations, as it facilitates the use of matrix separation techniques. Since sample loss will not affect the accuracy of the results once equilibration between the sample and the spike is established. Additionally, IDMS enables the smallest measurement uncertainties while providing traceability to the international system of units (SI).9,10 In combination with thermal ionization mass spectrometry (TIMS) IDMS has been applied to determine sulphur in metal samples11,12 and fossil fuel samples.13 The sample preparation of ID-TIMS consists of two major steps; the sample digestion and the formation of As2S3. During digestion sulphur is oxidized to sulphate. This sulphate then has to be reduced to H2S, transferred and finally converted into As2S3, which is laborious and time-consuming. Moreover, the relatively high blank values and their uncertainties strongly contribute to the combined uncertainties of the sulphur mass fraction.13
In this study, the IDMS concept is combined with ICP-MS analysis and a new approach for sulphur–copper separation is developed. The sample preparation including sample digestion, matrix removal by complexation and ion exchange chromatography, as well as critical points will be presented in detail. The aim of this research is to develop a reliable measurement procedure which enables low measurement uncertainties and SI traceability and which can be applied to the certification of reference materials, the assignment of reference values and the calibration of other analytical procedures. The target for the relative measurement uncertainty in this study is set to below 2%. In order to demonstrate the SI traceability, the unbroken chain of calibrations will be established.
2. Experimental
2.1 Materials and reagents
The determination of sulphur especially at the low μg g−1 level requires clean working conditions and specialized sample handling equipment to keep the blank contribution and contamination risks small. All samples were prepared in a class 10 clean room (Fed STD 209E). All plastic laboratory equipment was soaked in 10% HNO3 for at least 60 hours whereas PFA beakers were cleaned with a device using nitric acid vapours for cleaning and subsequent soaking in Milli-Q water for at least 12 h. Thereafter the laboratory equipment was dried by air flow in the cleanroom cabinets.To keep the sulphur blank as low as possible all reagents were used in the highest available purity. Nitric acid, which was used for sample digestion and sulphur–matrix separation, was purified by a two-stage sub-boiling procedure. Ammonia solution (Suprapur®) and hydrogen peroxide (Ultrapur®) were obtained from Merck KgaA (Darmstadt, Germany).
As the primary assay, used as the back-spike in IDMS, the sulphur standard solution NIST SRM3154 was employed. The enriched isotope 34S was obtained from Trace Sciences International Inc. (Delaware USA) in the form of solid sulphur, with a nominal enrichment of 99.8%. The 34S was dissolved in HNO3 to prepare the 34S enriched spike solution, which then was characterized using back-spike solutions. Sodium sulphide (Na2S·9H2O; purity >98%, ACS reagent; ACROS Organics; New Jersey; USA), sodium sulphite (NaSO3 anhydrous; purity >98%, ACS; Bernd Kraft der Standard; Duisburg; Germany) and sulphuric acid (H2SO4; SRM3154; NIST; USA) were used to investigate the effect of different sulphur species on the separation. Three different ion exchange resins were used in this study. The details of these resins are shown in Table 2. All resins were activated and cleaned before use.
| a Depends on the amount of copper (2 mL for copper ≤20 mg). | |||
|---|---|---|---|
| Property | AmberliteCG50 (ref. 14) | AG1X8 (ref. 15) | Chelex-100 (ref. 16) |
| Company | Sigma-Aldrich | Biorad labs | Biorad labs |
| Resin type | Weak cation exchange | Strong anion exchange | Weak cation exchange |
| Functional group | Carboxylic acid | Quaternary ammonium | Carboxylic acid |
| Ionic form | H+ | Cl− | Na+ |
| Size (mesh) | 100–200 | 200–400 | 200–400 |
| Total exchange capacity (mmol mL−1) | 3.5 | 1.2 | 0.4 |
| Selective to copper | High | None | Very high |
| Function in the separation procedure | Removes copper | Retains sulphur on the resin | Removes copper |
| Amount of resin (mL) | 2a | 1 | 1 |
The copper reference materials BAM-M385, BAM-M376a, BAM-228 and BAM-227, all produced by BAM, were selected to serve as well-defined samples for the development of the sulphur–matrix separation procedure (for details see Table 1).
2.2 Equipment and instrumentation
The samples were weighed by using an analytical balance (Mettler Toledo AX205, Giessen, Germany). When plastic labware was used for weighing, the labware was flushed with a nitrogen ion stream to remove electrostatic charges. Digestion was accomplished using a high pressure asher, HPA (Anton Paar GmbH Graz, Austria) equipped with a heating block holding 5 quartz digestion vessels of 90 mL volume. The digestion program lasted 4 h at a maximum temperature of 320 °C and a maximum pressure of about 130 bar. The temperature was increased from room temperature to 150 °C in 30 min and then up to 320 °C in 60 min. This temperature was kept for 150 min followed by a cooling step down to room temperature with a duration of 60 min.All mass spectrometric measurements were performed using a sector field ICP-MS instrument element 2 (Thermo-Fisher Scientific, Germany), unless stated otherwise. Table 3 shows the operating conditions of the instrument.
| Parameter | Setting |
|---|---|
| Instrument type | Element 2 |
| Autosampler | Cetac ASX 100 |
| Aspiration mode | Self-aspirating |
| Nebulizer | MicroMist 100 μL |
| Spray chamber | Cyclonic spray chamber |
| Interface | Jet interface |
| Cones | Ni sampler and skimmer X-cone |
| Cool gas flow rate | 16 L min−1 |
| Auxiliary gas flow rate | 0.8–1.0 L min−1 |
| Sample gas flow rate | 0.9–1.25 L min−1 |
| RF power | 1200 W |
| Guard electrode | On |
| Mass resolution mode | Medium |
| Acquisition mode | Pulse and analog mode |
| Runs/passes | 10/40 |
| Sensitivity in cps/(μg g−1) | 1 × 107 for 32S |
| Drift correction | Yes |
2.3. Sample preparation
The complete sulphur–copper separation process is visualized in Fig. 1. The newly developed procedure for sulphur–copper separation consists of three subsequent separation steps and is described in the following in detail. Approximately 1.0–1.5 g of the digested sample solution (light blue colour, b) was weighed into Savillex PFA beakers and was evaporated to dryness at 110 °C. The residue then was dissolved in 8 mL of Milli-Q water followed by the addition of conc. ammonia in excess at room temperature (at least >10 times the amount of ammonia as calculated on the basis of the chemical reaction per mole of copper, c). The colour of the sample solution turned a characteristic deep blue due to the formation of the copper–ammonia complex. To this solution CG50 resin (white) was added in excess (1 mL: 10 mg Cu, d), and the resulting suspension was mixed well by using an automatic shaker for 3 h. The resin turned light blue whereas the solution was clear and transparent. In the meantime, 1 mL of anion exchange resin (AG-1X8) was packed in a column (2 mL PP + PE column, I.D. 0.7 cm, e) and rinsed with Milli-Q water. The column was closed at the lower end with parafilm and was then loaded with the clear solution from above. After about 20 min the parafilm was removed to let the remaining matrix pass through. The remaining CG50 resin from above was rinsed with 4 mL Milli-Q water which was then loaded onto the AG 1X8 column (this was repeated 4 times). Thereafter, the sulphur fraction was eluted from the AG1X8 resin by addition of 12 mL of 0.25 M HNO3 onto the column. The eluted solution was evaporated to dryness at 110 °C overnight. Typically, the residue was blue or green (f) which means that remains from the copper matrix are still present. Therefore, the Chelex resin was used to remove the rest of the copper. Approximately 1 mL of Chelex resin was packed into the column (g), which was closed at the lower end with parafilm. In parallel, the residue was dissolved in 2 mL of Milli-Q water and was loaded onto the column. After 20 minutes, the parafilm was removed and the sulphur was eluted with 10 mL of Milli-Q water. Then the sulphur containing solution was evaporated to dryness at 110 °C. Finally, the residue was dissolved in 2% HNO3 (h) such that a final sulphur mass fraction of approximately 2 μg g−1 was obtained, based on the known sulphur mass fraction in the solid sample and the recovery. This was verified within ±20% for each sample by comparing to a 2 μg g−1 sulphur standard.
 | ||
| Fig. 1 Sulphur–copper separation process; (a) Cu sample, (b) digested sample solution, (c) copper–ammonia complex solution, (d) sample solution after adding weak action (CG50) resin, (e) separation with anion exchange (AG1X8) resin, (f) residue after separation by anion exchange, (g) separation with Chelex resin, (h) residue and final solution, and (i) ICP-MS measurement. | ||
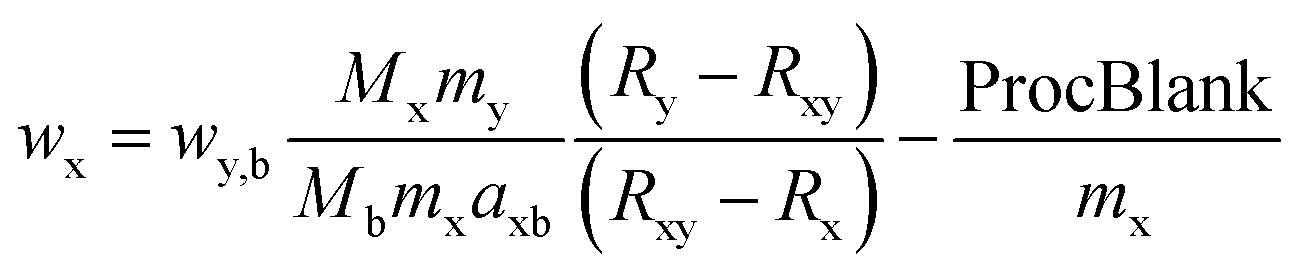 | (1) |
For each IDMS analysis, the ICP-MS measurement sequence started with the sulphur standard (also named back-spike), then the unspiked sample, sample blend, procedure blank and at the end the spike, with regularly measuring a standard or back-spike in between.
3. Results
Whenever uncertainties are provided they are expanded measurement uncertainties with k = 2 calculated from combined uncertainties via U = k × uc, unless stated otherwise.Sulphur measurement
The major challenges of sulphur measurement by ICP-MS are high background, high first ionization potential (10.36 eV),18 light mass causing high mass discrimination, and spectral interference at m/z 32 and 34. The main interferents are 16O16O+, 31P1H+, 14N18O+, 15N16O1H+, and 64Zn2+ on 32S+ and 16O18O+, 32S1H1H+, 16O16O1H1H+, and 68Zn2+ on 34S+. To separate all interferents the medium resolution mode (m/Δm > 4300) was required. The intensities of 64Zn+ and 68Zn+ were monitored (3 × 105 and 2 × 105 cps for 64Zn and 68Zn, respectively, <10 ng g−1) to observe any upcoming problems with high intensities of doubly charged Zn isotopes. 63Cu+ and 65Cu+ were used to investigate the matrix removal efficiency. The corresponding Cu signal intensities were 0.04–2 × 107 cps for 63Cu and 0.02–1.5 × 107 cps for 65Cu (<150 ng g−1) in the final solution.The signal intensities observed for 32S in 2% HNO3 (instrumental background) were in the range of 0.2–1.3 × 106 cps; for procedure blanks they were 0.4–2.5 × 106 cps and for sulphur standards at 2 μg g−1 they were 2–3 × 107 cps. As a consequence, the standard signal for 2 μg g−1 was 20 times above those of the blank in all cases, except the standard addition IDMS for NIST SRM494, where it was only 10 times above the blank level. The relative standard deviation of the sulphur isotope ratio over all back spike measurements within one sequence was around 1.3% relative. This number expressed not only the reproducibility of the ratio measurement, but also indicated instrumental drift and wash-out effects during the whole IDMS sequence.
S-Species vs. digestion/oxidation/equilibration conditions
Most copper is produced by mining and/or extraction from copper sulphide and thus sulphur impurities might also occur in the form of sulphides. Unfortunately, the anion exchange resin (AG1X8) is selective to sulphate and sulphite but less-selective to sulphide.15 Therefore, when quantifying total sulphur in copper, the different species of sulphur need to be oxidized to sulphate prior to the sulphur–matrix separation on the AG1X8 resin in order to avoid any measurement bias. Consequently, the recovery of the different sulphur species was checked for the applied separation procedure. Stock solutions of sulphate, sulphite and sulphide were gravimetrically prepared from sulphuric acid, sodium sulphite and sodium sulphide. Then portions of the species standards containing about 10 μg sulphur were oxidized separately in two different ways: (1) addition of 3 mol L−1 HNO3 and H2O2 (30%, w/w) and hotplate oxidation at 120 °C for 3 h; (2) addition of 3 mol L−1 HNO3 and H2O2 (30%, w/w) and digestion with HPA. After this oxidation, the samples were subjected to the AG1X8 separation procedure and subsequently spiked. The samples were measured by ICP-IDMS; the sulphur amount was calculated and subsequently the recovery for each sample was calculated referring to the gravimetrically determined input. The results are displayed in Table 4. For sulphate, of course quantitative recovery was obtained. In the case of sulphite, the hotplate oxidation is insufficient (79% recovery), whereas the HPA oxidation yields quantitative recovery (100%). In the case of sulphide, either oxidation experiment is insufficient yielding recoveries below 50%. Therefore, another oxidation experiment was carried out applying the HPA oxidation as described above but with concentrated nitric acid (65%, w/w) followed by AG1X8 separation, evaporation to dryness and redissolution in 2% nitric acid. This time, the samples were measured with ICP-MS applying external calibration. The obtained recovery for sulphide was 94% with an estimated expanded uncertainty of 10%. Thus, the oxidation of sulphide to sulphate can be considered complete with a quantitative recovery for the AG1X8 separation procedure.When applying the HPA oxidation with concentrated HNO3 and H2O2 a complete conversion from sulphide and sulphite to sulphate could be achieved. The recovery of all investigated sulphur species is quantitative within measurement uncertainties.
Sulphur–matrix separation
The performance of the separation procedure was evaluated mainly by checking the sulphur recovery and the efficiency of matrix removal.As a starting point the separation procedure used by Das et al.17 was applied. This separation procedure employs a strong anion exchange resin (AG1X8), which retains the sulphur on the column while the matrix elutes without retardation. The recovery of this procedure was checked with a sulphur standard solution (sulphate form) and was found to be 100 ± 2%. Then the procedure was applied to synthetic sample solutions (sulphur 8 μg g−1 and copper 24000 μg g−1). The recovery of sulphur dropped to 10–30%. An explanation for the low recovery could be the formation of copper(II) sulphate (CuSO4·(H2O)x) complexes by the reaction of sulphate with excess copper. Therefore, the complexation of copper by ammonia being a highly selective ligand for Cu(II) was explored. The formation of the tetraamine-copper(II) complex releases trapped sulphate and leads to increased recovery rates. The chemical reaction of ammonia and copper is well known and is shown in eqn (2) and (3). Upon ammonia addition, the sulphur recovery for the synthetic sample increased to 100 ± 3 (n = 4).
| CuSO4(aq) + 2NH3(aq) + 2H2O(l) → Cu(OH)2(s) + (NH4)2SO4(aq) | (2) |
| Cu(OH)2(s) + 4NH3(aq) → [Cu(NH3)4](OH)2(aq) | (3) |
This procedure was then applied to real world samples (solid copper metal). The recovery of sulphur dropped to 10–20% again. Possible reasons for this considerable decrease are the very high amount of the copper matrix compared to the sulphur mass fraction (approximately 3 times higher than in the synthetic sample) and the occurrence of different sulphur species in the sample. It was assumed that the removal of the major part of the copper prior to the AG1X8 separation should solve this problem. Unfortunately, most cation exchange resins being capable of separating copper are strong acidic cation resins containing sulphonated polystyrene as the exchange site. This however would lead to unacceptably high procedure blanks. When considering weakly acidic cation resins a suitable material could be identified: the resin CG50 does not contain sulphur groups and is capable of retaining copper.19,20 Together with its high capacity (3.5 mmol mL−1) this makes it highly suitable for the intended task. The new separation step was carried out by adding an excess amount of cation resin CG50 (1 mL: 20 mg Cu) into the sample solution. The deep blue solution turned into a clear and transparent solution, while the resin itself turned from white to blue. The clear solution was loaded onto the column which contains AG1X8 to further purify the sulphur fraction. After eluting the sulphur from the AG1X8 resin the solution was dried on a hot plate until dryness yielding a blue/green residue which still contains copper above 100 μg g−1. Therefore, another chelating ion exchange resin (chelex100) was employed to trap the remaining copper.21
The copper samples investigated in this study contain copper in the range of 0.85–0.99 kg kg−1 and zinc from <10 to 300 g kg−1. Approximately 0.10–0.25 g of these samples were used to perform the sulphur–copper separation. After applying the complete three stage separation procedure the mass fractions of both elements were significantly reduced to below 400 ng g−1 for copper and below 50 ng g−1 for zinc, respectively. Nearly the complete matrix (>99.999%) was removed which results in an extremely high matrix removal factor of above 105.
Measurement results of sulphur in copper by ICP-IDMS
The above described sulphur–matrix separation was applied and combined with the described ICP-IDMS procedure for the sulphur quantification in six different reference materials. The results are displayed in Table 5. Samples BAM-M385, BAM-M376a, BAM-228, BAM-227 and SRM494 were quantified by normal IDMS whereas SRM494 and SRM1034 were quantified by a combination of the standard addition technique and the IDMS technique because the content of sulphur was lower than the working range of the separation procedure. The obtained measurement results for samples BAM-M376a, BAM-228 and SRM494 agree well with the reference values (Table 1) whereas for BAM-M385 and BAM-227 the results differ from the certified values. Sample BAM-227 was certified in 1979 by photometrical methods only. At that time, metrological concepts such as measurement uncertainty and SI traceability were not clear or even not existing, and therefore the reference value was expressed in terms of precision only. In the case of sample BAM-M385 the IDMS result obtained within this work and the certified value are significantly different. The uncertainty of the certified value also represents only the dispersion of the inter-laboratory comparison.| List | BAM-M385 | BAM-M376a | BAM-228 | BAM-227 | NIST SRM494 | NIST SRM494a | NIST SRM1034a | |
|---|---|---|---|---|---|---|---|---|
| a Combined standard addition and IDMS technique. b For the type of reference refer to Table 1. | ||||||||
| Measurement value and MU (μg g−1), k = 2 | (37.72 ± 0.19) | (133.68 ± 0.86) | (385.50 ± 2.40) | (1376.60 ± 6.2) | (14.34 ± 0.09) | (14.97 ± 0.20) | (6.79 ± 0.36) | |
| Relative measurement uncertainty (%) | 0.5 | 0.6 | 0.6 | 0.5 | 0.7 | 1.34 | 5.30 | |
| Reference valueb | (31.2 ± 1.5) | (133 ± 19) | (360 ± 40) | (1220 ± 100) | (15 ± 3) | (15 ± 3) | (2.8 ± 0.2) | |
| Cu mass fraction in the final solution (ng g−1) | <100 | <150 | <100 | <150 | <150 | <260 | <370 | |
| Zn mass fraction in the final solution (ng g−1) | <10 | <50 | <10 | <10 | <10 | <10 | <10 | |
| Number of replicates | 8 | 8 | 8 | 8 | 4 | 6 | 6 | |
| Uncertainty budget | Type | % contribution | ||||||
| Observed ratio of the back spike | A | 48.1 | 58.5 | 65.5 | 33.7 | 41.8 | 11.1 | 2.8 |
| Mass fraction of the spike | B | 42.4 | 27.3 | 28.3 | 55.0 | 24.3 | 45.1 | 81.2 |
| Observed ratio of sample blends | A | 5.2 | 8.2 | 2.4 | 4.0 | 29.7 | 22.4 | 2.4 |
| Observed ratio of natural | A | 2.7 | 4.8 | 1.2 | 1.1 | 1.6 | 0.1 | <0.1 |
| Observed ratio of the spike | A | <0.1 | <0.1 | 0.8 | 0.4 | 1.8 | 2.0 | <0.1 |
| Weighing of samples | A | 0.4 | <0.1 | 1.2 | 4.7 | <0.1 | <0.1 | <0.1 |
| Weighing of spikes | A | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | 5.0 | 2.3 |
| Procedure blank | B | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| Mass fraction of the back spike (standard addition) | B | — | — | — | — | — | 13.2 | 10.7 |
| Weighing of the back spike (standard addition) | A | — | — | — | — | — | <0.01 | <0.01 |
| Others | — | 1.2 | 1.2 | 0.6 | 1.1 | 0.8 | 1.2 | 0.6 |
The quantification of sulphur in NIST SRM494 by conventional IDMS is hindered by the very high Cu/S ratio, which clearly affects the separation in a negative way: the recovery of sulphur dropped to about 30% for four replicates, while two further replicates even showed recoveries below 10%, revealing a non-linear recovery function with a threshold amount of sulphur which is lost during the whole separation procedure. The resulting samples show sulphur amounts too low for reliable ICP-MS measurements. To enable measurement without completely changing the separation procedure, an exact amount of sulphur was added prior to spiking, such that the sulphur mass fraction was shifted to the optimum working range of the separation procedure. So, exact amounts of sulphur were added prior to spiking to enhance the mass fraction of sulphur from 15 μg g−1 to 40 μg g−1, and then the IDMS analysis was performed as usual and finally the added sulphur amount was subtracted. The so obtained measurement result agreed well with the certified value within the uncertainties. The measurement uncertainties for the IDMS experiment with the addition of a known amount of sulphur were approximately twofold those obtained with the IDMS experiment without addition of a known amount of sulphur.
The same concept of the addition of a standard solution was applied to NIST SRM1034 by increasing the sulphur content from 3 μg g−1 to 40 μg g−1. However, in this case the measurement result was significantly different from the certified value. It has to be noted here that NIST SRM 1034 was certified by a single method only in 1982. This disagreement requires further investigation. The measurement results for NIST SRM494, however, proved that the standard addition technique combined with IDMS is a suitable tool to extend the working range of the separation procedure. This combination provides reliable results which are true within the stated uncertainties as shown for NIST SRM494.
The developed procedure for the quantification of low sulphur amounts in copper has been validated here via three different routes, which will be explained in the following chapter: first an inter-laboratory comparison at the highest metrological level (see the Method validation section), second a step-by-step validation by checking each single step of the procedure and third the setup of a complete uncertainty budget. Additionally, SI traceability is provided in the most direct way. These metrological properties make the IDMS results more likely to be the true values for the materials BAM-M385 and BAM-227 rather than the certified values.
Method validation
:1 (Na:S). The so obtained result shows an excellent agreement with the other results and demonstrates that the developed procedure enables sulphur measurements at the low μg g−1 level in complex matrices with sufficiently low measurement uncertainties (Fig. 2).22
 | ||
| Fig. 2 Results of the CCQM-K123 inter-laboratory comparison: the mass fraction of sulphur in biodiesel fuel displayed for the participating laboratories together with the reference value (all error bars represent expanded uncertainties, k = 2) based on the data from ref. 22. BAM's result was (7.39 ± 0.10) μg g−1 while the reference value was (7.38 ± 0.35) μg g−1.22 | ||
However, LOD and LOQ are more of a theoretical concept for reference measurements applying IDMS, because the applicability of IDMS procedures is more strongly defined by the working range, which itself is limited by the separation procedure and the measurement uncertainty aimed at. When applying the same separation procedure without any adaption and when aiming at relative measurement uncertainties of <2%, a working range from approximately 15 μg g−1 to 1500 μg g−1 could be established. For samples containing sulphur mass fractions of <15 μg g−1 the addition of an accurately weighed amount of standard is necessary, as explained above for the sample NIST SRM494.
Within this study ICP-IDMS was applied as a higher-order reference measurement procedure or in other terms a primary ratio method of measurement, where the measurement process is well understood and a measurement equation can be written down, permitting the calculation of the mass fraction of sulphur directly from the signal intensities. Consequently, measurement uncertainties were assessed based on the IDMS equation. The individual contributions to the measurement uncertainty listed for each sample type are displayed in Table 5. The main contribution to the uncertainty accounting to >30% derived from the observed isotope ratio in the back spike for conventional IDMS; this is caused by the relative standard deviation of the sulphur isotope ratio of the back spike which amounts to approximately 1.3% for a complete measurement sequence as mentioned before. The second largest contribution is made up by the mass fraction in the spike (>24%), followed by the observed isotope ratio in the sample blend, the unspiked sample and the spike (<10%). All other quantities do not contribute significantly (<2%). This also applies for the very low procedure blank (average value 14 ng). In the case of the modified IDMS, where back-spike is added to the sample before spiking, the main contribution to the measurement uncertainty is made up by the mass fraction of the spike and the back spike.
The relative expanded measurement uncertainties for conventional IDMS are below 1%. When applying the modified IDMS, where back-spike is added to the sample before spiking, the relative expanded measurement uncertainties are larger and amount to 1.34% and 5.30% as calculated for sample no. SRM494 and SRM1034, respectively.
The establishment of the metrological traceability of the sulphur content in copper samples is visualized in Fig. 3 for the example of sample no. BAM-M376a. The metrological traceability chain for double IDMS shows the course leading from the mol and/or kg down to the final sulphur mass fraction in the sample.
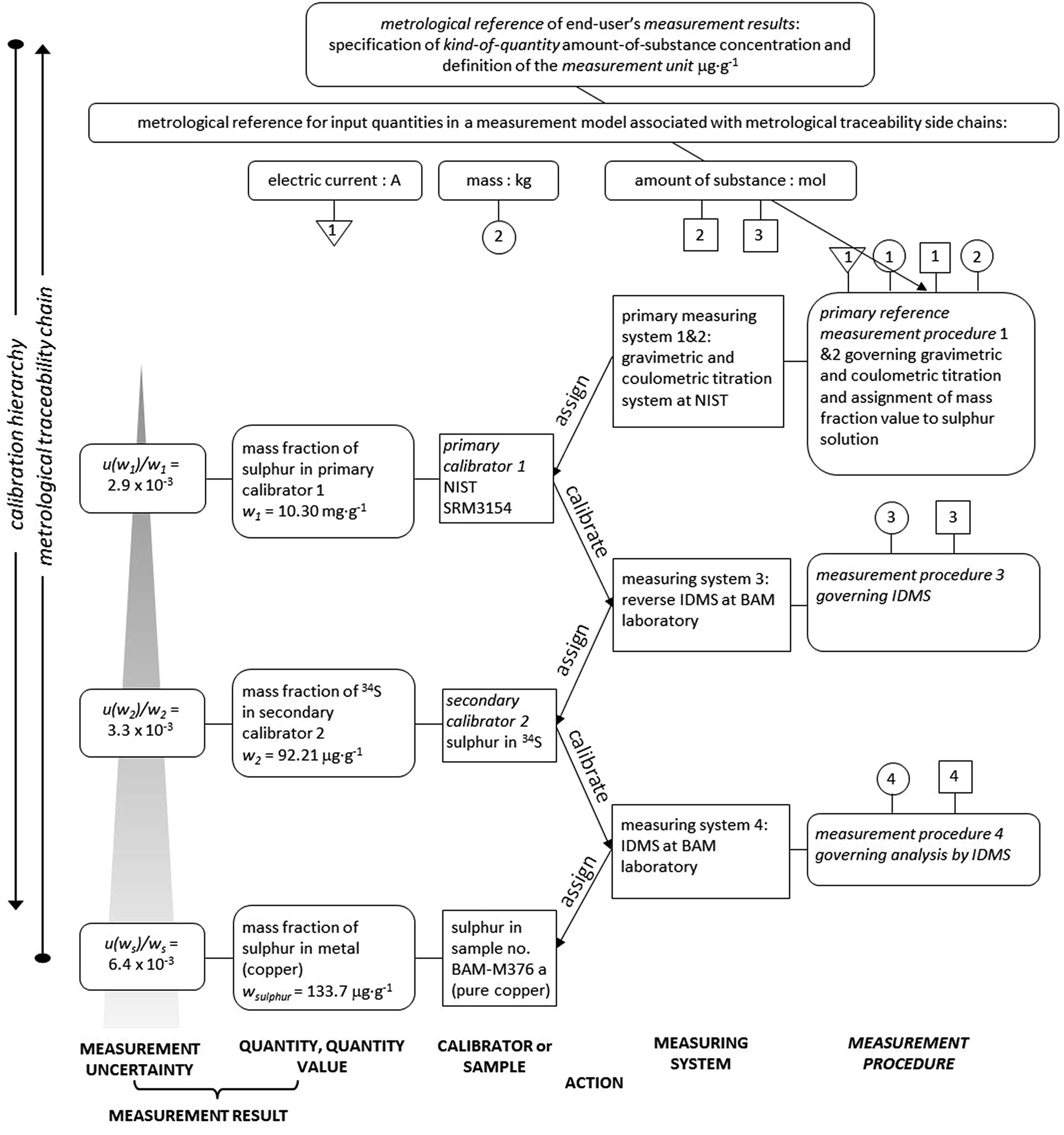 | ||
| Fig. 3 Metrological traceability chain of IDMS measurement results for the sulphur content in sample no. BAM-M376a. | ||
The boxes on the left-hand side express measurement results with measurement uncertainties of the calibrator or sample, while the boxes on the right-hand side show details of the measuring systems and the measurement procedures. The arrows in the middle of the chart express the action of calibrators and measuring systems.
From the bottom of the calibration hierarchy, the three boxes (from left to right) describe the measurement uncertainty, the measurement result, the sulphur mass fraction and the sample, here BAM-M376a. The measurement result was assigned by applying the IDMS approach at BAM (labelled measurement procedure 4 in Fig. 3) as described in this research. For the IDMS approach, the exact mass fraction of the 34S spike solution is required (second calibrator 2) which was obtained by reverse IDMS which is labelled measurement procedure 3 in Fig. 3. The reverse IDMS was applied by using the primary assay NIST SRM3154 as back-spike. NIST SRM3154 was certified by two measuring systems which were gravimetric and coulometric titration at NIST (the information is displayed in the certificate) and represents the primary calibrator 1 for the sulphur mass fraction. The sulphur mass fraction of the primary calibrator 1 is in turn metrologically traceable to the definition of the SI measurement unit mole through the quantity values for electric current and kilogram. This makes the sulphur mass fraction of sample BAM-M376a, as obtained by the here described IDMS procedure, traceable to the SI in the most direct way.
4. Conclusions
In this study, a new analytical procedure for the sample preparation and quantification of the sulphur mass fraction in copper and copper alloys by ICP-IDMS was developed. This includes the purification of sulphur by a three-stage separation procedure consisting of the complexation of copper with ammonia, cation exchange and anion exchange chromatography. The procedure features a high performance which is expressed in a matrix removal efficiency of above 99.999% while the overall sulphur recovery stays above 80%, which is more than sufficient for IDMS applications. Procedure blanks are in the range between the maximum values of 4 ng and 53 ng, with the majority of the blank values ranging from 9 ng to 16 ng. This is 7 times lower than those reported for ID-TIMS14 whereas LOD and LOQ values are 0.20 μg g−1 and 0.54 μg g−1, respectively being the same level as previous work but it has to be noticed that the matrices are different. The procedure blank obtained in this study contributed to the combined uncertainty below 0.1% whereas in ID-TIMS the contribution is 36–99%.13 As mentioned before, the LOD and LOQ are more theoretical concepts for IDMS including trace-matrix separation and have only limited practical use, because the working range of the analytical procedure is defined by the matrix separation procedure and the measurement uncertainty aimed at. With the presented sulphur–matrix procedure a working range from approximately 15 μg g−1 to 1500 μg g−1 can be achieved. In addition to the low detection limits, the here presented procedure offers a high matrix removal efficiency, low measurement uncertainties and SI traceability.The presented analytical procedure was successfully validated via three different routes, first a partial validation via an inter-laboratory comparison at the highest metrological level,22 second a step-by-step validation of the whole analytical procedure, and third the setup of a complete uncertainty budget. Additionally, most of the certified values of the analysed reference materials agree well with the results obtained by the IDMS procedure. Expanded relative measurement uncertainties were estimated to range below 1% while metrological traceability to the SI is clearly expressed. Therefore, the procedure is well suited to provide reference values for the total sulphur mass fraction in copper materials.
Thus, the procedure reported in this study is established as a reference procedure for sulphur measurement in copper being fit for the following purposes: certification of reference materials, assignment of reference values for inter-laboratory comparison and calibration of routine analytical methods such as GDMS, LA-ICPMS, X-ray fluorescence, and carrier gas hot extraction (CGHE) via matrix-matched standards calibrated by the here described analytical procedure.
Conflicts of interest
There are no conflicts to declare.List of abbreviations
| BAM | Bundesanstalt für Materialforschung und -prüfung (BAM) |
| BCR | Community Bureau of Reference |
| BE | Belgium |
| Cps | Count per second |
| CCQM | Consultative Committee for Amount of Substance |
| CENAM | The National Metrology Center, Mexico |
| CGHE | Carrier gas hot extraction |
| CRM | Certified reference material |
| Cu | Copper |
| DE | Germany |
| GDMS | Glow discharge mass spectrometry |
| HPA | High pressure asher |
| ICP-MS | Inductively coupled plasma-mass spectrometry |
| IDMS | Isotope dilution mass spectrometry |
| INMETRO | National Institute of Metrology, Quality and Technology, Brazil |
| K | Coverage factor |
| LA-ICPMS | Laser ablation inductively coupled plasma-mass spectrometry |
| MU | Measurement uncertainty |
| NIMT | National Institute of Metrology (Thailand) |
| NIST | National Institute of Standards and Technology |
| NMIJ | National Metrology Institute of Japan |
| RM | Reference material |
| S | Sulphur |
| SI units | International System of Units |
| SRM | Standard reference material |
| TIMS | Thermal ionization mass spectrometry |
| UME | National Metrology Institute, Turkey |
| UK | United Kingdom |
| US | United States |
| VIM | International Vocabulary of Metrology |
Acknowledgements
The authors thank D. Becker and M. Koenig for assistance and support with the lab work. Especially, the first author thanks the Federal Institute for Materials Research and Testing (Bundesanstalt für Materialforschung und -prüfung (BAM), Germany), the Office of the Civil Service Commission (OCSC, Thailand) and the National Institute of Metrology (NIMT, Thailand) for financial support.Notes and references
- Copper Development Association (CDA), http://copperalliance.org.uk/industry/economy, accessed, 19 Feb, 2017.
- R. Matschat, J. Hinrichs and H. Kipphardt, Anal. Bioanal. Chem., 2006, 386, 125–141 CrossRef CAS PubMed.
- L. Li and R. W. Messler, Weld. J., 1999, 78, 387s–396s Search PubMed.
- Y. S. Sayi, P. S. Shankaran, C. S. Yadav and G. C. Chhapru, Indian J. Chem. Technol., 2003, 10, 373–381 CAS.
- Laboratory of the government chemist (LGC), Copper reference materials, 2016 Search PubMed.
- J. G. Martinez-Sierra, O. G. San Bias, J. M. M. Gayon and J. I. G. Alonso, Spectrochim. Acta, Part B, 2015, 108, 35–52 CrossRef.
- M. C. R. Matschat, M. Hamester and S. Pattberg, Fresenius' J. Anal. Chem., 1997, 359, 418–423 CrossRef.
- B. Lange, S. Recknagel, M. Czerwensky, R. Matschat, M. Michaelis, B. Peplinski and U. Panne, Microchim. Acta, 2008, 160, 97–107 CrossRef CAS.
- M. Sargent, R. Harte and C. Harrington, Guidelines for Achieving High Accuracy in Isotope Dilution Mass Spectrometry (IDMS), Royal Society of Chemistry, 2002 Search PubMed.
- J. Vogl and W. Pritzkow, Mapan-J Metrol Soc I, 2010, 25, 135–164 Search PubMed.
- W. R. Kelly and P. J. Paulsen, Talanta, 1984, 31, 1063–1068 CrossRef CAS PubMed.
- W. R. Kelly, L. T. Chen, J. W. Gramlich and K. E. Hehn, Analyst, 1990, 115, 1019–1024 RSC.
- W. Pritzkow, J. Vogl, R. Koppen and A. Ostermann, Int. J. Mass Spectrom., 2005, 242, 309–318 CrossRef CAS.
- Rohm and Haas Company, Specification's product, 2007 Search PubMed.
- Bio-Rad Laboratories, Instruction Manual: Strong Anion Exchange Resin AG 1, AG MP-1 and AG 2 Search PubMed.
- Bio-Rad Laboratories, Instruction Manual: Chelex® 100 and Chelex 20 Chelating Ion Exchange Resin Search PubMed.
- A. Das, C. H. Chung, C. F. You and M. L. Shen, J. Anal. At. Spectrom., 2012, 27, 2088–2093 RSC.
- https://en.wikipedia.org/wiki/Ionization_energy, accessed, 24 June, 2017.
- R. Biesuz, M. Pesavento, G. Alberti and F. Dalla Riva, Talanta, 2001, 55, 541–550 CrossRef CAS PubMed.
- M. Pesavento and E. Baldini, Anal. Chim. Acta, 1999, 389, 59–68 CrossRef CAS.
- M. Pesavento, R. Biesuz, A. Profumo and T. Soldi, Environ. Sci. Pollut. Res. Int., 2003, 10, 317–320 CrossRef CAS PubMed.
- T. Kuroiwa, Y. Zhu, K. Inagaki, S. Long, S. Christopher, M. Puelles, M. Borinsky, N. Hatamleh, J. Murby, J. Merrick, I. White, D. Saxby, R. C. de Sena, M. D. de Almeida, J. Vogl, P. Phukphatthanachai, W. Fung, H. Yau, T. O. Okumu, J. N. Kangiri, J. A. Salas Téllez, E. Z. Campos, E. C. Galván, N. Kaewkhomdee, S. Taebunpakul, U. Thiengmanee, C. Yafa, N. Tokman, M. Tunç and S. Z. Can, Metrologia, 2017, 54(Tech. Suppl.), 08008 CrossRef.
- JCGM200:2012, JCGM, 3rd edn, 2012, ch. 2008, version with minor corrections Search PubMed.
- P. De Bievre, R. Dybkaer, A. Fajgelj and D. Brynn Hibbert, Pure Appl. Chem., 2011, 83, 1873–1935 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |