
Isolation of circulating plasma cells from blood of patients diagnosed with clonal plasma cell disorders using cell selection microfluidics†
 acd
Steven A.
Soper
*cdghij
acd
Steven A.
Soper
*cdghij
Abstract
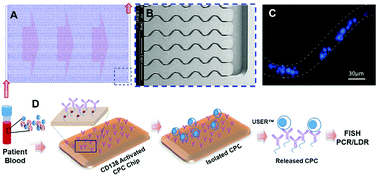
Blood samples from patients with plasma cell disorders were analysed for the presence of circulating plasma cells (CPCs) using a microfluidic device modified with monoclonal anti-CD138 antibodies. CPCs were immuno-phenotyped using a CD38/CD56/CD45 panel and identified in 78% of patients with monoclonal gammopathy of undetermined significance (MGUS), all patients with smouldering and symptomatic multiple myeloma (MM), and none in the controls. The burden of CPCs was higher in patients with symptomatic MM compared with MGUS and smouldering MM (p < 0.05). FISH analysis revealed the presence of chromosome 13 deletions in CPCs that correlated with bone marrow results. Point mutations in KRAS were identified, including different mutations from sub-clones derived from the same patient. The microfluidic assay represents a highly sensitive method for enumerating CPCs and allows for the cytogenetic and molecular characterization of CPCs.
 Please wait while we load your content...
Please wait while we load your content...