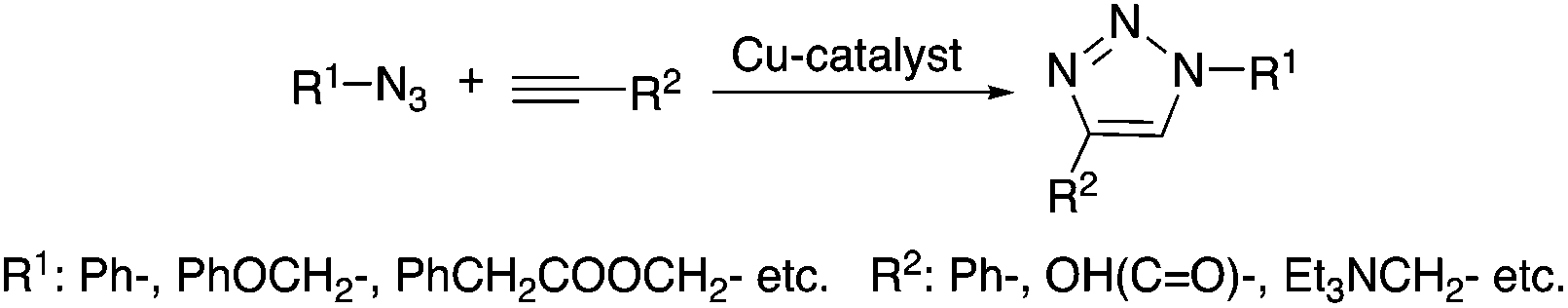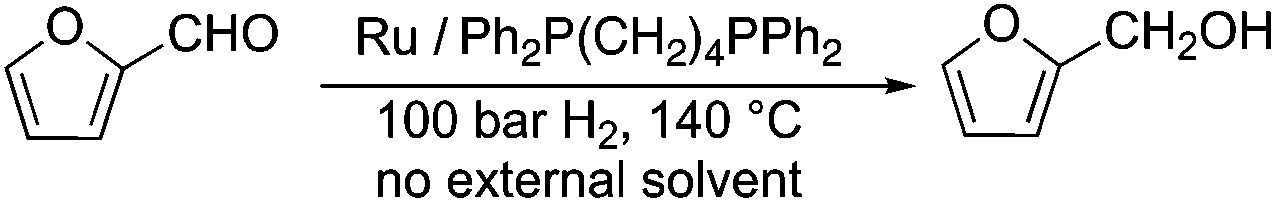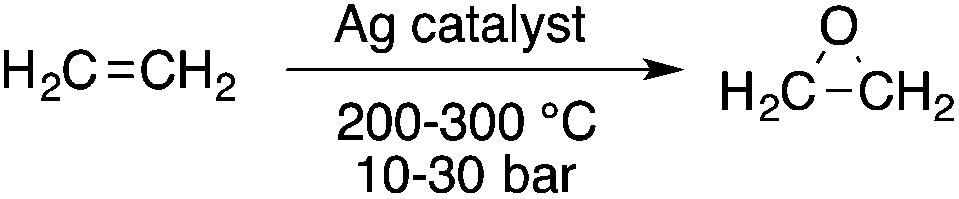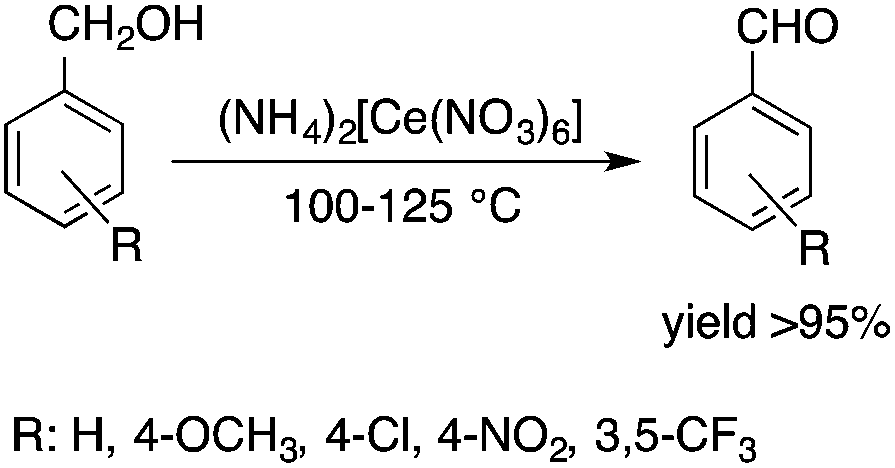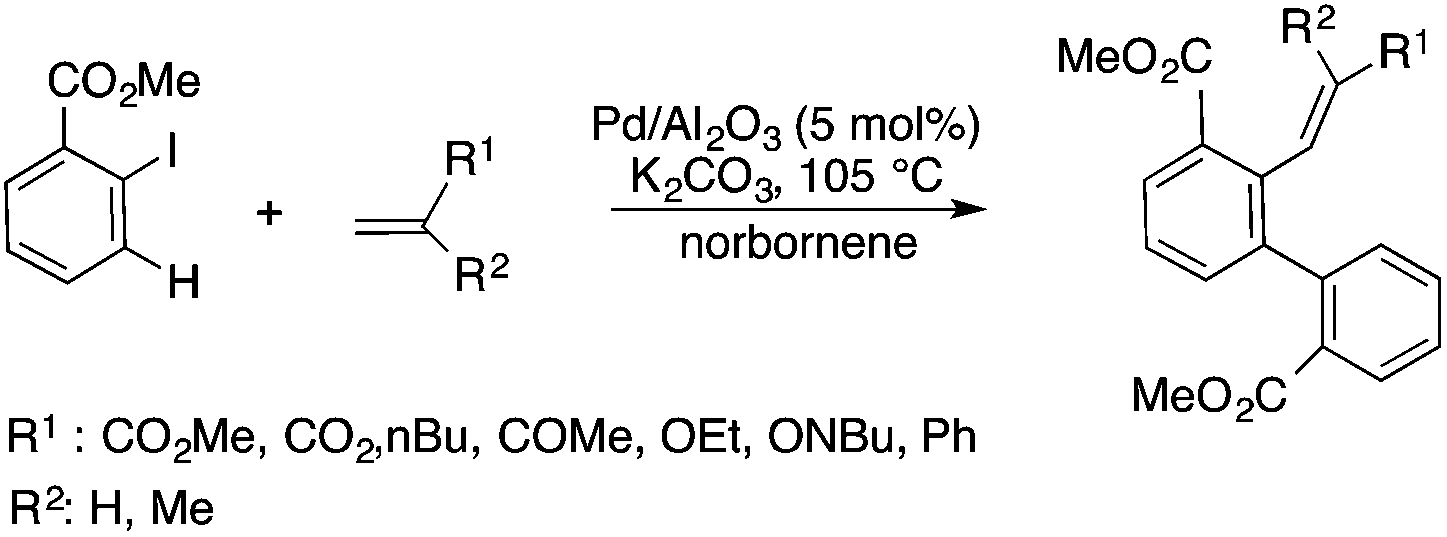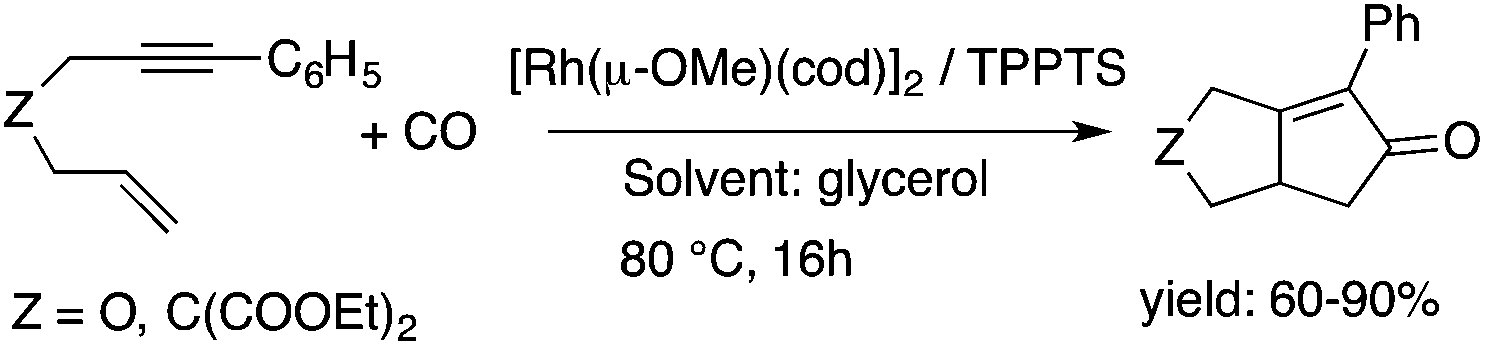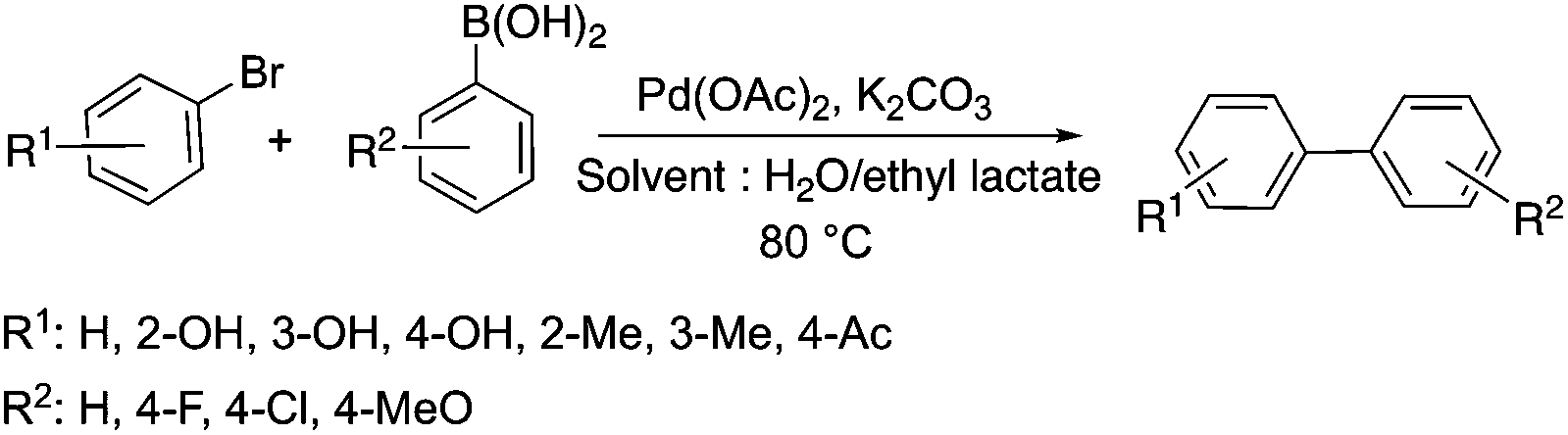DOI:
10.1039/C8GC00514A
(Tutorial Review)
Green Chem., 2018,
20, 2171-2191
Conservative evolution and industrial metabolism in Green Chemistry†
Received
13th February 2018
, Accepted 23rd April 2018
First published on 23rd April 2018
Abstract
To date, chemistry has provided more than a hundred million new compounds, most of which are non-natural species that have never existed before and are a source of an enormous amount of pollution. With the increased amount of these “man-made” chemicals being synthesised and environmental concerns of chemical processes, chemists have become very aware of these potential risks and dangers. As an answer to this challenge, Green Chemistry emerged and brought with it a list of necessary measures to be followed in the laboratory that has been gradually completed. Herein, we present evidence for the manifestation of two related principles within Green Chemistry: (i) conservative evolution, which refers to the observation that throughout the history of the universe, old construction blocks, such as elementary particles, amino acids, or living cells remained conserved while the world evolved in its complexity, and (ii) the practice of industrial metabolism that is a manifestation of conservative evolution in human activity referring to the application of biological metabolism in the production of goods for our civilization. Related concepts of Green Chemistry, such as the atom economy, as a metric of green chemistry, the application of safer chemicals and solvents, the utilization of catalysis, the utilization of renewable resources, and in situ spectroscopy, are discussed, providing examples to students and researchers from academy to industry involving the work of Professor István T. Horváth.
 Gábor Náray-Szabó | Gábor Náray-Szabó graduated in chemistry in Budapest. He had positions in the pharmaceutical industry, and later he was appointed as professor of chemistry at the Eötvös Loránd University, Budapest, Hungary. He is a member of the Hungarian Academy of Sciences, Académie des Arts, des Sciences et des Lettres, Academia Scientiarium et Artium Europaea, Doctor honoris causa at the Universitatea de Vest, Timisoara. He formulated the principle of electrostatic enzyme catalysis in collaboration with Arieh Warshel. Author of 250 scientific publications and 8 books in the field of theoretical chemistry, computer-aided molecular design, structural biology and sustainability. |
 László T. Mika | László T. Mika received his Ph.D (organic- and organometallic chemistry) under the supervision of Professor István T. Horváth at Eötvös Loránd University, Budapest, Hungary in 2010. He worked as an assistant professor at the Institute of Chemistry until 2012, when he became an associate professor and head of the Laboratory of Catalysis at the Budapest University of Technology and Economics. He became a head of the Department of Chemical and Environmental Process Engineering in 2016. His research activity documented by about 40 scientific papers covers different areas of green chemistry (biomass conversion, applications of alternative reaction media, and the design of new catalytic systems). |
Introduction
Evolution can be defined as a process that leads to an increased and viable complexity.1 By accepting this definition, it can be definitely stated that humans, as part of the biosphere, also contribute to evolution by creating new constructions involving processes and materials synthesized using chemical methods. Today, millions of products in the chemical industry, such as transportation fuels, fertilizers, polymers and composites, pharmaceuticals, detergents, food additives, dyes, agrochemicals, etc., are in widespread use, maintaining, or even increasing, the living standards of our society. So far, more than 130 million artificial chemical substances have been produced, which have previously not existed in Nature. In addition, this production is highly based on fossilized carbon resources, i.e. crude oil, natural gas, and coal, making our society fossil fuel dependent. While this number continuously increases by ca. 1 million per year, limited information is available on the impact of new compounds on living organisms and the environment. By releasing these compounds into the biosphere, these materials could interact with natural substances and a very small portion may be embedded in existing living systems without harm. The rest has to be considered as more or less toxic, threatening life, which is manifested in the harmful interaction of xenobiotics with biomolecules. Moreover, several serious environmental and health problems have been caused by chemicals over the last 50 years. For example, chemical disasters have occurred in Seveso (Italy, 1976)2 and Bhopal (India, 1985),3 and the large scale utilization of hazardous substances such as DDT (1940–1970),4 chlorofluorocarbons (1960s–1994),5,6 or the direct use of chiral substances such as thalidomide on humans (1960s),7 just to name a few, have resulted in global and/or local issues, which have directed the attention of researchers towards the development of environmentally friendly methodologies for the synthesis of new chemicals and the use of less harmful substances.
The evolution of Green Chemistry, as a collection of guiding concepts, can be considered as one of the first responses for these purposes, focusing on materials and processes that could operate in harmony with Nature and minimize their risk on the environment.8,9 Although it is indeed hard to apply all of the principles of Green Chemistry together at the same time, the more of them that result in the realization of alternative processes, the more that environmentally benign processes can be realized.9
As an analogy to biological metabolism, that involves the elimination of superfluous substances from an organism, industrial metabolism was born as a concept, and realizes the importance of material and energy integration and the turnover of industrial systems.10 Therefore, a generalization of industrial metabolism is a concept of a circular economy, representing a regenerative system, where resource input and waste and emissions, as well as energy leakage, are minimized by optimizing materials and energy loops.11
The concept of conservative evolution refers to the observation that during the history of the universe only new construction blocks survived, which were based on ones that already existed.12 The proton, the neutron and the electron appeared very early after the Big Bang and these particles conserved their properties and have been the basic constituents of molecular matter up until the very present. A variety of different molecules was formed and though these can be decomposed under specific conditions, some of them conserved their role over billions of years. The most important groups are amino and nucleic acids, which have been the basic molecular building blocks of living systems for more than three billion years. Water has a specific role in life as a safe and very effective solvent. Thus, the vast majority of biochemical processes take place in an aqueous environment and this has not changed since the first living cell appeared on Earth. Furthermore, cells, though differentiated into dozens of forms, have remained the basic constituents of living systems throughout the history of life. Catalysis plays a fundamental role in the efficient transformation of molecules and energy production in living organisms. Life has evolved through the harmonious utilization of renewable resources and treatment of waste. Lots of further examples can be given for the manifestation of conservative evolution. Consequently, the adaption of these fundamental observations could lead to the establishment of greener and cleaner chemical processes.
Herein, we call attention to the chemical substances and processes that play a key role in the twelve principles of Green Chemistry representing conservative evolution and/or industrial metabolism.8 Our examples primarily refer to the work of Professor István T. Horváth, whose contribution to this field is spectacular.
Results and discussion
The atom economy
The manifestation of industrial metabolism can be exemplified by the second principle of Green Chemistry. As Barry M. Trost has proposed, synthetic methods should be designed to maximize the incorporation of all atoms used in the reaction into the final product, which allows the minimization or even elimination of useless or harmful wastes.13 Atom economy can be considered an excellent “green metrics” of transformations at the molecular level and it is able to define a reaction to be “green”. However, process chemistry usually needs auxiliary substances to assist in the establishment of conditions for a 100% atom economic reaction. Therefore, environmental aspects should be subsequently evaluated by the fate of auxiliary substances after the isolation and purification of the product(s). For this purpose, Sheldon introduced the Environmental factor (E-factor)14 as a metric applicable for the assessment of the performance of a complete production process. Recently, it has been generally accepted and used by the chemical industry. The E-factor is defined as the mass ratio of waste to the desired product. Both the E-factor and atom economy are strongly related to the greenness of a chemical process from the laboratory to industrial scale and represent widely accepted measures of the environmental impact of such processes. Here we provide some representative examples of reactions exhibiting 100% atom economy.
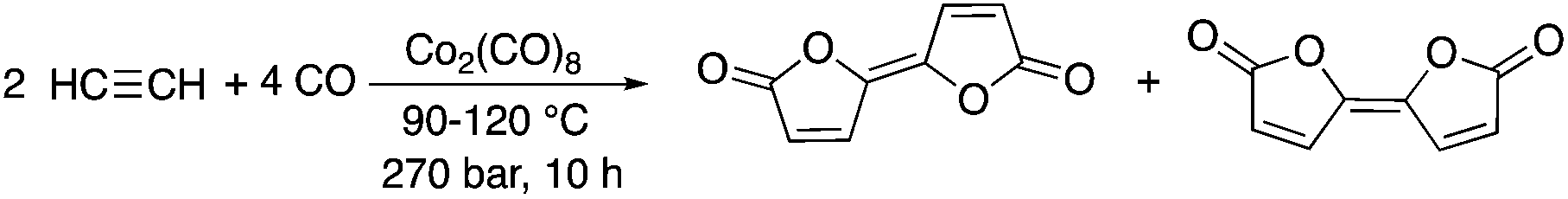
It goes without saying that addition reactions are excellent examples of transformations representing 100% atom economy. One of the earliest papers by Várady, Horváth, and Pályi et al. described the conversion of carbon monoxide and acetylene to bifurandiones, which can be considered as attractive compounds to the polymer industry. The Co2(CO)8-catalyzed reaction represents 100% atom economy; however, the efficiency of the transformation is rather low, indicated by only a 4–6% product yield (Scheme 1).15 It was shown that the addition of tertiary trialkylphosphines or trialkylphosphites resulted in a considerable improvement in the yields, reaching 30–72% depending on the type of alkyl substituents present on the phosphorous ligand. A yield of 72% was achieved by conversion of acetylene and CO in the presence of P(nBu)3 at 120 °C.16
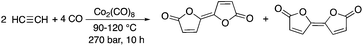 |
| | Scheme 1 Synthesis of bifurandiones. | |
Several addition reactions can be considered as a part of “click-chemistry”, which represents a powerful and highly reliable tool for the synthesis of useful organic, and usually biologically active, compounds.17 Since the seminal papers by Sharpless18 and Meldal,19 highly regioselective copper-catalyzed azide–alkyne cyclo-addition (Cu-AAC, Scheme 2) has received significant interest as a novel synthetic methodology. Accordingly, several improvements were made in order to increase the efficiency of the reaction and to widen its applicability. Heaney et al. reported a simplified protocol to obtain 1,2,3-triazoles using an alkynylcopper(I) polymeric precatalyst without the necessity of the addition of a ligand in a microwave-assisted reaction at 100 °C.20 Díez-Gonzáles showed that commercially available CuBr(PPh3)3 acts as a catalyst for the preparation of diverse 1,2,3-triazoles in a strict click reaction. The catalyst operates at room temperature and at a low (50 ppm–0.5 mol%) catalyst loading, resulting in the formation of triazoles in generally high (>85%) yields. It is worth noting that this system also allowed “neat” conditions to be used.21 The Cu-AAC mediated addition of acetylene to both aliphatic and aromatic azides to form the corresponding 1-substituted-1,2,3-triazoles was demonstrated by Liang. The reactions were carried out in the presence of triethylamine as a base in DMSO under 1 bar of acetylene at room temperature22 and the isolated yields were between 86–97% depending on the type of azide used. Lim applied β-cyclodextrin as a phase transfer catalyst for the preparation of 1,2,3-triazoles with excellent yields (90–99%) in water, as an environmentally benign reaction medium, at room temperature, which is a useful protocol from both an economic (e.g. using a cheap CuSO4·5H2O catalytic precursor) and an environmental point of view, as well as for the practical convenience of not having to handle flammable anhydrous organic solvents, and toxic and expensive reagents.23
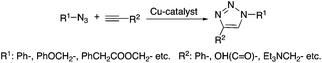 |
| | Scheme 2 Azide–alkyne cycloaddition (Cu-AAC). | |
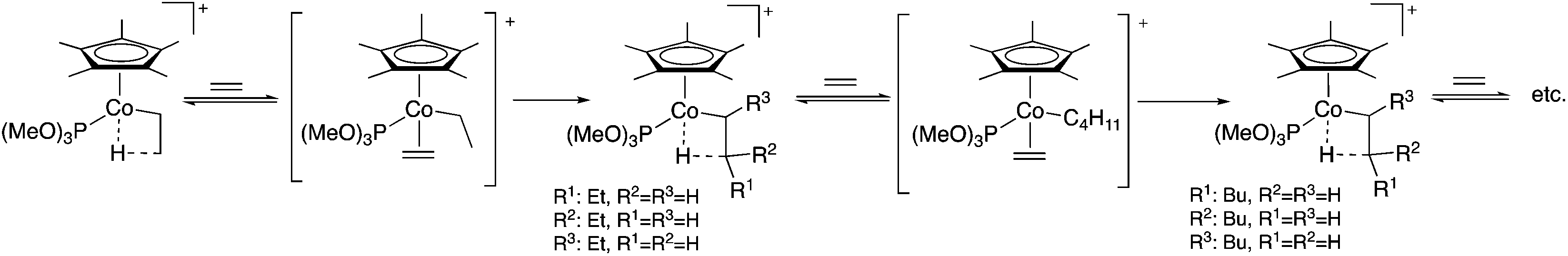
Polymerization reactions were used to produce more than 300 million tons of materials in 2014 worldwide and assist in the everyday life of human society, from plastic bags, clothing, and cutlery, automobiles to healthcare, and electronics and smart materials and they are also excellent examples of 100% atom economy. The mechanism of the transition metal-catalyzed polymerization of ethylene (or another olefin), which has a distinguished role in polymer chemistry, starts by the coordination of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to the activated metal center of the metallocene catalyst and followed by the insertion of a coordinated olefin into the metal–carbon bond of the growing polymer chain. According to the Cossée-Arlman mechanism, a β-migratory insertion step, taking place via an intermediate alkyl-olefin complex, was proposed. However, its presence was not detected until 1990, when the first observation of Co-alkyl complexes in the [C5Me5(P(OMe)3)CoCH2CH2-μ-H]+-catalyzed polymerization of ethylene was reported by Brookhart and Horváth et al. A pioneering in situ high-pressure NMR study using 13C-labelled ethylene evidenced the existence of these complexes and clearly established the chain growing mechanism (Scheme 3).24 A similar study was reported in collaboration with Prof. Nozaki revealing the mechanism of the alternating copolymerization of propylene with carbon monoxide in the presence of a Pt-(R,S)-BINAPHOS catalyst. It was shown that the cis/trans isomerization from (SP-4-4)-[Pt(CH3)(CO)((R,S)-BINAPHOS)] [B(3,5-(CF3)2C6H3)4] to the more stable SP-4-3 isomer is faster than the formation of its acyl derivatives via CO insertion. For the Pd-(R,S)-BINAPHOS-catalyzed copolymerization, there exist at least two major resting states, most probably the acyl palladium species, (SP-4-3)-[Pd(C(O)R)((R,S)-BINAPHOS)(L2)][B(3,5-(CF3)2C6H3)4] (L2 = CH3CN, CO), and the alkyl palladium species, [Pd{CH2CH(CH3)C(O)R} ((R,S)-BINAPHOS)][B(3,5-(CF3)2C6H3)4].25
C bond to the activated metal center of the metallocene catalyst and followed by the insertion of a coordinated olefin into the metal–carbon bond of the growing polymer chain. According to the Cossée-Arlman mechanism, a β-migratory insertion step, taking place via an intermediate alkyl-olefin complex, was proposed. However, its presence was not detected until 1990, when the first observation of Co-alkyl complexes in the [C5Me5(P(OMe)3)CoCH2CH2-μ-H]+-catalyzed polymerization of ethylene was reported by Brookhart and Horváth et al. A pioneering in situ high-pressure NMR study using 13C-labelled ethylene evidenced the existence of these complexes and clearly established the chain growing mechanism (Scheme 3).24 A similar study was reported in collaboration with Prof. Nozaki revealing the mechanism of the alternating copolymerization of propylene with carbon monoxide in the presence of a Pt-(R,S)-BINAPHOS catalyst. It was shown that the cis/trans isomerization from (SP-4-4)-[Pt(CH3)(CO)((R,S)-BINAPHOS)] [B(3,5-(CF3)2C6H3)4] to the more stable SP-4-3 isomer is faster than the formation of its acyl derivatives via CO insertion. For the Pd-(R,S)-BINAPHOS-catalyzed copolymerization, there exist at least two major resting states, most probably the acyl palladium species, (SP-4-3)-[Pd(C(O)R)((R,S)-BINAPHOS)(L2)][B(3,5-(CF3)2C6H3)4] (L2 = CH3CN, CO), and the alkyl palladium species, [Pd{CH2CH(CH3)C(O)R} ((R,S)-BINAPHOS)][B(3,5-(CF3)2C6H3)4].25
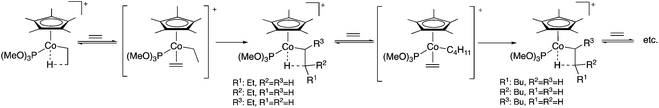 |
| | Scheme 3 Mechanism of the polymerization of ethylene. Adapted with permission from ref. 24. Copyright of the American Chemical Society (1990). | |
Selective hydrogenation reactions can also exemplify atom economic processes. Among these transformations, the conversion of an initial platform chemical furfural (FAL)26 to furfuryl alcohol (FOL) is one of the most industrially important hydrogenation reactions. It takes place in either the liquid or gas phase in the presence of a highly toxic copper-chromite catalyst on a large scale.27 Therefore, in order to reduce the environmental risk of this transformation, several attempts have been made to develop alternative catalyst systems.28 Recently it was shown that a Ru/Ph2P(CH2)4PPh2 catalyst was able to convert FAL to FOL in the absence of any external solvent or other auxiliary materials, opening up an environmental friendly route (Scheme 4) to this reaction. The catalyst was found to be recyclable over twelve consecutive runs without any decrease in its activity and selectivity.29
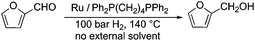 |
| | Scheme 4 Selective hydrogenation of FAL to FOL. | |
The oxidation of ethylene to ethylene oxide is also an important reaction in the large-scale chemical industry. The original reaction, the so-called “chlorohydrin process” exhibited several serious environmental concerns including the loss of chlorine gas that was used in the formation of calcium chloride and the generation of unwanted chlorine-containing by-products. Since the discovery of a heterogeneous Ag-based catalyst by Lefort in 1931 that allows the direct oxidation of ethylene to ethylene oxide, the manufacturing process has theoretically become a 100% atom economic process (Scheme 5); however, the practical selectivity of the industrial-scale process is between 80–90% depending on the type of catalyst system used, i.e. the type of solid support used, the presence of promoters, etc. It should be noted that no other metal has been found that can compete with Ag for this purpose.
 |
| | Scheme 5 Oxidation of ethylene to ethylene oxide. | |
Safer chemicals
According to the fourth principle of Green Chemistry, chemical products should be designed to affect their desired function while minimizing their toxicity. This is again a manifestation of the principle of industrial metabolism.
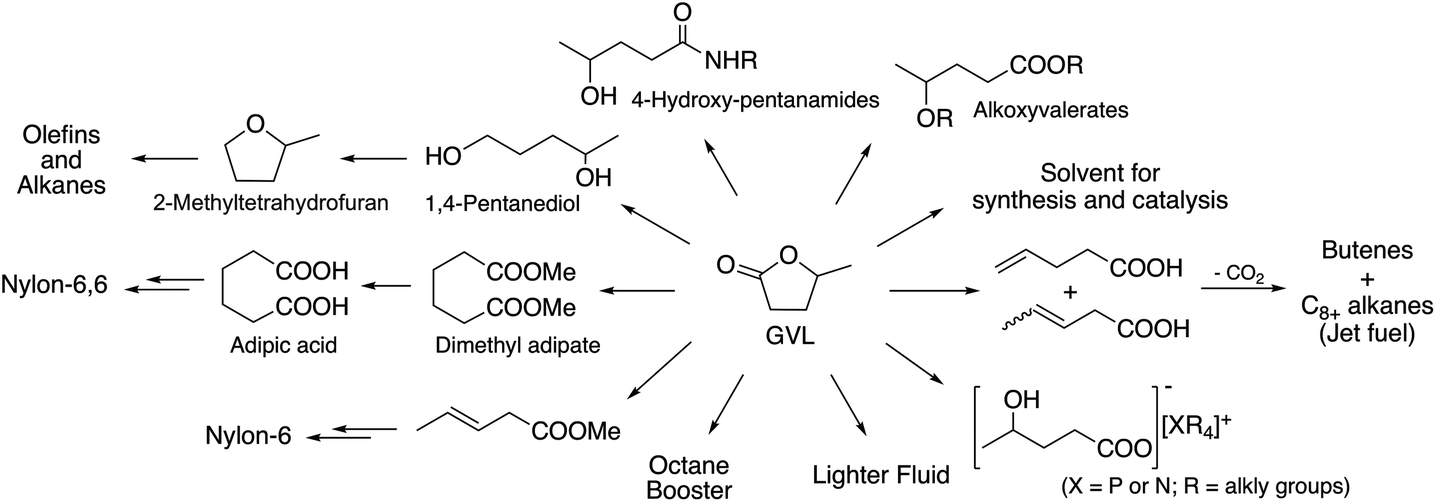
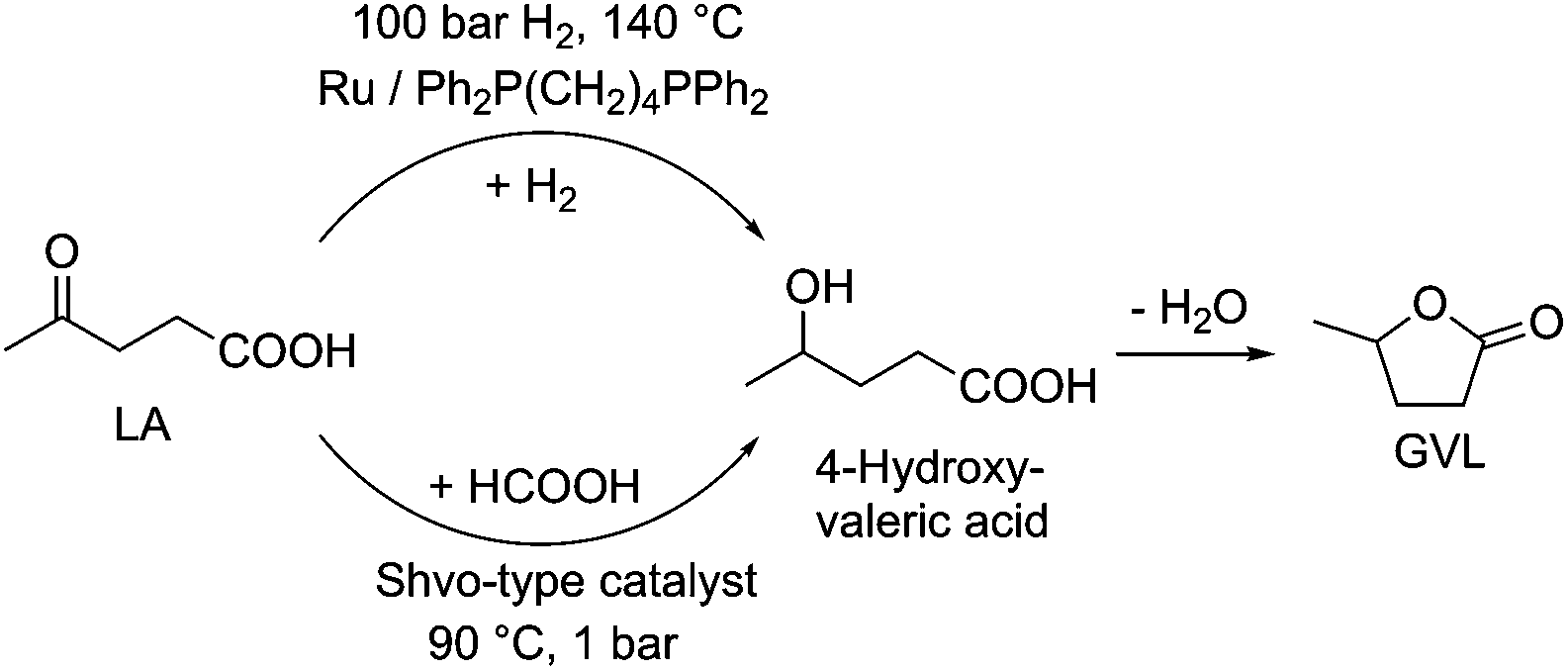
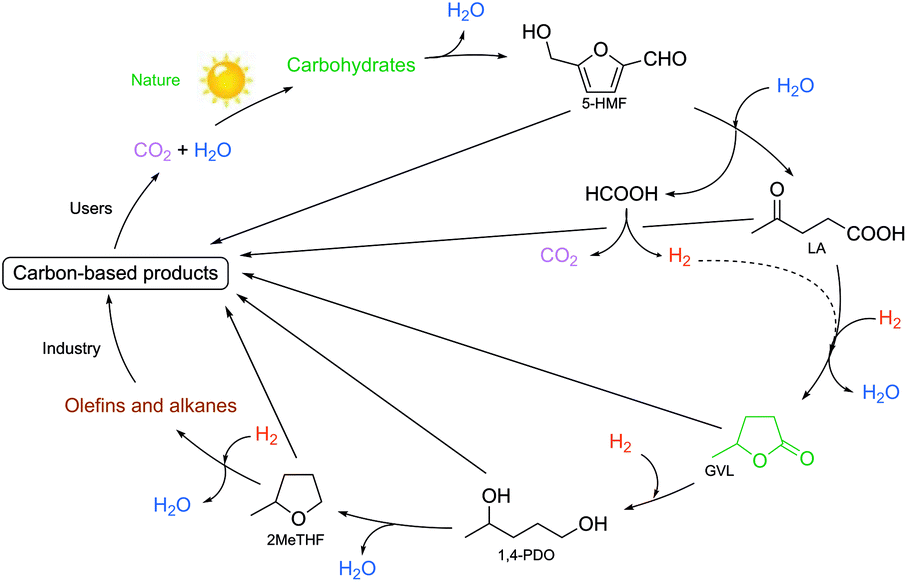
One of the excellent examples of a safer chemical is γ-valerolactone (GVL), which was first proposed as a sustainable liquid for the production of energy and carbon-based chemicals by Horváth in 2008.30 This compound is a component of fruit and it is frequently used as a food additive. It is a liquid in the temperature range of −31–207 °C, has a high open cup flash point (96 °C), a definite smell, low toxicity (LD50,rat = 8800 mg kg−1)30 and is fully miscible with water, assisting biodegradation. It is important to note that GVL does not form any measurable amount of peroxides under air in weeks,31 contrary to the behaviour of 2-methyltetrahydrofurane (2Me-THF) or ethers, e.g. diisopropyl-ether, making it a rather safe compound for large scale use. GVL has a low vapor pressure, preventing its emission into the environment, and can be separated from water and alcohols, without the formation of an azeotropic mixture.32,33 The multistep conversion of biomass from sucrose to alkanes involving the synthesis of GVL was first demonstrated by Horváth's group in 2008.34 Obviously, the most effective protocol to synthesize GVL is the selective hydrogenation of levulinic acid in the presence of a catalyst. Reduction leads to the formation of 4-hydroxyvaleric acid, which readily undergoes ring closure to form GVL. For this purpose, external solvent-free methods were developed, including the transfer hydrogenation protocol.35–37 Since GVL was proposed as a platform molecule, several innovative applications have been demonstrated, opening up a renewable-based route to produce carbon-based chemicals. It can be reduced to form 1,4-pentanediol and 2Me-THF by a tertiary phosphine, e.g. P(nBu)3,34 P(nOct)3, or a triphos (C(CH3)P(CH2PPh2)3)-modified Ru-based catalyst system.37 It should be noted that 2Me-THF can be subsequently reduced in the presence of a Pt-based catalyst to form olefins and alkanes.34 GVL can be converted to butenes by its decarboxylation in the presence of a SiO2/Al2O3 catalyst, and can then be oligomerized to from industrially important higher olefins.38,39 As Horváth proposed, GVL could be a replacement for fossil-based hydrocarbons in igniting and lighter fluids, as well.40 Alkoxyvalerates and valerate-based ionic liquids, which could be considered and used as partially bio-ionic liquids, can also easily be obtained from GVL.41,42 Their applications as reaction media for hydrogenation reactions involving transfer hydrogenation protocol42,43 and Ullmann-type cross-coupling reactions44 were recently demonstrated. Biomass-based polymer precursors such as hydroxyamides can also be easily synthesized by the ring-opening of GVL under mild conditions.45 Moreover, the synthesis of dimethyl adipate or adipic acid from GVL could open up alternative routes as a feedstock for the Nylon industry.46 Since optically-active lactones occur naturally and can be used as chiral building blocks for the synthesis of biologically active compounds, GVL has been proposed as a chiral biomass-based building block.47 A summary of GVL-based chemicals is presented in Scheme 6.
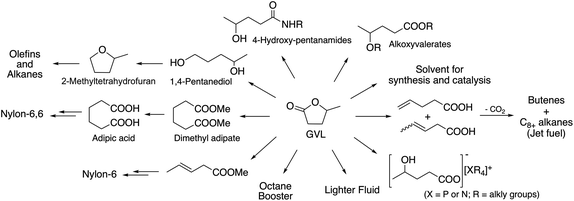 |
| | Scheme 6 GVL-based chemicals. | |
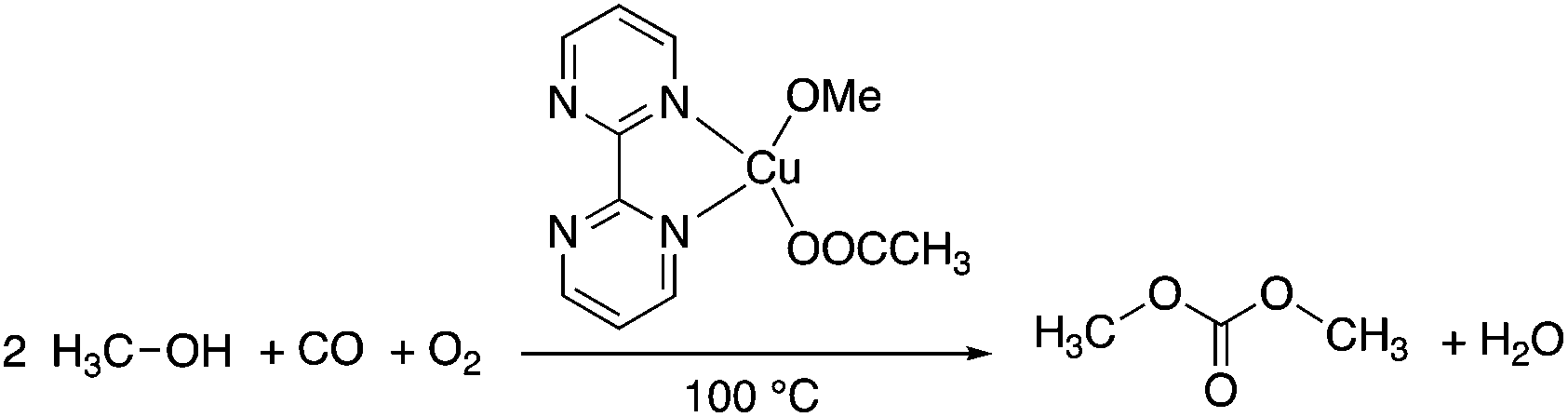
Dimethyl carbonate (DMC), the simplest organic carbonate, has an important role in the chemical industry and moreover, its low toxicity (LD50,rat = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg kg−1)48 for human health and other life forms makes it a safe, non-corrosive green chemical for sustainable processes. The conventional production of DMC is based on the reaction of methanol with highly toxic phosgene using a chlorinated solvent. The first phosgene-free alternative process was based on the oxidative carbonylation of methanol in the presence of Cu(I)-chloride. However, due to the formation of wet HCl by the hydrolysis of the chloride containing Cu-species, the process is extremely corrosive and all of the commercial equipment exposed to the CuCl–MeOH–H2O–HCl mixture must therefore be glass-lined. Furthermore, the formation of chlorinated organic side-products could also result in an environmental issue. Because the catalyst precursor is the only source of chlorine in the system, application of a chlorine-free catalyst overcomes the drawbacks of the process. Horváth and Mehnert et al. developed a chlorine free-catalyst, which is prepared in situ from Cu(II)-acetate and 2,2′-bipyrimidine for the oxidative carbonylation of methanol (Scheme 7), which was investigated under both batch and semi-continuous conditions.49In situ high-pressure IR and NMR spectroscopic studies suggested the formation of [Cu(2,2′-bipyrimidine)(CO)(OMe)] as one of the key intermediates. The catalytic performance of the novel catalyst system was found to be similar to the CuCl-based one under identical conditions. The chlorine-free catalyst was immobilized by co-polymerizing 5-vinyl-2,2′-bipyrimidine with styrene.49
000 mg kg−1)48 for human health and other life forms makes it a safe, non-corrosive green chemical for sustainable processes. The conventional production of DMC is based on the reaction of methanol with highly toxic phosgene using a chlorinated solvent. The first phosgene-free alternative process was based on the oxidative carbonylation of methanol in the presence of Cu(I)-chloride. However, due to the formation of wet HCl by the hydrolysis of the chloride containing Cu-species, the process is extremely corrosive and all of the commercial equipment exposed to the CuCl–MeOH–H2O–HCl mixture must therefore be glass-lined. Furthermore, the formation of chlorinated organic side-products could also result in an environmental issue. Because the catalyst precursor is the only source of chlorine in the system, application of a chlorine-free catalyst overcomes the drawbacks of the process. Horváth and Mehnert et al. developed a chlorine free-catalyst, which is prepared in situ from Cu(II)-acetate and 2,2′-bipyrimidine for the oxidative carbonylation of methanol (Scheme 7), which was investigated under both batch and semi-continuous conditions.49In situ high-pressure IR and NMR spectroscopic studies suggested the formation of [Cu(2,2′-bipyrimidine)(CO)(OMe)] as one of the key intermediates. The catalytic performance of the novel catalyst system was found to be similar to the CuCl-based one under identical conditions. The chlorine-free catalyst was immobilized by co-polymerizing 5-vinyl-2,2′-bipyrimidine with styrene.49
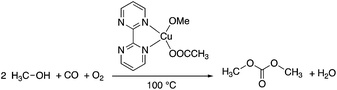 |
| | Scheme 7 Oxidative carbonylation of methanol to dimethyl carbonate. | |
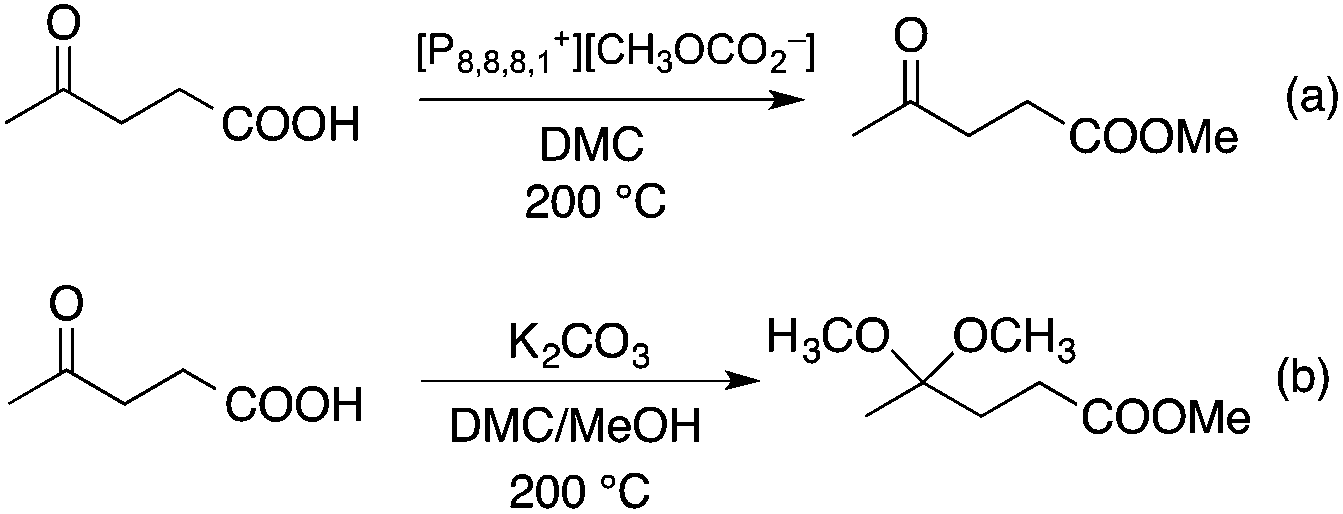
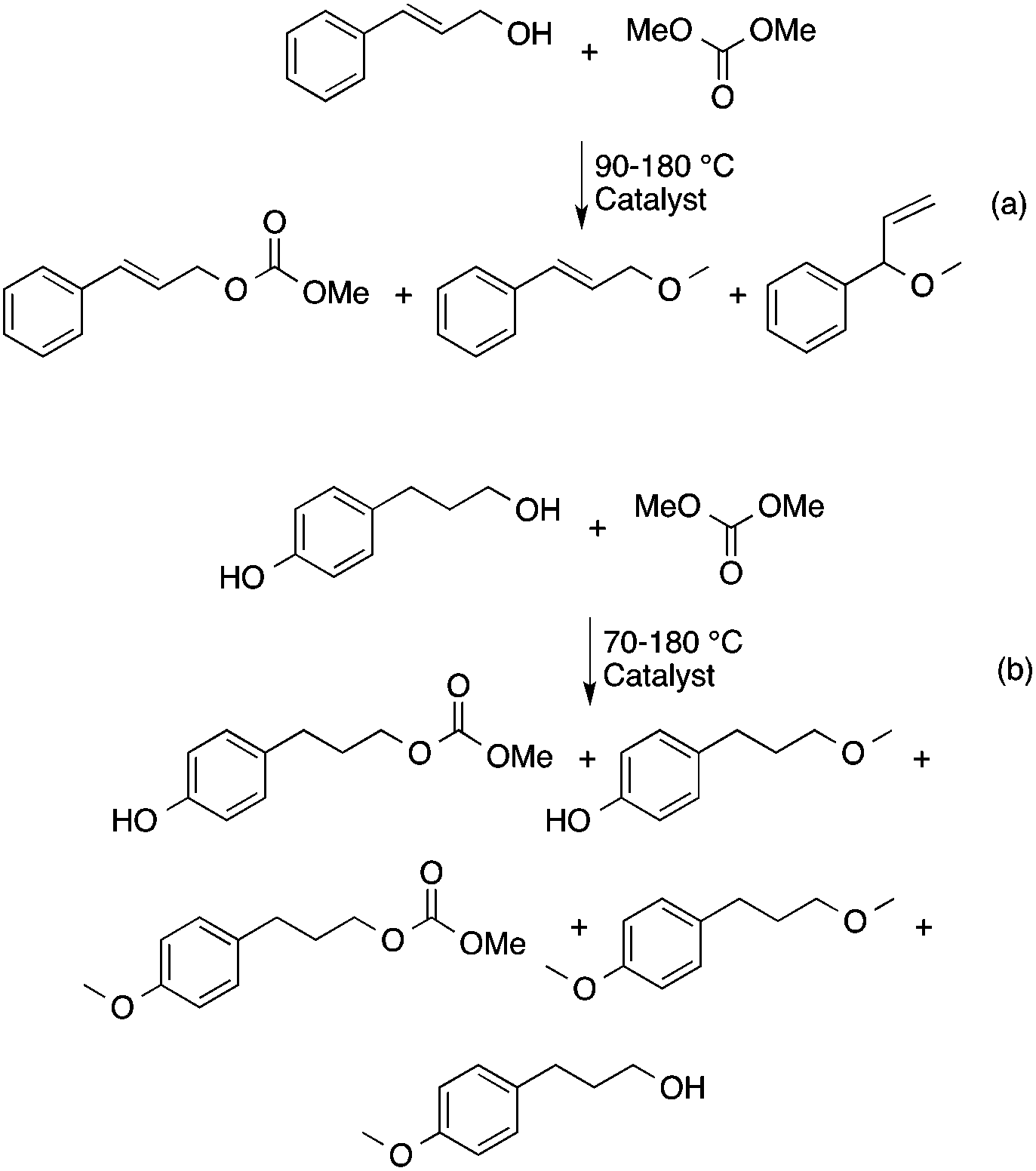
DMC is a safe, non-corrosive, and environmentally friendly alternative building block for carbonylating, carboxymethylating and methylating agents, phosgene, methoxycarbonyl chloride, dimethyl sulfate, and methyl halides, respectively.50–53 Recently, DMC has been introduced as an alkylating agent in the conversion of platform chemicals obtained from renewable feedstocks or biomass-based waste streams. Perosa's group investigated the reaction of levulinic acid (LA) with DMC in the presence of a different catalyst. Complete conversion of LA and an exceptional yield of methyl levulinate (96%) was detected by using a 20-fold excess of DMC and [P8,8,8,1][CH3OCO2] as a base/catalyst at 200 °C for 6 h (Scheme 8). By addition of methanol as a co-solvent, the formation of the dimethyl-ketal of methyl levulinate in the presence of K2CO3 was detected.54 The same group demonstrated that cinnamyl alcohol and 4-(3-hydroxypropyl)phenol, the two compounds resembling the lignin building block p-coumaryl alcohol, could be selectively converted by catalytic methodologies based on DMC as the green reagent (Scheme 9). It was revealed that selectivity of the catalytic system could be tuned by changing the reaction temperature and the nature of the catalyst. While basic catalysts, e.g. K2CO3, [P8,8,8,1][CH3OCO2] ([P8,8,8,1]: a trioctyl methylphosphonium cation) and CsF/Al2O3 promote transesterification reactions at lower temperatures (ca. 90 °C), amphoteric solids, such as alkali–metal exchanged faujasites, selectively accelerate the formation of alkyl ethers at higher temperatures (165–180 °C).55 As an extension of this study, the bis-O methylation of 1,2- and 1,4-dihydroxybenzene derivatives, which can also be found in the product spectrum of lignin depolymerization to the corresponding bis-methyl ethers, was investigated in the temperature range of 100–170 °C using microwave irradiation as a heating method. Both stoichiometric as well as catalytic amounts of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were effective for bis-methylation at relatively mild temperatures. An almost quantitative conversion of catechol and hydroquinone toward the corresponding bis-methylated products were achieved in excellent isolated yields at 170 °C.56
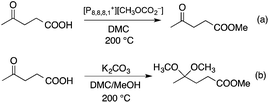 |
| | Scheme 8 Alkylation reactions of levulinic acid with dimethyl carbonate. | |
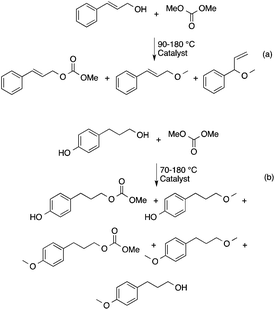 |
| | Scheme 9 Alkylation of cinnamyl alcohol and 4-(3-hydroxypropy)phenol with DMC. Adapted from ref. 55 with permission from the Royal Society of Chemistry. | |
Safer solvents
Solvents are an intrinsic part of many chemical reactions and “solvent-friendly chemical thinking” has evolved due to many advantages in both laboratory and industrial operations, including the simple regulation of temperatures (reflux), moderation of exothermic reactions, dissolution of solids to get molecules into a common phase, facilitation of mixing, lowering of viscosity and density, etc. Although “external solvent-free” transformations could offer environmental friendly solutions, in reality, many thousands, if not millions of chemical reactions, can only operate in liquid media. As an outcome, the industrial activities involving solvents result in the release of volatile organic compounds (VOCs), including conventional solvents, into the environment. For example, for the EU28 alone, over 6 million tons of VOC are released annually,57 some of which are leading to serious environmental concerns. Consequently, the elimination or replacement of conventional organic solvents with environmentally benign or even biomass-based alternatives with a low vapor pressure, low or no toxicity, low flammability and a limited negative impact on the environment is a crucial part of the development of greener and cleaner chemical technologies.
Obviously, the complete elimination of solvent(s) as auxiliary material(s) from reactions could result in an ideal “external solvent-free” or so-called “neat” reaction condition offering the best solution for the solvent issue. When one of the substrates is liquid under reaction conditions, it also acts as an “internal solvent”. Consequently, the term “solvent-free” cannot be stated for these cases and the use of the term “external solvent(s)-free” has to be preferred. In addition, the composition of the reaction environment continuously changes from the substrate(s) to product(s) during the reaction. This can easily be demonstrated by the “neat” hydrogenation of LA to 4-hydroxyvaleric acid (4-HVA) in the presence of a catalyst in situ generated from Ru(acac)3 and Ph2P(CH2)4PPh2 (Scheme 10).34,35,37 At the beginning of the reaction, 100% of LA is present as a solvent and at the end of the conversion GVL:water = 1:1 (n/n) has to be considered as the reaction medium. It should also be noted that the transfer hydrogenation of LA by the use of HCOOH can also be carried out under “neat” conditions (Scheme 10) resulting in a mixture of GVL, H2O, and HCOOH. This elegant approach was reported by Horváth in 2014.36
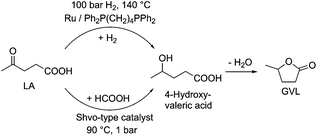 |
| | Scheme 10 External solvent-free (neat) hydrogenation of LA to 4-hydroxyvaleric acid. | |
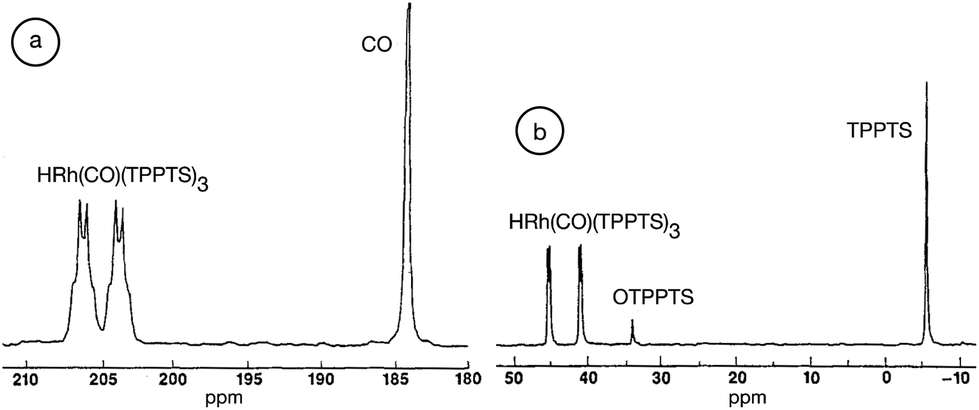
Although “neat” transformations could offer environmentally friendly alternatives, most reactions can only be operated in the presence of solvents. Because chemical reactions take place under aqueous conditions in living organisms, there is no doubt that water could be an excellent reaction medium as a readily available, cheap, nonflammable and nontoxic liquid. Water is one of the basic and irreplaceable components of living systems and other solvents, especially VOCs, seem to not be able to compete with its evolutionary advantages. Accordingly, several efforts have been made to develop catalytic systems that operate in water, of which organic analogues have been well-known for a long time. An outstanding example is the aqueous Ruhrchemie/Rhône–Poulenc biphasic hydroformylation process of propylene in the presence of HRh(CO)(TPPTS)3 (TPPTS: the sodium salt of m-trisulfonated-triphenylphosphine).58 The process was developed for the hydroformylation of propylene to butanal and the ratio of normal- to iso-butanal is about 25, which is surprisingly high compared with that used in the conventional triphenylphosphine-modified (HRh(CO)(PPh3)3) hydroformylation system under similar conditions. It was proposed that the coordination of the olefin to the coordinatively unsaturated {HRh(CO)(phosphine)2} or {HRh(CO)2(phosphine)} species results in a high or low normal- to iso-butanal ratio, respectively. A pioneering high-pressure in situ NMR study by Horváth and co-workers established that only HRh(CO)(TPPTS)3 was detectable in water even under high syngas (CO:H2 = 1:1) pressures of up to 200 bar (Fig. 1). In the case of PPh3, only the presence of the known species HRh(CO)2(PPh3)2 was observed at a low (30 bar, CO:H2 = 1:1) syngas pressure making the coordinatively unsaturated HRh(CO)2(PPh3) species easily accessible for the olefin. The activation energy of the dissociation of TPPTS from HRh(CO)(TPPTS)3 was found to be significantly higher, 11 ± 1 kcal mol−1, than that of the dissociation of PPh3 from HRh(CO)(PPh3)3.59 The higher activation energy of the dissociation of TPPTS from HRh(CO)(TPPTS)3 was attributed to the intramolecular association of –SO3− of the neighboring TPPTS ligands via hydrogen bonding in aqueous medium, representing one of the first examples of outer sphere control.
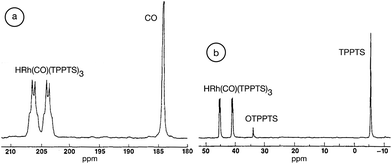 |
| | Fig. 1
13C NMR (a) and 31P NMR (b) spectra of HRh(13CO)(TPPTS)3 in the presence of a 3 molar excess of TPPTS under a 200 bar pressure of 13CO:H2 = 1:1 in D2O. Modified with permission from ref. 59. Copyright of Springer (1989). | |
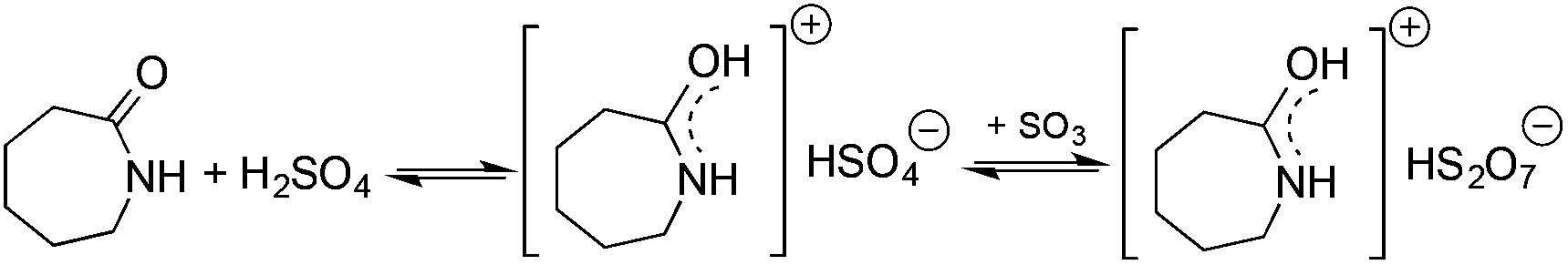
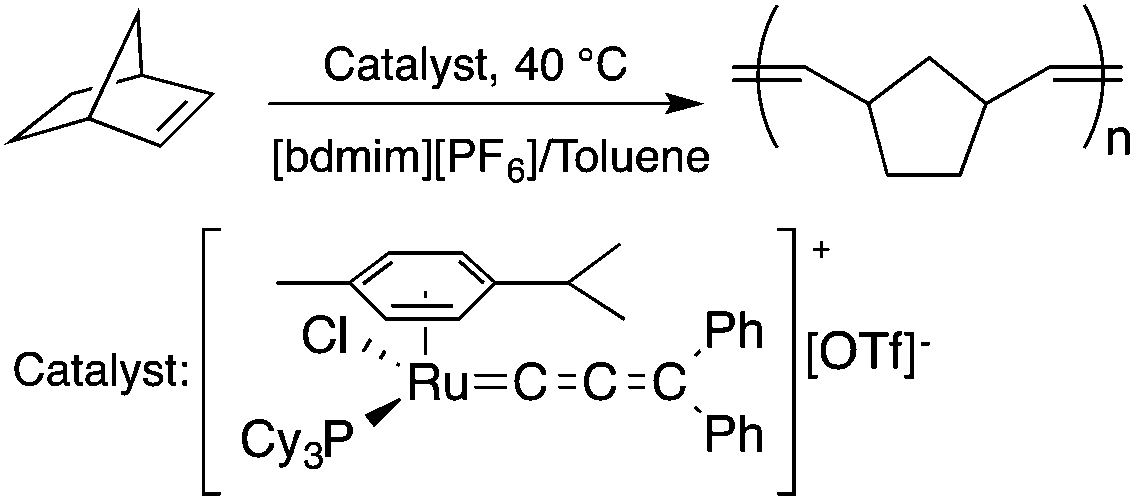
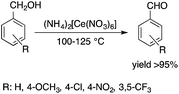
Ionic liquids (ILs) have attracted considerable attention as alternative reaction media for a huge variety of chemical transformations, due to their extremely low vapor pressure, good solvating properties, reasonable thermal stability, and easily tunable chemical (e.g. acidity, basicity, and polarity) and physical properties (e.g. viscosity, melting point or vapor pressure).60–62 However, few applications of ILs as reaction media have been realized on an industrial scale. While a huge variety of ILs have been utilized on a small laboratory scale, the large scale application of an ionic liquid was realized during an investigation of the mechanism of a well-known Beckmann-rearrangement reaction of cyclohexanone oxime to ε-caprolactam in H2SO4 or H2S2O7.63 Horváth first recognized that the industrially applied “rearrangement mixture”, used by all ε-caprolactam manufacturers, is actually a ε-caprolactamium hydrogen cation with a HSO4− and/or HS2O7− anion (Scheme 11) depending on the presence of sulfur trioxide. It is important to note that formation of an ε-caprolactamium-type ionic liquid that could hold SO3 very strongly up to 140 °C results in a very low vapor pressure of the reaction environment providing safer conditions for this very exothermic reaction. The first ring-opening metathesis polymerization of norbornene as a cyclic olefin in the ionic liquid [bdmim][PF6] (1-butyl-2,3-dimethylimidazolium hexafluorophosphate) in the presence of the cationic ruthenium allenylidene precatalyst [(p-cymene)RuCl(PCy3)(CCCPh2)][OTf] was reported by Horváth and Dixneuf et al.64 When norbornene was reacted in [bdmim][PF6], a 98% polymer yield was isolated after its extraction with toluene and precipitation using methanol. However, the catalyst activity decreased after recycling the catalyst containing the IL, presumably due to either decomposition or removal of the catalyst. By applying a [bdmim][PF6]-toluene biphasic system to assist in the in situ extraction of the polymer, excellent yields (>96%) were obtained over three consecutive runs (Scheme 12). Although several examples of rare-earth-mediated organic reactions in ILs have been described, the first reaction involving cerium(IV)-salts in IL was reported by Horváth and Binnemans in 2007 (Scheme 13).65 The Ce(IV)-mediated oxidation of benzyl alcohol as a model reaction was demonstrated in 1-ethyl-3-methylimidazolium triflate [C2mim][TfO]. It was found that the selectivity of the reaction of cerium(IV)-triflate with benzyl alcohol in dry ionic liquids depended on the degree of hydration of the cerium(IV)-triflate as follows: anhydrous cerium(IV) triflate transforms benzyl alcohol into dibenzyl ether, whereas hydrated cerium(IV) triflate afforded benzaldehyde as the main reaction product. Investigation of the oxidation of a series of benzyl alcohol derivatives in a temperature range of 100–125 °C showed that an increase in the reaction temperature leads not only to a faster reaction but also to a higher selectivity toward the desired product. Quantitative conversion of 5-hydroxymethylfurfural (5-HMF) to 2,5-furandicarboxaldehyde and 1,4-hydroquinone to 1,4-quinone were also achieved at 100 °C and room temperature, respectively.65 Recent developments in the chemistry of ionic liquids, including their characterization, synthesis, and applications, were reviewed in a themed issue of Chemical Reviews in 2017.60
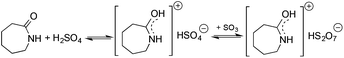 |
| | Scheme 11 Formation of ε-caprolactamium-type ionic liquids. | |
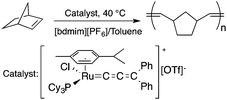 |
| | Scheme 12 Ring-opening metathesis polymerization of norbornene. Adapted from ref. 64 with permission from the Centre National de la Recherche Scientifique (CNRS) and the Royal Society of Chemistry. | |
 |
| | Scheme 13 Oxidation of benzyl alcohol and its derivatives. | |
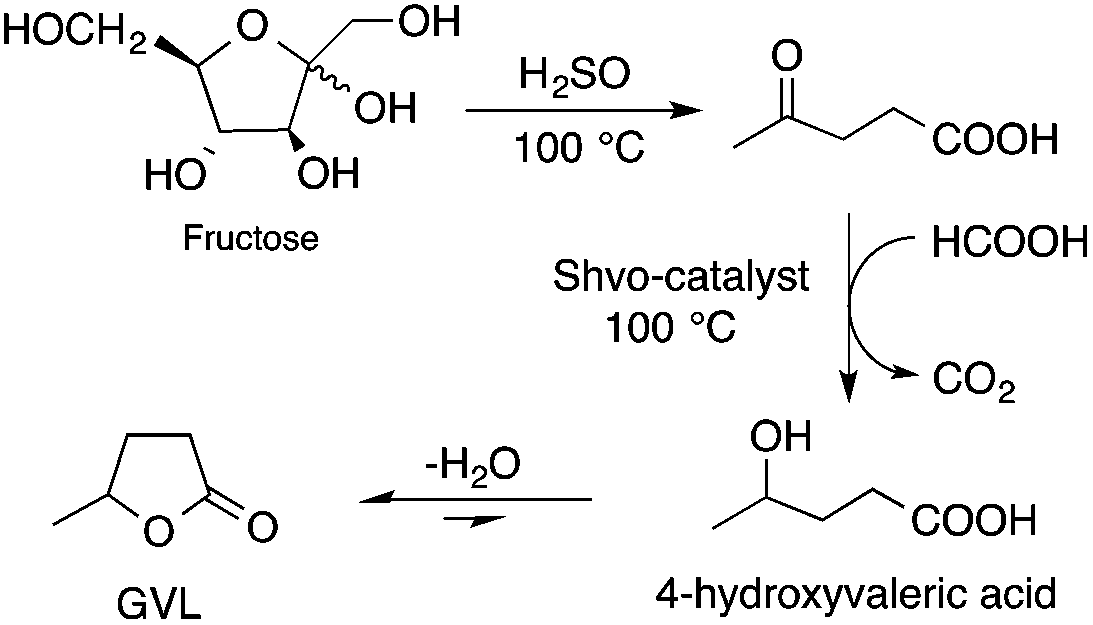
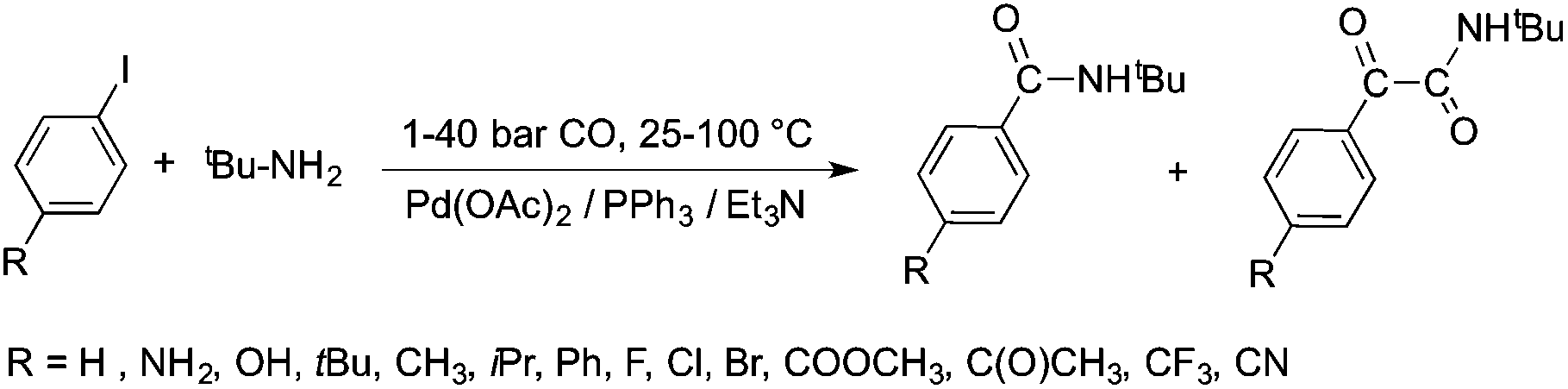
Bioliquids derived from a renewable feedstock represent a novel replacement for conventional organic solvents in well-established chemical transformations. Although some of them, i.e. ethanol, have been well-known as reaction media for a long time, the characterization of new candidates and possible applications have come into the focus of interest and have been the subject of recent research. GVL was first proposed as a biomass-based solvent by Horváth in 2008.66 Because it is an aprotic and dipolar molecule, it can dissolve several types of substrates and catalysts. However, some limiting factors have to be considered e.g. (i) the presence of water in GVL results in the formation of 4-HVA, which could be a reactant in unwanted side reactions, and (ii) at higher temperatures the thermal stability of GVL is a limiting factor for its applicability. Despite these drawbacks, several successful applications for both homogeneous and heterogeneous catalysis have already been demonstrated. Horváth used GVL as a reaction medium for the acid-catalyzed conversion of fructose to levulinic acid and its subsequent selective reduction via a transfer hydrogenation reaction to 4-hydroxyvaleric acid using a Shvo-type catalyst (Scheme 14). The 13C5-GVL derived from 13C6-fructose with an overall yield of 55% and the GVL solvent was distinguished with ease using a 13C-labelling technique. GVL can also be successfully utilized as a reaction medium for the homogeneous Pt- (Scheme 15) and Rh-catalyzed enantioselective hydroformylation of styrene,67,68 as well as the Pd-catalyzed aminocarbonylation (Scheme 16) of iodoaromatic compounds.69 In general, slightly lower activities were observed in GVL compared to in conventional organic solvents, however, the selectivities were similar, allowing its application as an alternative reaction environment. For heterogeneous catalysis, Vaccaro and co-workers demonstrated that synthetically important coupling reactions such as the Heck,70 Sonogashira-,71 Hiyama-,72 and Catellani-reactions (Scheme 17)73 could be successfully performed in GVL. The same group investigated C–H arylation reactions, which were accomplished with a user-friendly heterogeneous palladium catalyst in GVL. The protocol was characterized by an ample substrate scope and high functional group tolerance in the C–H arylation of 1,2,3-triazoles, and the palladium catalyst could be recycled and reused in the process.74
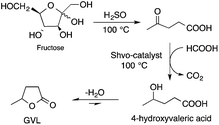 |
| | Scheme 14 Conversion of D-fructose to GVL in GVL. | |
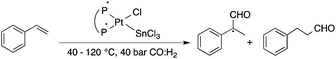 |
| | Scheme 15 Enantioselective hydroformylation of styrene in GVL. | |
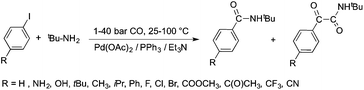 |
| | Scheme 16 Aminocarbonylation of iodobenzene and its derivatives in GVL. | |
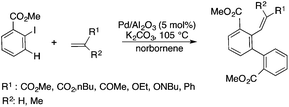 |
| | Scheme 17 The Catellani reaction in GVL. | |
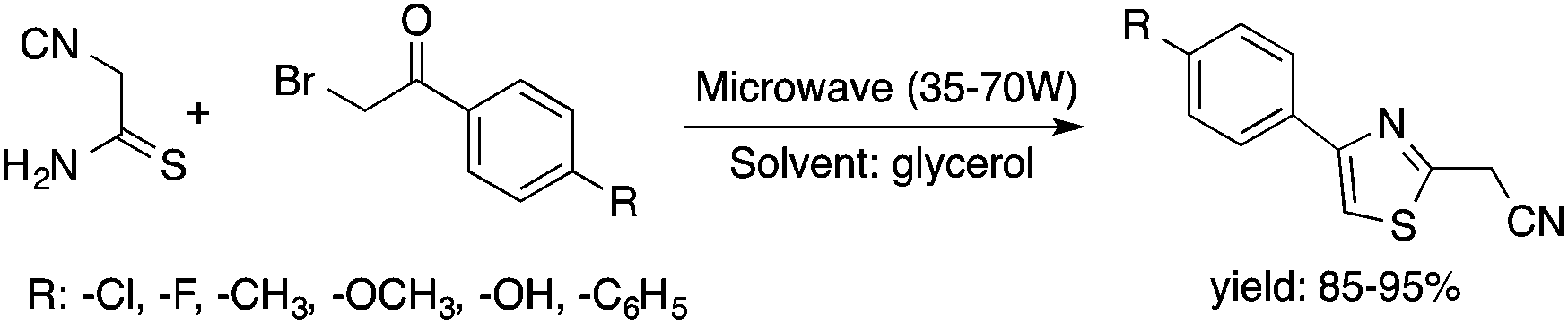
Glycerol is a readily available, biodegradable, and environmentally benign solvent obtained as a by-product of the large-scale transesterification of a triglyceride in the production of natural fatty acid derivatives on an industrial scale. Its toxicity (LD50,rat = 12600 mg kg−1)75 is much lower than that of methanol (LD50,rat = 5600 mg kg−1)76 or ethanol (LD50,rat = 7000 mg kg−1).77 In addition, the increasing demand for biodiesel fuel production also results in it being produced in an increased amount. However, the production of solvent quality glycerol requires high-energy consuming purification processes. Wolfson first reported the application of glycerol as a reaction medium for the Baker's Yeast catalyzed asymmetric reduction of ketones under mild conditions. From a series of stoichiometric organic transformations, Jérôme and co-workers demonstrated that the aza-Michael reaction of p-anisidine and butyl acrylate as a model reaction can be performed at 100 °C.78 While over 80% yield was obtained in glycerol, in water and conventional organic solvents such as toluene, dimethylsulfoxide (DMSO) or DMF less than 5% and zero conversions were detected, respectively. Substrate screening revealed that both electron donating and withdrawing groups were tolerated resulting in product yields of between 60–90% (Scheme 18). The addition of indol and its derivatives to β-nitrostyrene also proceeded under similar conditions. Deligeorgiev reported on an environmentally benign, microwave-assisted procedure for the synthesis of substituted 2-cyanomethyl-4-phenylthiazoles from 2-bromoacetophenones in glycerol (Scheme 19). Excellent yields were obtained over very short (3.5–4.5 min) reaction times and the products were also isolated with very high purity. This approach can be applied to the preparation of a variety of 2-cyanomethyl-4-phenylthiazoles.79 Glycerol has also received increased interest as a reaction medium for transition metal catalyzed reactions. Gómez and co-workers demonstrated the efficient Rh-catalyzed Pauson–Khand carbocyclizations of 1,6-enynes to bicyclo[3.3.0]octenones in neat glycerol. Interestingly, the reaction can be performed by [Rh(μ-OMe)(cod)]2 in the absence of any ligand under an atmospheric CO pressure at 80 °C (Scheme 20). However, TPPTS can enhance the catalyst performance at low Rh/TPPTS (1:1 or 1:2) ratios.80 It should be noted that no or negligible activities were detected in toluene or tetrahydrofuran as reference organic solvents. The catalytic activity associated with [Rh(μ-OMe)(cod)]2 is in contrast to the lack of activity observed with the chlorine-bridged [Rh(μ-Cl)(cod)]2, which can be attributed to the Brønsted-basic character of the methoxide group in a protic medium such as glycerol. Glycerol has been proposed as an ideal alternative to isopropanol as both hydrogen source and solvent in transfer hydrogenation reactions. Crotti showed that organoiridium derivatives of the type Ir(diene)(N–N)X (diene: 1,5-hexadiene or 1,5-cyclooctadiene; N–N = 2,2′-bipyridine, 1,10-phenanthroline and its substituted derivatives; X = Cl, I) in the presence of a base, such as K2CO3, was able to catalyze the hydrogen transfer reaction from glycerol to acetophenone, yielding dihydroxyacetone and phenylethanol in a temperature range of 100–120 °C.81 At the same time, Wolfson and co-workers demonstrated the activity of [RuCl(μ-Cl)(μ6-p-cymene)]2 in the reduction of benzaldehyde to benzyl alcohol, using KOH or NaOH as bases, with a yield of 99%.82 When ultrasonic (US) conditions were applied, the catalyst loading and reaction time could be reduced to 1 mol% and 3 h, respectively. Both Ir(II) and Ir(III) complexes bearing N-heterocyclic carbene (NHC) ligands also showed catalytic activity in the transfer hydrogenation reactions of various aldehydes to the corresponding alcohols in glycerol. The catalytic behaviour of Ir complexes was explored under microwave (MW) irradiation and US conditions allowing significant reduction in the catalyst loading and reaction time from 15–24 h to 0.5–1 h.83,84 Beyond these selected examples, several solvent applications of glycerol, for example in the reduction of nitroarenes and nitriles, in Heck and Sonogashira coupling, in Suzuki reactions, in the enantioselective reduction of aromatic ketones, in the acid catalyzed dehydration of fructose, etc., have also been demonstrated.85–87
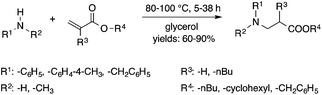 |
| | Scheme 18 Aza-Michael reactions in glycerol. | |
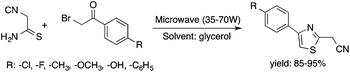 |
| | Scheme 19 Synthesis of substituted 2-cyanomethyl-4-phenylthiazoles. | |
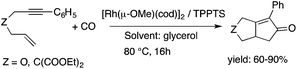 |
| | Scheme 20 The Rh-catalyzed Pauson–Khand carbocyclization of enynes. | |
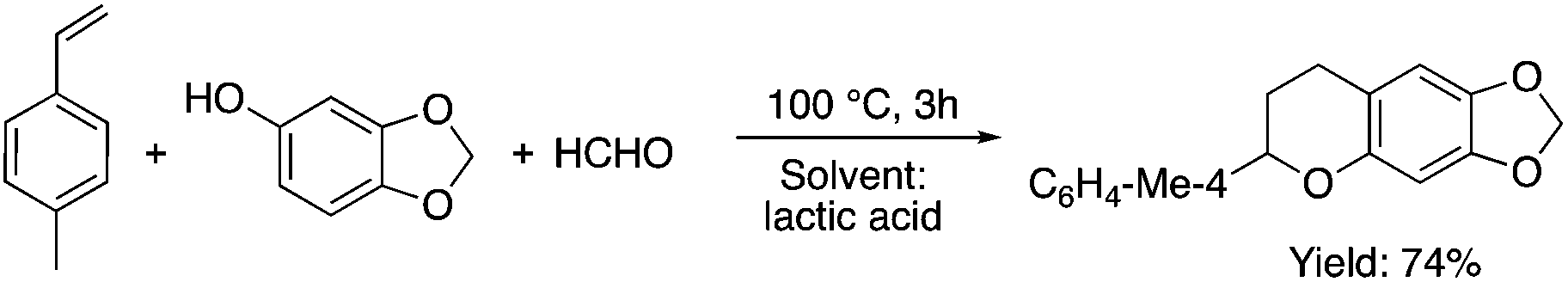
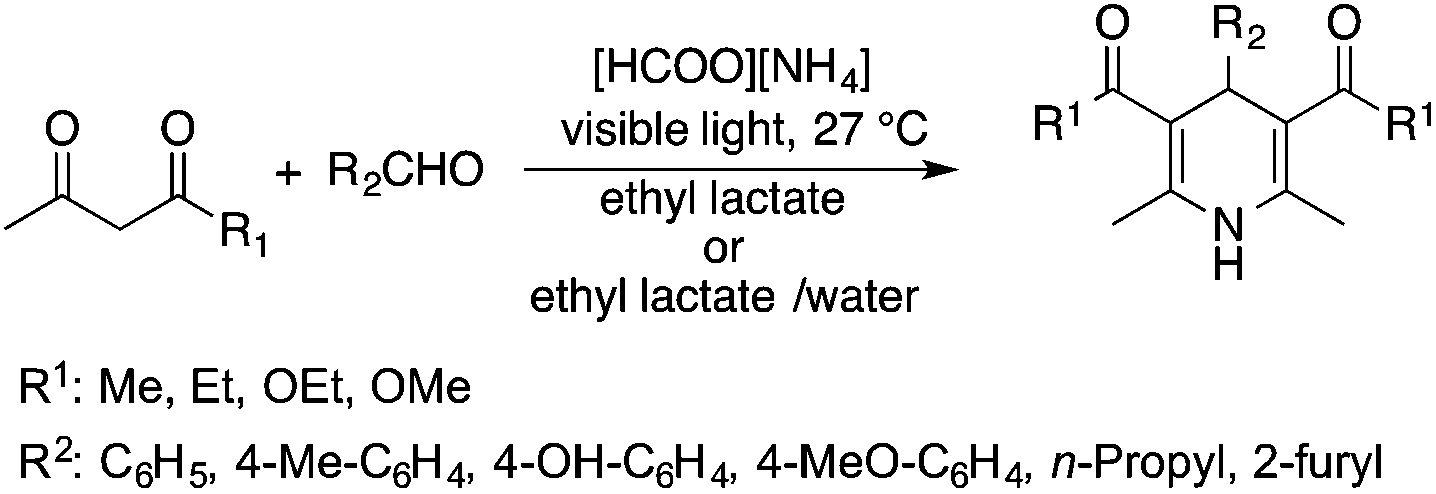
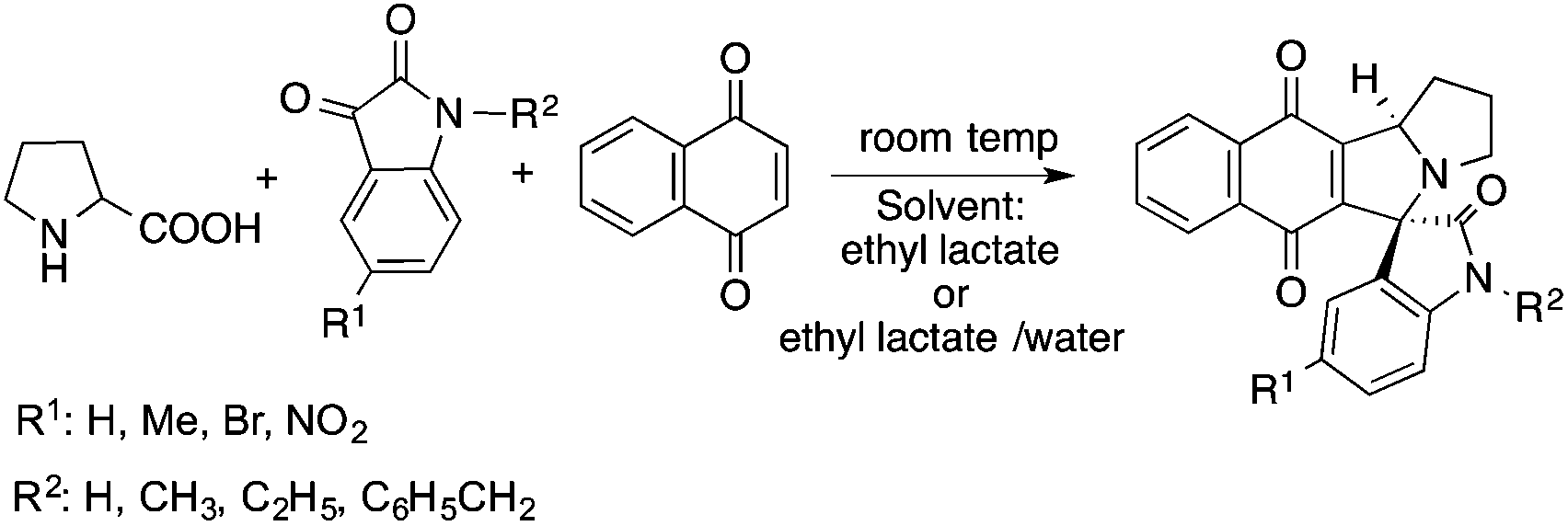
Lactic acid and its esters are cheap and readily available bio-based solvents and can be industrially produced on a large scale by fermentation. Lactic acid has been used for the first time to promote organic reactions, such as the three-component reactions of styrenes, formaldehyde and an active phenol derivative (Scheme 21). The three-component reactions of diethyl acetylenedicarboxylate, anilines and aromatic aldehydes, aniline-catalyzed condensations between salicylaldehydes and diethyl acetylenedicarboxylate, and the synthesis of substituted quinolines through the Friedländer reaction between 2′-aminoacetophenone and 1,3-dicarbonyl compounds resulting in excellent yields in several cases have also been demonstrated.88 Ligand-free Pd-catalyzed Suzuki–Miyaura cross coupling reactions have been performed in a tunable solvent system comprising water and ethyl lactate (Scheme 22). The protocol tolerated the reactions of several substituted aryl halides and aryl boric acids under mild conditions, representing a low environmental impact and a non-toxic methodology. The chemoselectivity of the reaction was shown for the synthesis of bromo-substituted biaryls, which could have significant synthetic relevance.89 1,4-Dihydropyridines, representing an important class of N-heterocyclic scaffolds, could act as ligands for biological receptors in the field of medicine. A highly efficient, environmentally-friendly one-pot protocol was reported for their manufacture, including biologically active substrates in ethyl lactate under thermal and light mediated conditions (Scheme 23). Although a good isolated yield (75%) was obtained in ethyl lactate, it can be increased up to 92% by addition of water as a co-solvent.90 The heterocyclic spirooxindole skeleton is of the utmost importance in organic and medicinal chemistry because of its potency across a wide spectrum of biological activities: these compounds have antifungal, anti-microbacterial, anti-tumor, anti-oxidant, anti-microbial, and anti-malarial effects, and can also serve as synthons for naturally occurring alkaloids and pharmaceutically important drug molecules. Excellent isolated yields (>88%) were achieved for spiro[benzo[f]pyrrolo[2,1-a]isoindole-5,30-indoline]-20,6,11-trions in both ethyl lactate and ethyl lactate water mixtures at room temperature over 1 h (Scheme 24).91
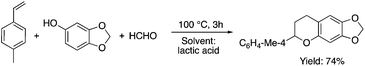 |
| | Scheme 21 Three component reaction in lactic acid. | |
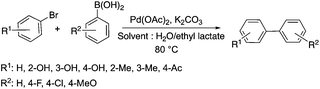 |
| | Scheme 22 Cross-coupling reactions in a water/ethyl lactate mixture. | |
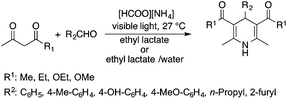 |
| | Scheme 23 Synthesis of 1,4-dihydropyridines. | |
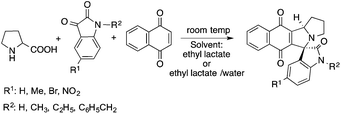 |
| | Scheme 24 Synthesis of spiro[benzo[f]pyrrolo[2,1-a]isoindole-5,30-indoline]-20,6,11-trions. | |
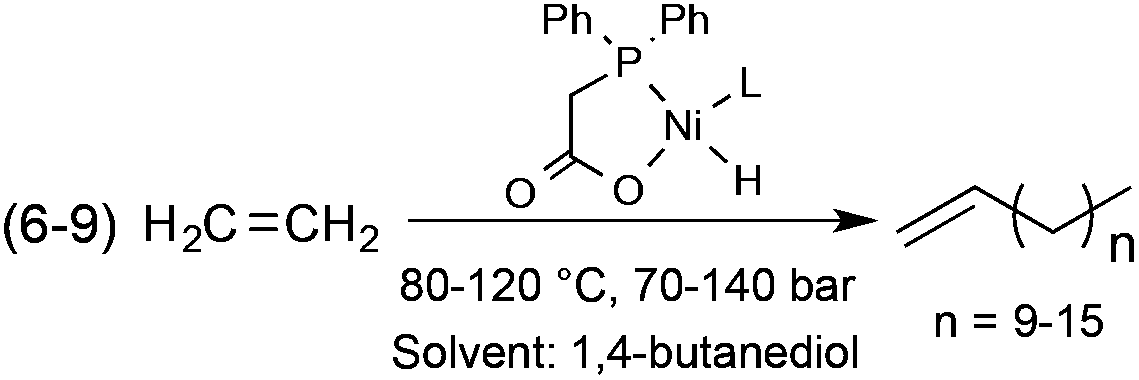
Alcohols represent a family of easily tuneable solvents via the variation in the number of polar and protic hydroxyl functional group(s) on the apolar and non-protic alkyl group(s). This property can be spectacularly demonstrated in the Shell Higher Olefin Process (SHOP), which manufactures higher α-olefins from ethylene via controlled oligomerization (Scheme 25). While 1,4-butanediol serves the Ni-based catalyst containing phase, where the oligomerization takes place, the limited solubility of α-olefins results in facile product separation.92 It is important to note that the SHOP process was the first realization of an industrial biphasic catalytic system.
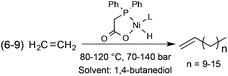 |
| | Scheme 25 Oligomerization of ethylene (the SHOP-process). | |
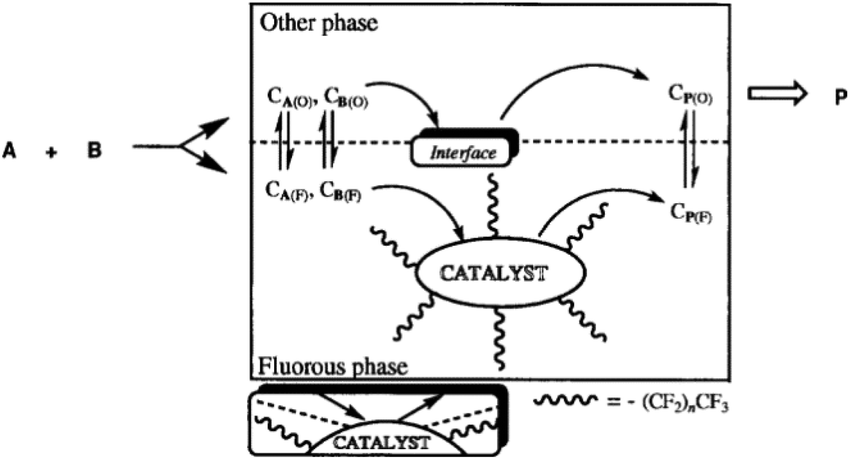
Fluorous solvents such as perfluoronated alkanes, dialkylethers, and trialkylamines are extremely non-polar reaction media. Miscibility of these compounds, even with common organic solvents such as toluene, acetone, tetrahydrofuran, and alcohols is low at room temperature. Consequently, these liquids could easily form biphase systems. The term “fluorous” was first introduced by Horváth in a seminal paper published in 1994,93 as an analogue to the term aqueous, to emphasize the fact that one of the phases of the biphase system is richer in fluorocarbons than the other. Fluorous biphase systems (FBS) can be used in a huge variety of chemical transformations to immobilize catalysts. A typical fluorous catalyst system consists of a fluorous phase containing a preferentially fluorous soluble catalyst and a second product phase, which may be made up of any organic or non-organic solvent with limited solubility in the fluorous phase. The original image of the fluorous concept is shown in Fig. 2. The most effective fluorocarbon moieties are linear or branched perfluoroalkyl chains with a high number of carbon atoms that may contain other heteroatoms. The great potential of the fluorous-liquid/liquid concept for catalyst recovery was first demonstrated for the hydroformylation of olefins. The fluorous soluble P[CH2CH2(CF2)5CF3]3 modified Rh catalyst system is excellent for the hydroformylation of decene-1 at 100 °C under 11 bar syngas pressure (CO:H2 = 1:1) in CF3C6F11 and the aldehydes can be easily separated from the fluorous catalyst, which was subsequently tested under semi-continuous and continuous conditions. The first investigation focused on the hydroformylation of decene-1 with the catalyst Rh/P[(CH2)2(CF2)5CF3]3. For nine consecutive reaction/separation cycles, a total turnover of more than 35000 was achieved with a loss of 1.18 ppm of Rh per mol of undecanals. The same system was then tested under continuous hydroformylation of ethylene using FC-70, which allows the continuous removal of propanal at 110 °C. The long-term stability of the catalyst was found to be better than that of the classical Rh/PPh3 catalyst, representing the first system which could be used for the hydroformylation of both low- and high-molecular weight olefins providing facile catalyst separation for both low and high molecular weight aldehydes.94 After the successful introduction of “fluorous catalysis” several transition metal complexes bearing fluorous ligands were synthesized in collaboration with Gladysz including HRh(CO){P[CH2CH2(CF2)5CF3]3}3, the fluorous Wilkinson's catalyst ClRh{P[CH2CH2(CF2)5CF3]3}3,95 the Ir-based fluorous Vaska complex ClIr(CO){P[CH2CH2(CF2)5CF3]3}2,96 and its rhodium analogue,97 binuclear [Ru(μ-O2CCH3)(CO)2{P[CH2CH2(CF2)5CF3]}3]2,98 fluorous porphyrins with Co, Fe, and Mn metal centers,99 and fluorous cyclopentadienyl complexes with Mn, Re, Fe, and Co metal centers.100 Since 1994, fluorous chemistry has become a well-established, individual area of chemistry, providing a complementary approach to other biphasic systems. In the case of need, the catalytic decomposition of perfluoroalkanes is available.101 Overviews in the recent developments in fluorous chemistry have been published presenting excellent examples of its application.102–105
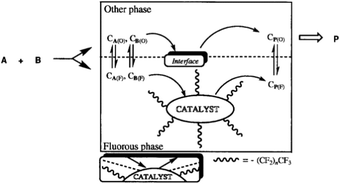 |
| | Fig. 2 Original drawing of the fluorous biphase concept for the catalytic conversion of substrates A and B to product P. Adapted from ref. 93. Copyright of The American Association for the Advancement of Science (1994). | |
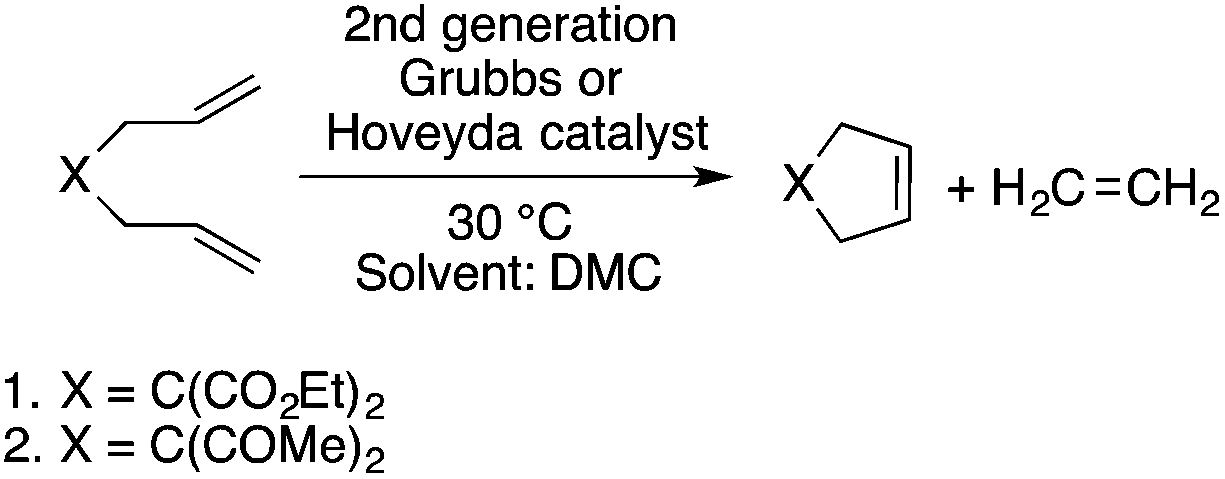
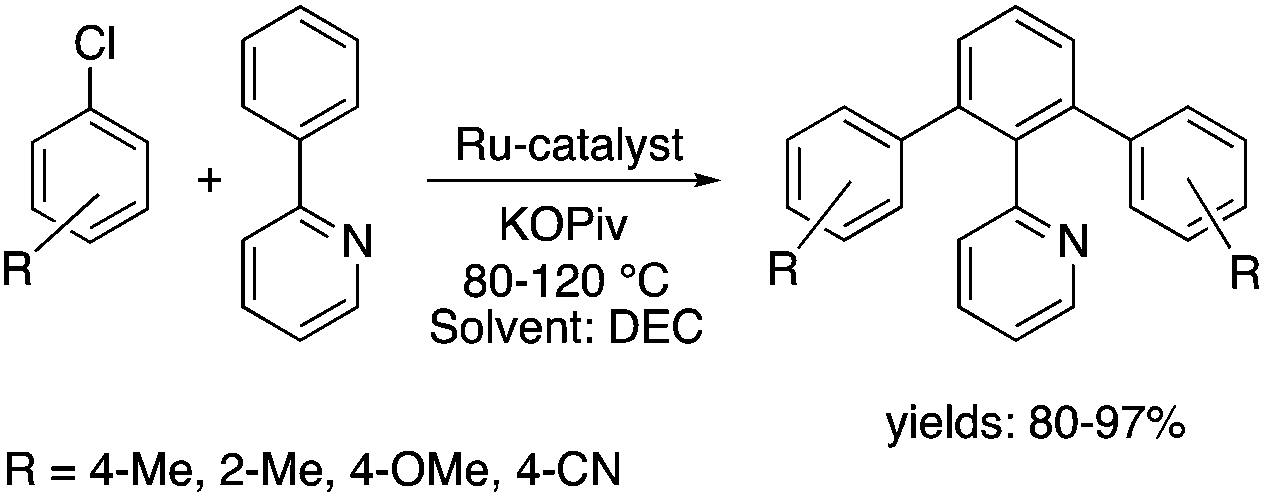
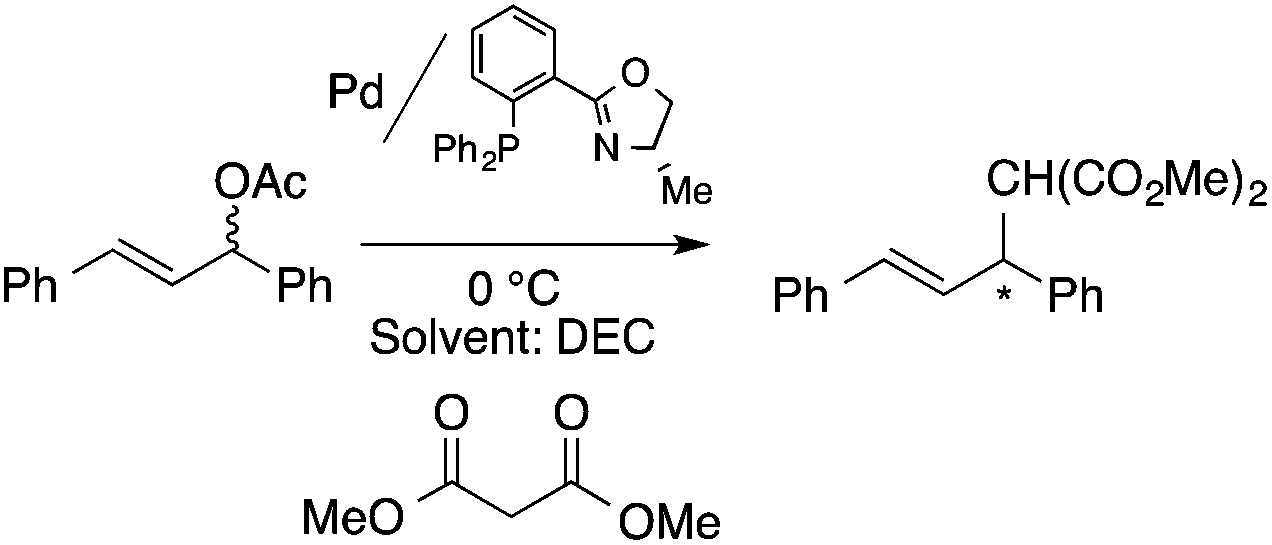
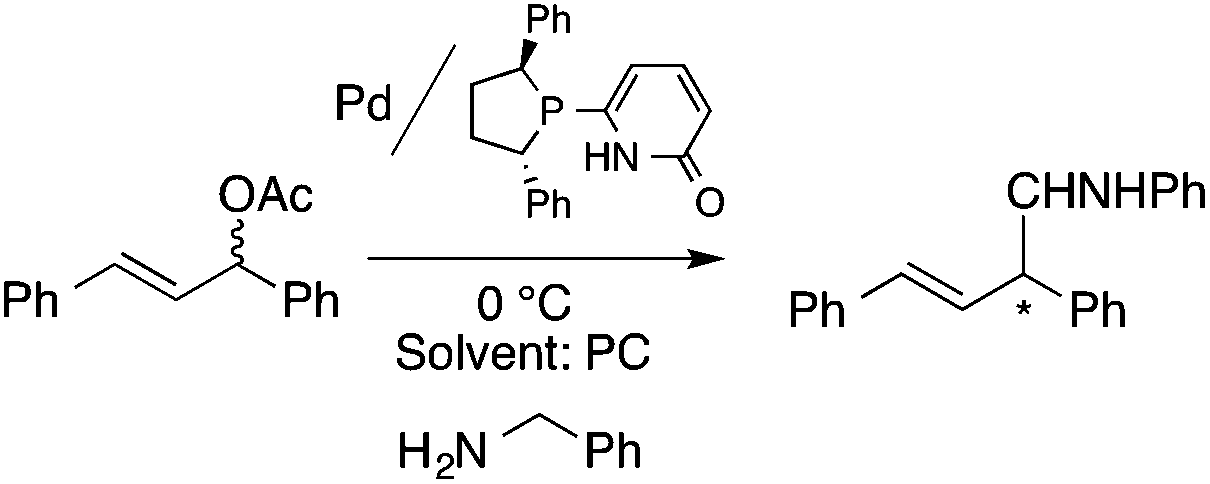
Organic carbonates, such as dimethyl carbonate (DMC), are excellent solvents for several catalytic transformations. Fischmeister demonstrated that DMC could be used as an environmentally-friendly alternative to conventional organic solvents (such as dichloromethane, benzene, toluene, chlorobenzene) used for the metathesis reaction. When 2nd generation Grubbs or Hoveyda catalyst (Scheme 26) mediated ring-closing metathesis (RCM) reactions of functionalized dienes (Scheme 22) were compared in CH2Cl2 and DMC, almost identical reaction profiles and isolated yields were achieved. The cross-metathesis of decene-1 and methyl acrylate also proceeded equally in DMC, yielding H17C7CHCHCO2Me. It should be noted that sterically-hindered disubstituted olefins have been subjected to RCM, and outstanding product yields were achieved in DMC, due to the increased (90 °C) reaction temperature. As a subsequent development, the Hoveyda catalyst was immobilized on a silica or zirconia surface and tested in model RCM reactions and the re-use of the catalyst was demonstrated over several consecutive runs.106,107 The same group utilized diethyl carbonate (DEC) as a replacement for conventional N-methyl-2-pyrrolidone in the direct functionalization of arene sp2 C–H bonds by aryl halides. The catalytic system based on [RuCl2(p-cymene)]2/KOPiv was found to be active in the selective arylation reaction of phenylpyridine at 80–120 °C depending on the type of chloroaromatic substrate used (Scheme 27).108 DEC was also successfully utilized as a reaction medium for the palladium-catalyzed asymmetric allylic alkylation of 1,3-diphenyl-3-acetoxyprop-1-ene with dimethyl malonate (in comparison with CH2Cl2 as a conventional solvent) by Börner and co-workers (Scheme 28).109 Using Me-phox as a ligand, no change in yield and enantiomeric excess was detected. The same group demonstrated the asymmetric allylic amination of 1,3-diphenyl-3-acetoxyprop-1-ene with benzyl amine in propylene carbonate (PC) at 0 °C (Scheme 29). Similarly, methylene chloride (CH2Cl2) can be replaced by an eco-friendly solvent, resulting in a superior yield (91%) and enantioselectivity (57%).109
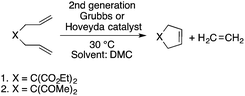 |
| | Scheme 26 Ring-closing metathesis in DMC. | |
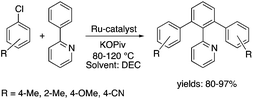 |
| | Scheme 27 Ru-Catalyzed C–H bond functionalization in DEC. | |
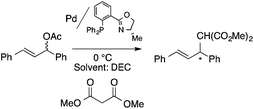 |
| | Scheme 28 Pd-Catalyzed asymmetric alkylation in DEC. | |
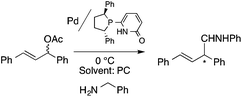 |
| | Scheme 29 Pd-Catalyzed asymmetric alkylation using a chiral phosphine catalyst. | |
Supercritical carbon dioxide (ScCO2) should also be noticed as one of the frequently utilized alternative reaction media for chemical synthesis. It has a critical temperature Tc = 31.0 °C, and critical pressure pc = 73.8 bar with a respective density of dc = 0.446 g mL−1. It can be used as a low cost and readily available solvent either by itself or in combination with other solvents such as hydrocarbons, ionic liquids, or fluorous solvents. Due to its physical properties, no product contamination has to be considered in case of its accidental release into the immediate environment or even in final product separation. The latter allows the specific utilization of scCO2 in the food and nutrient industry, in which the extraction of caffeine from coffee bean represents a long-term industrial application.110 Over the last few decades, the successful introduction of scCO2 as a solvent in chemical processes has been widely demonstrated from the laboratory to industrial scale involving dry cleaning processes and metal degreasing,111 synthesis and catalysis,112,113 and materials science.114 Furthermore, in the pharmaceutical industry, where residual solvent traces are crucial and could result in serious health issues, the separation of enantiomers using scCO2 could also offer a benign alternative to conventional resolution techniques.115 For example, the separation of racemic ibuprofen that frequently uses model compounds by applying in situ diastereomeric salt formation between racemic ibuprofen and (R)-(1)-phenylethylamine, followed by its precipitation via a gas antisolvent method, resulted in up to 80% ee of (S)-ibuprofen.116,117
Use of renewable feedstocks
Currently, more than 90% of the energy needs of mankind, as well as the production of carbon-based chemicals, are produced from resources originating from fossils. The exact date of the depletion of crude oil, the main carbon resource of the chemical industry, is very hard to estimate and has been the subject of active debate. In 2014, the estimated reserves of crude oil were calculated to be 1700.2 billion barrels, which is ca. 600 billion barrels more than the amount calculated 20 years ago. This value accounts for more than 50% of the reserves that were proven to be in the Middle East at the end of 2015.118
Despite this increase in the amount of crude oil, global efforts to reduce CO2 emission according to the Kyoto Protocol, and the possible production of carbon-neutral end products have directed researchers’ attention towards identifying and developing innovative solutions for the replacement of fossil-based resources by renewable alternatives. The contribution of hydropower, wind, and photovoltaic energy119,120 to our overall energy demand and utilization of carbon dioxide have emerged over ca. 30 years. For the chemical industry, the utilization of biomass as a readily and globally available feedstock has come into focus and has been proposed as a most promising alternative to the use of carbon resources.26,121–128 Considering the increasing population of the Earth, the selection and consumption of appropriate resources have become controversial issues due to the dramatic increase in the use of edible resources. Thus, the production of energy and carbon-based chemicals from biomass cannot be seen to compete either directly or indirectly with food or feed production. On the other hand, this issue involves the availability or even an increase in the demand of land use, which gives the amount of soil (given in hectares) required to produce 1 ton of primary agricultural products. Obviously, the proper solution could be the vaporization of lignocellulosic rich waste streams,129 because of their very similar composition.130 A series of potential candidates obtained from carbohydrates, such as 5-HMF from cellulose, FAL from hemicellulose, as well as LA, 3-hydroxypropionic acid, furandicarboxylic and fumaric acids etc. was collected by Werpy and Petersen in 2004,131 and updated by Bozell in 2010.124 In 2008, GVL was also additionally proposed as a safe platform chemical by Horváth.30
Although biomass could be an ideal alternative, its sustainable utilization primarily depends on whether we have enough resources to cover the increasing needs of fuels and chemicals. To improve the evaluation of biomass utilization from a sustainability point of view, Horváth and co-workers proposed a new definition of sustainability using two fundamental evolutionary principles and mechanisms of Nature as follows (i) “resources including energy should be used at a rate at which they can be replaced naturally” and (ii) “the generation of waste cannot be faster than the rate of their remediation”.132 It should be emphasized that this novel approach addresses the changes in time (or the kinetics required to reach sustainable equilibria) and offered the opportunity to calculate the upper limits of sustainability per capita (or the parameters of sustainable equilibria). To calculate the sustainability of fuels (gasoline and jet fuels) and basic chemicals (ethylene, propylene, benzene, etc.) a new metric, the ethanol equivalent (EE) was introduced and used to calculate the land requirements of the biomass-based production of the corresponding products.132 As a subsequent development of this theory, three indicators, i.e. the sustainability value of the resource replacement (SVrep), the sustainability value of the fate of the waste (SVwaste), and the sustainability indicator (SUSind) were defined as the sustainability metrics of carbon-based chemicals.133 This methodology introduced by Horváth allows a calculation and comparison of the sustainability indexes of materials and processes relating to the chemical industry.
The first approach of the utilization of a complex biomass feed could be the “biorefinery concept”, which involves the integration of conversion processes to produce fuels, power, heat, and value-added chemicals by using up all of the carbon atoms of the resource. However, due to the complexity of lignocellulosic material, this has only been partially realized by transformation of its carbohydrate content. As a novel concept in this huge research field, Horváth achieved the multistep catalytic conversion of carbohydrates to alkanes and alkenes involving (i) dehydration of sucrose to LA via 5-HMF in the presence of sulfuric acid at 140 °C; (ii) aqueous reduction of LA to obtain GVL using either TPPTS or a PBu3 modified Ru catalyst under a 70 bar pressure of H2 at 135–140 °C for 12 h and 8 h, respectively; (iii) hydrogenation of LA to GVL and 1,4-PDO in the presence of the catalyst [(η6-C6Me6)Ru(bpy)(H2O)][SO4] and HCOONa under aqueous conditions at 70 °C for 18 h; (iv) hydrogenation of GVL to 2Me-THF using a Ru/PBu3/NH4PF6 catalyst system under a 75 bar pressure of H2 at 200 °C for 20 h; and (v) reduction of 2Me-THF to alkanes and alkenes by applying a Pt(acac)3/CF3SO3H catalyst at a 75 bar pressure of H2 at 150 °C for 15 h. As a result, ca. 53% C4-alkanes, 19% C5-alkanes, 7% C8-alkanes, and 6% C9-alkanes were identified by NMR.34 The multistep conversion is depicted in Scheme 30.
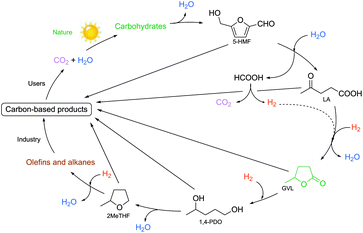 |
| | Scheme 30 Multistep conversion of the carbohydrate content of biomass wastes. Modified with permission from ref. 34. Copyright of Springer (2008). | |
Recently, several production routes and successful applications of platform chemicals were demonstrated and numerous reviews have been published summarizing these results and innovations.26,121,123,134–137 Thus, we provide selected examples regarding platform chemicals that are presented in Scheme 26.
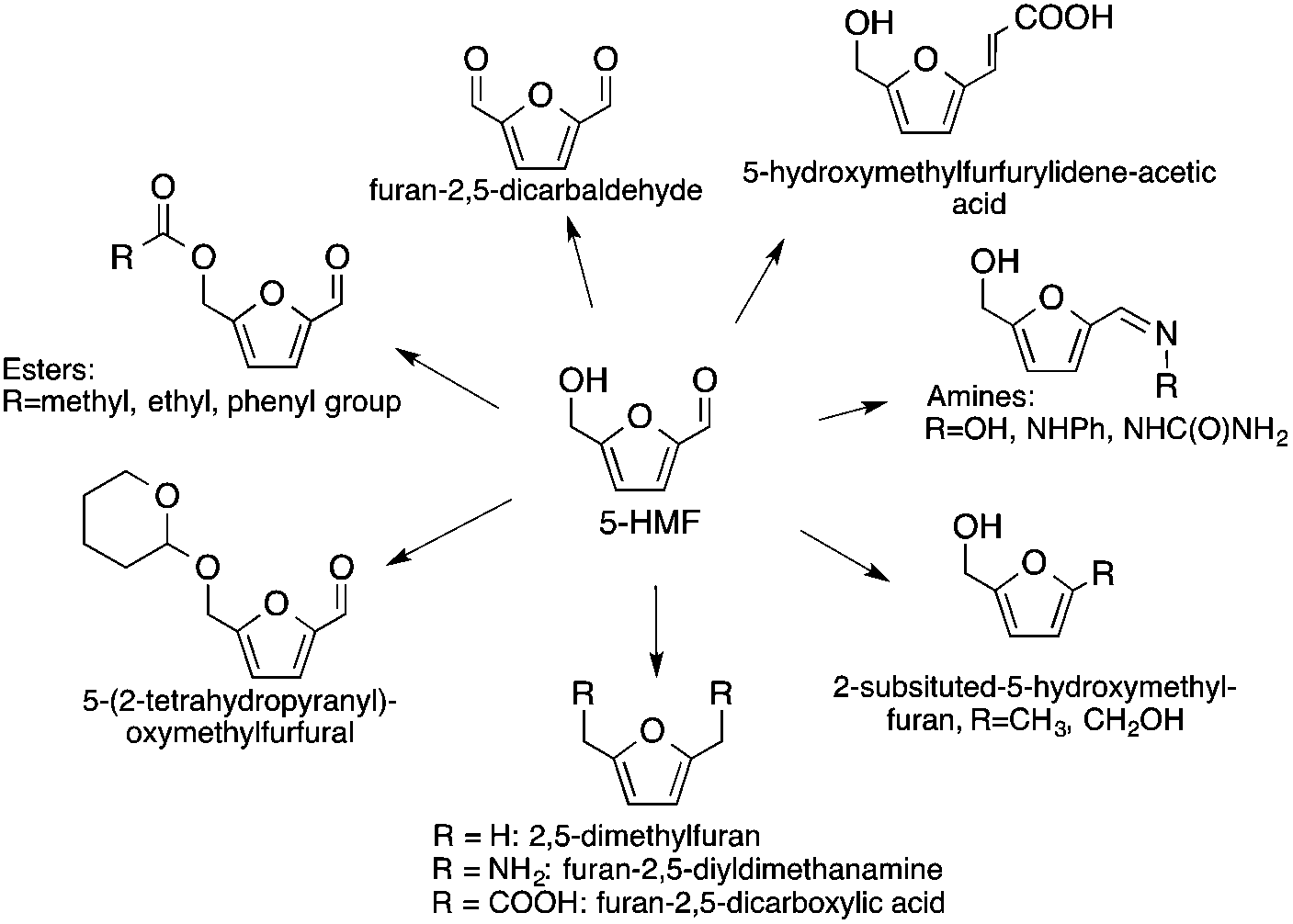
5-Hydroxymethylfurfural has been identified as a promising C6-building block serving as a renewable alternative for polymers, pharmaceuticals, agrochemicals, flavors and fragrances, macro- and heterocycles, and natural products (Scheme 31).26,135 It has also been proposed as a precursor for fuel components. Furan-2,5-dicarboxylic acid, that can be obtained by its oxidation, was also highlighted by the US Department of Energy as being a key bio-derived basic chemical that represents a starting point for the synthesis of several molecules, i.e. succinic acid, 2,5-furandicarboxylic acid dichloride, and 2,5-furandicarboxylic acid dimethyl ester. It is also a potential substitute for terephthalic or isophthalic acids in the manufacture of polyamides, polyesters and polyurethanes.126 It can also be applied as a cross-linking agent for poly(vinyl alcohol) in battery separators and as a component for foundry sand binders. Selective hydrogenation of 5-HMF leads to the formation of either 2,5-bis(hydroxymethyl)furan or 2,5-bis(hydroxymethyl)-tetrahydrofuran, which could be used in the manufacture of polyurethane foams or polyesters. The hydrogenolysis of 5-HMF results in the formation of 2,5-dimethylfuran, which has been targeted as a transportation fuel.
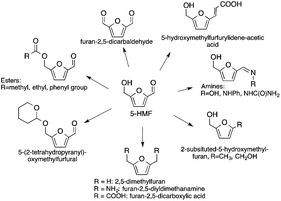 |
| | Scheme 31 Chemicals derived from 5-HMF. | |
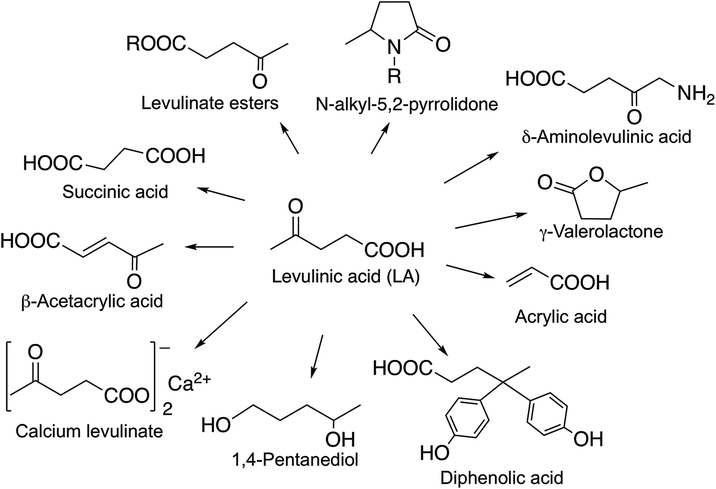
Levulinic acid has been identified as a non-toxic multipurpose C5 initial platform chemical26 for the synthesis of functionalized C3–C6 chemicals (Scheme 32). Its various ester derivatives may be used as blend components in fuels as well as also having significant potential as renewable replacements for kerosene as a home heating oil and as a fuel for the direct firing of gas turbines for electrical generation.138 A Bisphenol-A analogue, diphenolic acid (4,4-bis-(4′-hydroxyphenyl)pentanoic acid) derivative of LA prepared by the reaction of LA with two equivalents of phenol could be a renewable substitute for the synthesis of polycarbonates, epoxy resins, and polyarylates.136 LA also has numerous other uses, including applications in lubricants, adhesives and paints, while its sodium salt is used as an anti-freezing agent, and calcium levulinate can be applied orally or intravenously as a calcium ion carrier.139 δ-Aminolevulinic acid is a natural active ingredient in a series of environmentally benign, highly selective, broad-spectrum herbicides that can be used as insecticides.140,141
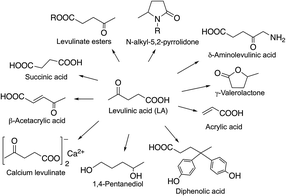 |
| | Scheme 32 Chemicals derived from LA. | |
Gamma-valerolactone, a naturally occurring C5-cyclic ester in fruits and a frequently used food additive, was discussed above.
2-Methyltetrahydrofuran has been listed as a renewable fuel additive and a component of the alternative P-fuels, which can be used alone or may be mixed in any proportions with petroleum.142 2-MeTHF can be added in amount of up to 30 vol% with petroleum with no adverse effects on the performance, and engine modifications are not required. Vehicle tailpipe and evaporative emissions tests carried out by the Environmental Protection Agency revealed that the P-Series formulations reduced the ozone forming potential of the fuels and resulted in reduced emissions of non-methane hydrocarbons and total hydrocarbons: ca. 30% of those formed from the use of commercial reformulated gasoline. It has been also estimated that when biomass-based 2-MeTHF and EtOH are utilized, the full fuel-cycle greenhouse gas emission levels could be between 45 and 50% below the levels seen using reformulated gasoline. 2Me-THF could also be used as a renewable solvent, however, its ability to form peroxides has to be noted. Horváth showed that the peroxide number of 2Me-THF can reach 716 over 12 days, which actually means that it must be handled as a dangerous material.31
Catalysis
The ninth amendment of Anastas and Warner to the reach goals of Green Chemistry, describes the use of catalytic reagents, which are superior to stoichiometric ones. By definition, a catalyst is a substance that accelerates a chemical reaction without being consumed; therefore, it can be recovered at the end of the process. Selective acceleration of a chemical process allows a reduction in the amount of byproducts, thus, chemical catalysis is a form of industrial metabolism.
Transition metal-assisted homogeneous catalysis offers a very efficient tool for the transformation of a huge variety of organic substrates over their heterogeneous counterparts. Their tunable and therefore significantly higher chemoselectivity, regioselectivity, enantioselectivity and milder operating conditions have to be emphasized. However, the catalyst separation and even recycling have been a great challenge and the subjects of recent research. Over the last few decades, several attractive solutions have been developed. In order to overcome the separation difficulties of homogeneous catalysts, several attractive solutions have been developed such as biphasic catalysis143 or the heterogenization of homogeneous catalysts.144 Among them, an elegant solution is the immobilization of homogeneous catalysts dissolved in a liquid phase on a solid carrier. The first stationary liquid phase catalyst (SLP, see Fig. 3), applied to the hydrogenation of arenes (benzene, toluene, o-xylene, and naphthalene), was reported by Horváth in 1991. The BF3·H2O-Pt catalyst was immobilized on CPG-240 or on clay.145 A catalyst prepared from BF3·H2O and Pt(Cl)2(CH3CN)2 was active in the reduction of a mixture of benzene and its alkyl-derivatives, such as toluene, o-xylene, and 1,2,4-trimethylbenzene under a 27.6 bar pressure of H2 at 22 °C. It was shown that the catalyst operated in the aqueous acid phase and that the solid did not chemically participate in the reaction. Recently, Horváth demonstrated the immobilization of a Shvo-type catalyst on silica using both covalent anchoring and sol–gel methods to prepare a recyclable transfer hydrogenation catalyst (Scheme 33).146
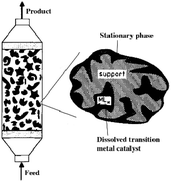 |
| | Fig. 3 A diagram showing supported liquid phase catalysis. Modified with permission from ref. 145. Copyright of John Wiley and Sons (1991). | |
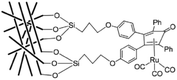 |
| | Scheme 33 A heterogenized Shvo-type catalyst. Modified with permission from ref. 146. Copyright of Elsevier (2017). | |
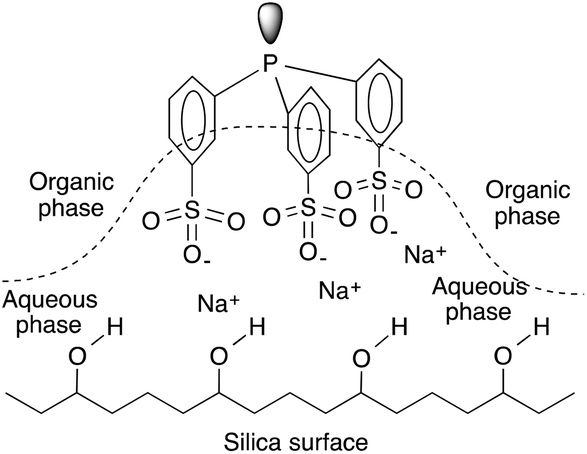
The Ruhrchemie/Rhône–Poulenc reaction offers the excellent recycling of a HRh(CO)(TPPTS)3 catalyst dissolved in water. The process is based on the significant differences between the high and low solubility of propylene and C4-aldehydes, respectively, in the aqueous phase. However, it is an intrinsic characteristic of the hydroformylation reaction that the reaction rate declines upon an increase in the chain length of the olefin under similar conditions.147 In the case of aqueous systems, it was suggested that the reaction under biphasic conditions with water-insoluble higher olefins was mass transfer limited.148 This fact is attributed to the decreasing solubility of higher olefins in the aqueous catalyst solution, which correspondingly leads to low olefin concentrations and thus reduced reaction rates.149 One of the most intriguing approaches for overcoming the solubility limitation is the immobilization of an aqueous solution of HRh(CO)(TPPTS)3 onto a high surface area hydrophilic silica support.150 It was later shown by Horváth using in situ infrared (IR) spectroscopy that the SAP (Supported Aqueous Phase) catalyst lost water at higher temperatures, significantly lowering the amount of water available on the support as well as that of the hydrophilic support holding the water-soluble phosphines by hydrogen bonding of the hydrated sodium-sulphonate groups to the surface and that the actual catalytic reaction occurs in the organic phase (Fig. 4).
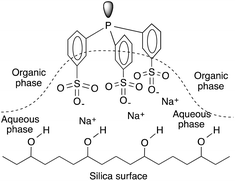 |
| | Fig. 4 Schematic representation of the immobilization of TPPTS on silica. Modified with permission from ref. 149. Copyright of Springer (1990). | |
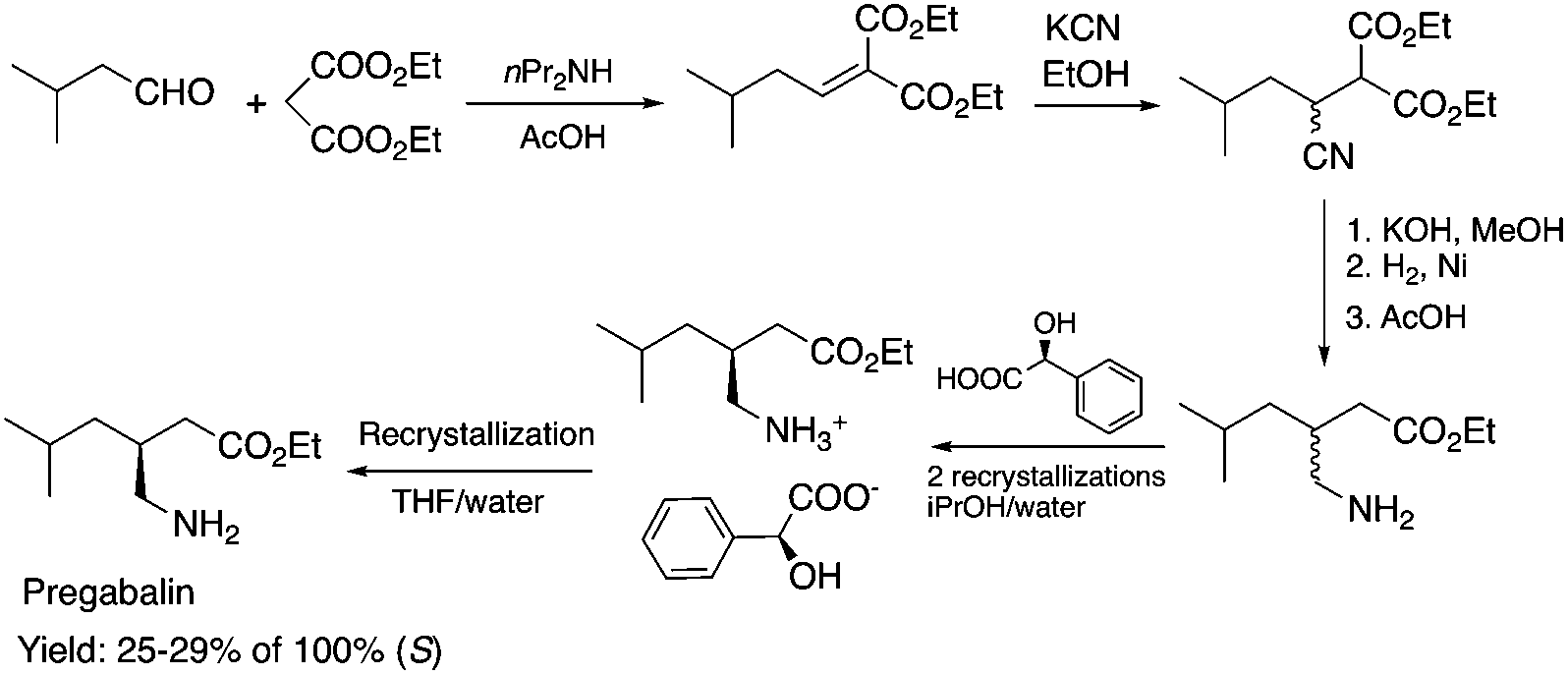
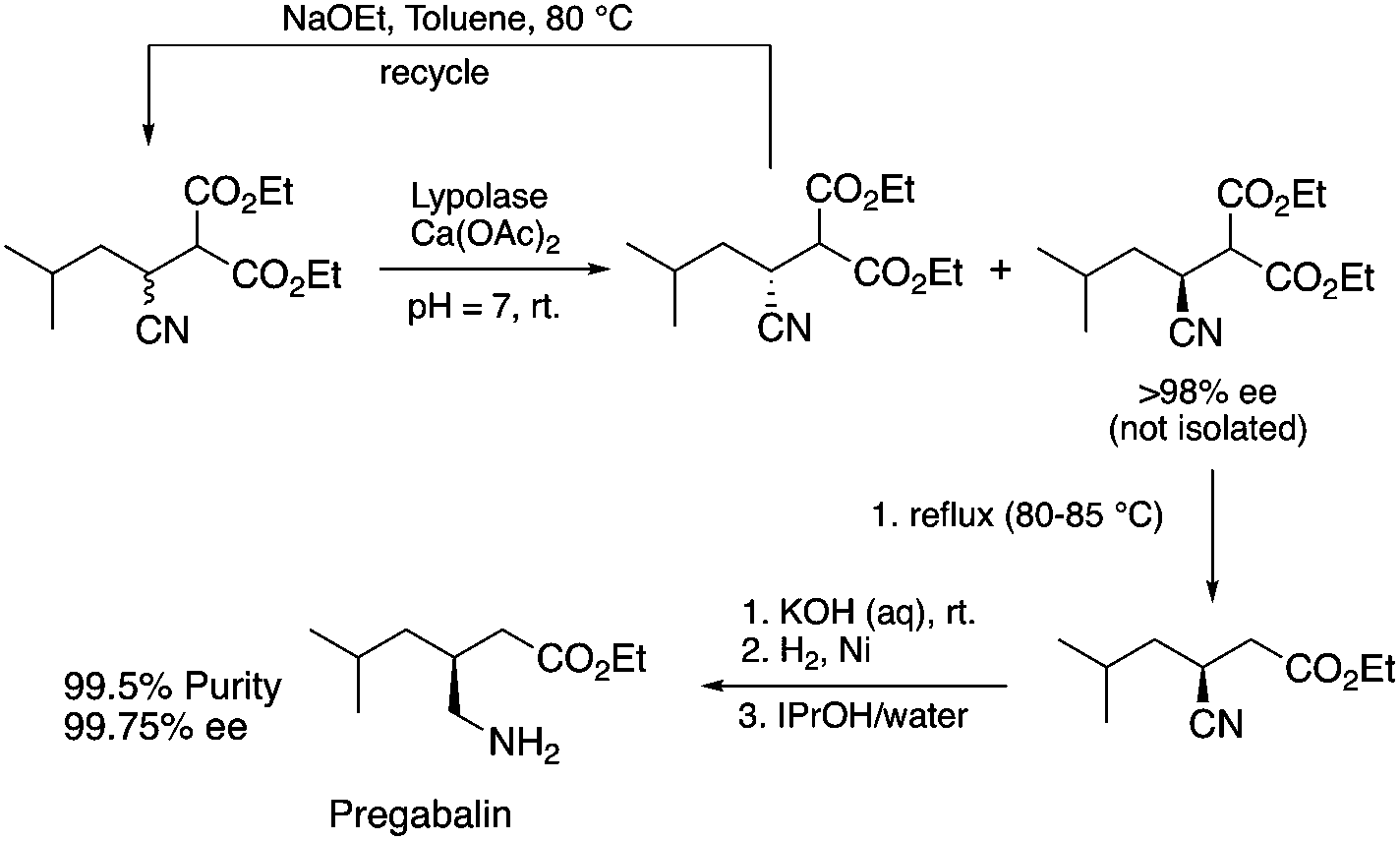
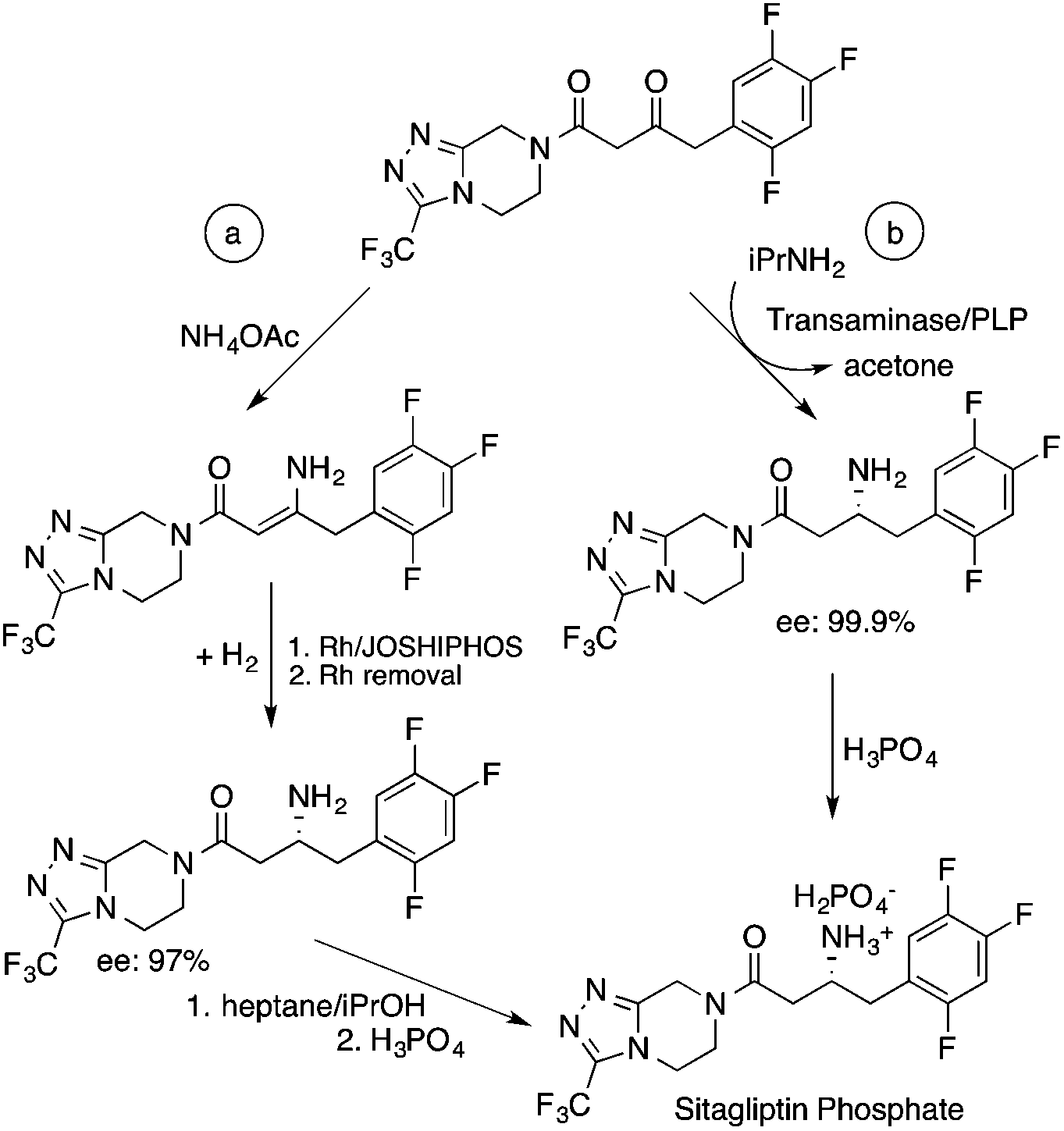
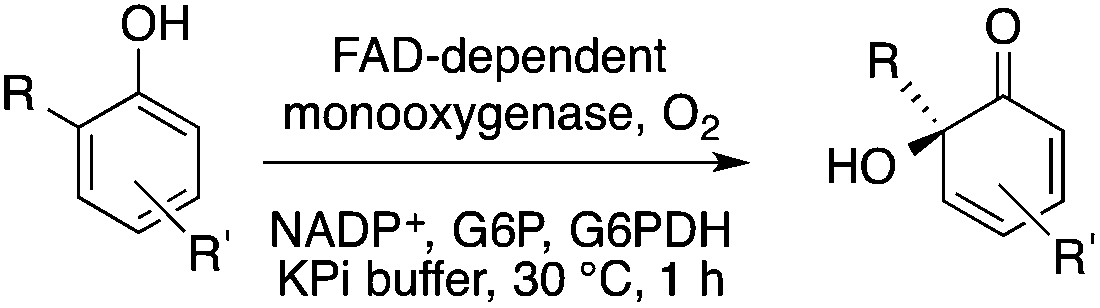
Biocatalysis offers an excellent tool for the manufacturing of primarily biologically active compounds that usually have one or more chiral center. This was recognized by pharmaceutical companies that have integrated this green and sustainable methodology with traditional medicinal chemistry. There is no doubt that the modern pharmaceutical industry cannot be operated without biotechnology. While a large number of enzymes, the catalysts of biochemical transformations, have been known for a long time, protein and gene engineering has enabled the optimization or improvement of the efficiency of existing biocatalysts followed by their implementation into new biocatalytic transformations, which were previously unknown in Nature. Because of this possibility, applications, and recent achievements in the field of biocatalytic transformations have been widely reviewed.151–156 Herein, we provide only selected examples for the benefit of biocatalytic conversion. In the pharmaceutical industry, the application of biocatalysis could reduce the number of synthesis steps required in the synthesis of products, as well as resulting in higher purities and product yields. For example, Pfizer have developed an efficient route for the synthesis of (S)-3-(aminomethyl)-5-methylhexanoic acid, Pregabalin, which is an antiepileptic drug used for central nervous system disorders. The original commercial synthesis route starts with a Knoevenagel condensation, followed by cyanation that results in a racemic mixture, hydrolysis, decarboxylation, and hydrogenation. A classical chiral resolution in the presence of (S)-(+)-mandelic acid was then applied to form a diastereomeric salt, which was separated by recrystallization in iPrOH/water. The pure ingredient is isolated after subsequent recrystallization from THF/water (Scheme 34).157 Pfizer's eventual solution, based on the enzyme-catalyzed kinetic resolution hydrolysis of one of the esters of β-cyano-diester significantly reduced the amount of waste generated and increased the final yield of the target species (Scheme 35). This development resulted in the reduction of the E-factor of the process from 86 for the original process to 17 for the enzyme-based route. It has to be noted that the authors also reported a reduction in solvent use from 50 kg kg−1 product to 6.2 kg kg−1.158 The biocatalytic asymmetric synthesis of chiral amines that have opened is a very efficient and safe way to manufacture Sitagliptin, a treatment for type 2 diabetes, and can be highlighted as another excellent example of biocatalysis. Merck's process applies a transaminase enzyme for the amination of the prositagliptine diketone precursor in the presence of iPrNH2 (Scheme 36).159 The process was made more environmentally friendly and industrially viable by avoiding the Rh/JOSIPHOS-catalyzed high-pressure hydrogenation step to result in a ca. 12% increase in the overall yield and ca. 50% increase in the total productivity. It should also be noted that the engineered transaminases also enabled access to chiral amines that heretofore could not be synthesized either enzymatically or chemically to yield chiral trifluoromethyl substituted and chiral pyrrolidine products. Narayan very recently reported the site- and enantioselective oxidative dearomatization of phenols, a chemical transformation that rapidly builds up in molecular complexity from simple starting materials and cannot be accomplished with high selectivity using existing catalytic techniques (Scheme 37). Using enzymes from biosynthetic pathways, a method to produce a series of ortho-quinols from corresponding phenols was developed and additionally, the scalability and robustness of this novel multi-enzyme and chemo-enzymatic cascade method was demonstrated.160
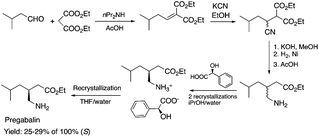 |
| | Scheme 34 Classical (first generation) synthesis of Pregabalin.157 | |
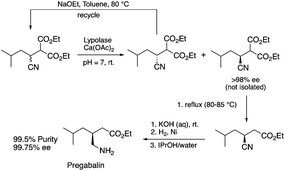 |
| | Scheme 35 Enzymatic synthesis of Pregabalin.158 | |
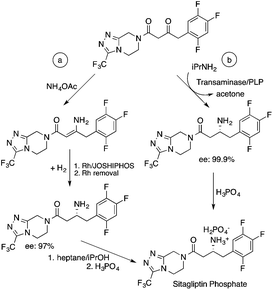 |
| | Scheme 36 Chemocatalytic (a) and biocatalytic (b) synthesis of Sitagliptin. Adapted with permission from ref. 159. Copyright of The American Association for the Advancement of Science (2010). | |
 |
| | Scheme 37 Oxidative dearomatization of phenols. A large variation of R and R′ is presented in ref. 160. | |
Heterogeneous catalysis represents the heart of industrial catalysis. More than 95% of industrial chemical conversions involves catalysts, of which more than 90% are heterogeneous systems. Their heavy use is due to their robustness and easy separation from the product(s). While homogeneous systems exhibit high activity and selectivity, as mentioned above, heterogeneous ones have less selectivity and activity, and therefore can operate at a higher pressure and temperature. There is a huge number of applications of heterogeneous catalysis from basic academic research to large-scale industry. Their importance has been highlighted in recent reviews,161–163 so therefore, it does not need to be reiterated herein. However, an industrially important heterogeneously catalyzed conversion of toluene to xylenes and benzene via a disproportionation reaction (STDP: Selective Toluene Disproportionation Process) should be noted as an excellent example of a “neat” heterogeneous transformation. The reaction is controlled by shape selectivity of the zeolite catalysts and yielded p-xylene in over 87%, with full conversion of toluene.164 Despite the outstanding importance of heterogeneous catalysis in the chemical industry, due to the use of usually heavy and even toxic metals as catalysts, for instance copper chromite for the reduction of furfural to furfuryl alcohol, in the long term, biocatalysis seems to be more viable.
In situ analysis
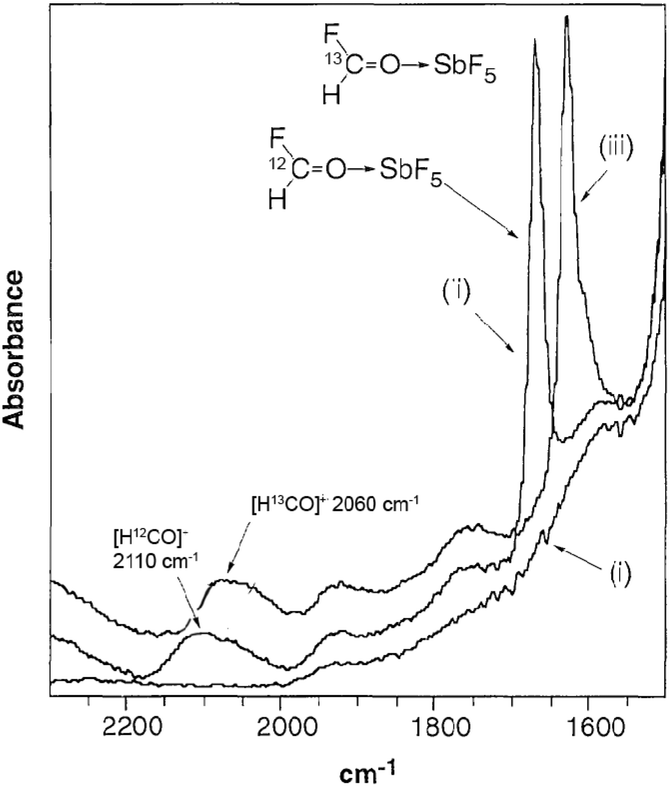
In situ analysis is one of the fundamental processes of life. For example, in the cells in our body the DNA is continuously attacked and damaged by external influences such as UV radiation and carcinogenic chemicals, including free radicals. During cell division, DNA is replicated, however, the cell recognizes and corrects errors that occur. This discovery was honoured by the Nobel Prize in Chemistry in 2015.165 Without these mechanisms, life on Earth could not be possible. Biological evolution has had millions of years to develop precise in situ analytical processes to monitor biochemical transformations in living organisms. In chemical research, however, gathering molecular level information from chemical transformations, which is fundamentally important for a better understanding of reaction mechanisms, and the design of more selective catalysts that reduce the formation of by-products at the molecular level, has still been a great challenge. The application of in situ spectroscopy plays a key role in discovering the mechanism of reactions, involving the characterization of intermediates in real time. On the other hand, the verification of the existence of a species that was proposed to explain a known reactivity or even lead to the discovery of new intermediates could lead to different mechanistic explanations being revealed for well-known reactions. For example, the formyl cation [HCO]+ has long been proposed as a key species in the chemistry of CO under acidic conditions. However, its presence in solution was first established by IR and NMR spectroscopy by Horváth at Exxon in collaboration with Prof. John A. Gladysz in 1997. High-pressure IR measurements on a reaction mixture of HF-SbF5 and CO established the presence of [HCO]+ (Fig. 5) and additionally, the study also revealed additional equilibrium reactions in the superacidic environment.166 A similar technique was used to monitor the carbonylation of CH4 with carbon monoxide in the superacids HF/SbF5 or HSO3F/SbF5. It was shown that the reaction leads to the exclusive and quantitative formation of the acylium ion ([CH3CO]+[SbF6]−) with the concomitant stoichiometric formation of SbF3.167
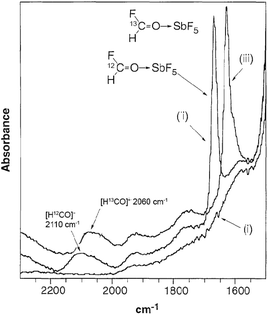 |
| | Fig. 5 IR spectra of (i) HF-SbF5, (ii) HF-SbF5 + 12CO, and (iii) HF-SbF5 + 13CO. Modified with permission from ref. 166. Copyright of The American Association for the Advancement of Science (1997). | |
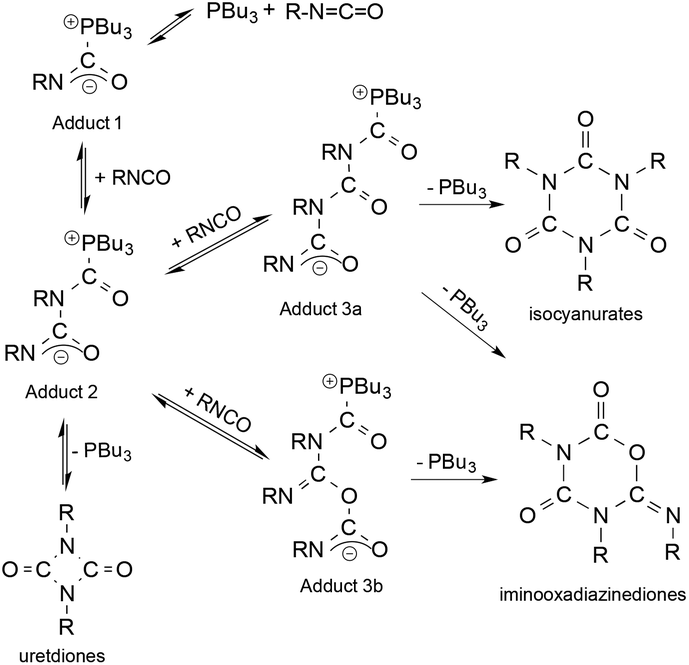
Another excellent study focused on the chemistry of polyurethanes. These chemicals, manufactured from aliphatic isocyanates, represent an important class of high-performance materials. Accordingly, their efficient synthesis that takes place via catalytic processes is of utmost importance. Horváth and Richter investigated the mechanism of the tri-n-butylphosphine-catalyzed cyclooligomerization reactions of alkyl isocyanates (Scheme 38) and structurally characterized one of the key catalytic intermediates, Adduct 2, for the first time. The in situ IR and NMR measurements established the equilibrium formation of Adduct 1, which readily reacts with another isocyanate to yield Adduct 2. It can undergo a ring closure reaction accompanied by the elimination of the catalyst PBu3 or act as an O- or N-nucleophile resulting in the formation of Adduct 3 and 4, respectively. While Adduct 3 was proposed to be responsible for the formation of isocyanurates, the ring closure of Adduct 4 resulted in the formation of iminooxadiazinediones.168 This example can also be considered as an atom economic transformation under “neat” conditions.
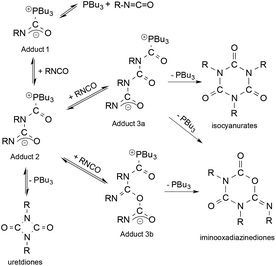 |
| | Scheme 38 Proposed mechanism of the tri-n-butylphosphine-catalyzed oligomerization of isocyanates. Modified with permission from ref. 168. Copyright of Wiley-VCH (2005). | |
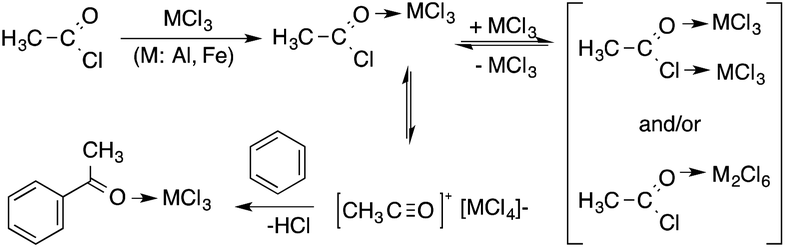
Pioneering in situ analysis of the Friedel–Crafts acylation of aromatic compounds, a reaction that has been of utmost importance to the pharmaceutical industry, showed that no differences between reaction mechanisms could be observed, when conventional and toxic 1,2-dichloroethane was replaced by non-volatile 1-methyl-3-butylimidazolium chloride. The analysis established the presence of the key intermediates of the reaction, i.e. the acetylium ion [CH3CO]+[MCl4]−.169 The proposed mechanism is depicted in Scheme 39. In addition, it was revealed that replacing the hazardous solvent with an environmentally benign one had no influence on the reaction mechanism.
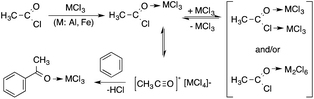 |
| | Scheme 39 Proposed mechanism of the Friedel–Crafts acylation of benzene. Adapted with permission from ref. 169. Copyright of the Royal Society of Chemistry (2001). | |
The in situ IR and NMR investigation of the mechanism of the pyridine modified cobalt-catalyzed hydromethoxy-carbonylation of 1,3-butadiene to methyl-3-pentenoate revealed that the initial step of the reaction sequence is the disproportionation of Co2(CO)8 to a [CoPy6][Co(CO)4]2 salt, followed by its reaction with methanol to form acidic HCo(CO)4. Because the equilibrium between HCo(CO)4 and [MeOH2][Co(CO)4] in MeOH is shifted towards the direction of the latter, the addition of [Co(CO)4]− to protonated butadiene results in the formation of the alkyl complex CH3CHCHCH2Co(CO)4. This species can undergo reversible decarbonylation to form allylcobalt-tricarbonyl ((η3-C4H7)Co(CO)3) or facile CO-insertion to yield acyl-Co tetracarbonyl (CH3CHCHCH2(CO)Co(CO)4), of which methanolysis by nucleophilic attack of [CH3O]− yields methyl-3-pentenoate and regenerates the catalyst (Scheme 40).170
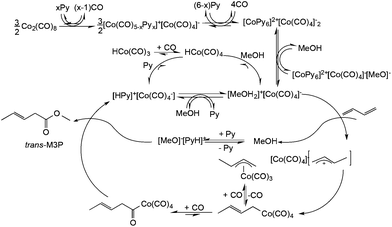 |
| | Scheme 40 Proposed catalytic cycle of the hydromethoxycarbonylation of butadiene by pyridine-modified cobalt catalysts in methanol. Adapted with permission from ref. 170. Copyright of the American Chemical Society (2011). | |
In very recent work by Horváth, the conversion mechanism of D-fructose to 5-HMF was mapped using a 13C isotope labelling technique in DMSO. Identification and characterization of the intermediates of the cyclic pathway of this conversion were reported by assigning the corresponding 13C-NMR peaks (Scheme 41).171 It was first established that all five isomers of D-fructose were detected in the solution and that the five-membered ring fructosyl oxocarbenium ion that formed via protonation and dehydration of D-fructofuranose could undergo deprotonation to form either 3,4-DIOL or 2,6-anhydro-β-D-fructofuranose. While carbaldehyde-5 can easily be dehydrated to 5-HMF, the 2,6-anhydro-β-D-fructofuranose as a key species in the reaction mixture could also form a six-membered ring fructopyranosil oxocarbenium ion as a starting material for the difructose dianhydrides (DFAs) and 3,4,5-TRIOL, which can then be converted to carbaldehyde-6 via dehydration. It is important to note that 3,4,5-TRIOL can also be derived from the D-fructopyranose form. Consequently, the equilibrium reactions between the fructose isomers, five- and six-membered oxocarbenium cations, and the presence of 2,6-anhydro-β-D-fructofuranose divide the conversion pathway into two parallel directions. While the first one, which is in excellent agreement with the proposed cyclic pathway, leads to the formation of 5-HMF, the second one leads to the formation of oligomers and humins as byproducts. These results can help to design an efficient and selective catalyst for this important conversion in biomass utilization.
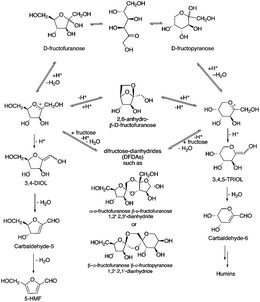 |
| | Scheme 41 Proposed intermediates of the acid-catalyzed dehydration of D-fructose to 5-HMF via a cyclic pathway, adapted with permission from ref. 171. Copyright 2012 of the Royal Society of Chemistry. | |
Conclusions
Chemistry plays a fundamental role in our modern society and has contributed a lot to technological evolution by allowing the invention of millions of chemical reactions that have led to the formation of new materials, which never existed before in the history of the universe. New substances, if interacting with living systems in a sufficient quantity, may be harmful or even poisonous, and this is one of the reasons why chemistry has a bad image in the public eye. Green Chemistry strives to reduce, or even eliminate, the danger due to the appearance of novel materials in Nature by following certain rules. These correspond to the principle of conservative evolution, namely that only new construction blocks, emerging along the long history of the universe, may survive, which are well anchored on already existing ones. Successful construction blocks survive even over billions of years, while materials or processes, which do not fit into the natural order, disappear within a limited time. In this review, a tribute to the work of Professor István T. Horváth, we provide a series of examples for the successful manifestation of conservative evolution and one of its consequences, industrial metabolism, by the principles of Green Chemistry.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
László T. Mika is grateful for the support of a József Varga Research Scholarship from the Faculty of Chemical and Biochemical Engineering, Budapest University of Technology and Economics. This work was also funded by the National Research, Development and Innovation Office – NKFIH (PD 116559).
References
-
P. Teilhard de Chardin, Le Phénomène humain, Seuil, Paris, 1955 Search PubMed.
- B. Eskenazi, P. Mocarelli, M. Warner, L. Needham, D. G. Patterson, S. Samuels, W. Turner, P. M. Gerthoux and P. Brambilla, Environ. Health Perspect., 2003, 112, 22–27 CrossRef.
- E. Broughton, Environ. Health, 2005, 4, 1–6 CrossRef PubMed.
- B. Eskenazi, J. Chevrier, L. G. Rosas, H. A. Anderson, M. S. Bornman, H. Bouwman, A. Chen, B. A. Cohn, C. de Jager, D. S. Henshel, F. Leipzig, J. S. Leipzig, E. C. Lorenz, S. M. Snedeker and D. Stapleton, Environ. Health Perspect., 2009, 117, 1359–1367 CrossRef PubMed.
- M. J. Molina and F. S. Rowland, Nature, 1974, 249, 810–812 CrossRef.
- R. G. Prinn, R. F. Weiss, P. J. Fraser, P. G. Simmonds, D. M. Cunnold, F. N. Alyea, S. O'Doherty, P. Salameh, B. R. Miller, J. Huang, R. H. J. Wang, D. E. Hartley, C. Harth, L. P. Steele, G. Sturrock, P. M. Midgley and A. McCulloch, J. Geophys. Res., 2000, 105, 17751–17792 CrossRef.
- J. H. Kim and A. R. Scialli, Toxicol. Sci., 2011, 122, 1–6 CrossRef PubMed.
-
P. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018 10.1039/C8GC00482J , in press.
-
R. U. Ayres, Industrial metabolism: Theory and policy, in Industrial Metabolism: Restructuring for Sustainable Development, ed. R. U. Ayres and U. K. Simonis, United Nations University Press, Tokyo, 1994 Search PubMed.
-
D. W. Pearce and R. K. Turner, Economics of Natural Resources and the Environment, Johns Hopkins University Press, Baltimore, 1989 Search PubMed.
-
G. Náray-Szabó, Conservative Evolution, Sustainability, and Culture. CLCWeb: Comparative Literature and Culture 2014, 16.1 (2014): DOI:10.7771/1481-4374.2316 (accessed on November 2, 2017).
- B. M. Trost, Science, 1991, 254, 1471–1477 Search PubMed.
- R. A. Sheldon, Chem. Ind., 1992, 903–906 Search PubMed.
- G. Albacti and M. Tovqlieri, Chim. Ind., 1959, 41, 189 Search PubMed.
- G. Váradi, I. T. Horváth, J. Palágyi, T. Bak and G. Pályi, J. Mol. Catal., 1980, 9, 457–460 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- B. Buckley, M. Figueres, A. Khan and H. Heaney, Synlett, 2015, 27, 51–56 CrossRef.
- S. Lal and S. Díez-González, J. Org. Chem., 2011, 76, 2367–2373 CrossRef PubMed.
- L.-Y. Wu, Y.-X. Xie, Z.-S. Chen, Y.-N. Niu and Y.-M. Liang, Synlett, 2009, 1453–1456 Search PubMed.
- J.-A. Shin, Y.-G. Lim and K.-H. Lee, J. Org. Chem., 2012, 77, 4117–4122 CrossRef PubMed.
- M. Brookhart, A. F. Volpe, D. M. Lincoln, I. T. Horvath and J. M. Millar, J. Am. Chem. Soc., 1990, 112, 5634–5636 CrossRef.
- K. Nozaki, T. Hiyama, S. Kacker and I. T. Horváth, Organometallics, 2000, 19, 2031–2035 CrossRef.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef PubMed.
-
H. E. Hoydonickx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, Furfural and Derivatives, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, DOI:10.1002/14356007.a12_119.pub2.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádabaand and J. M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 Search PubMed.
- J. M. Tukacs, M. Bohus, G. Dibó and L. T. Mika, RSC Adv., 2017, 7, 3331–3335 RSC.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- V. Fábos, G. Koczó, H. Mehdi and L. Boda, Energy Environ. Sci., 2009, 2, 767–769 Search PubMed.
- D. Havasi, P. Mizsey and L. T. Mika, J. Chem. Eng. Data, 2016, 61, 1502–1508 CrossRef.
- D. Havasi, G. Pátzay, Z. Kolarovszki and L. T. Mika, J. Chem. Eng. Data, 2016, 61, 3326–3333 CrossRef.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49–54 CrossRef.
- J. M. Tukacs, M. Novák, G. Dibó and L. T. Mika, Catal. Sci. Technol., 2014, 4, 2908–2912 Search PubMed.
- V. Fábos, L. T. Mika and I. T. Horváth, Organometallics, 2014, 33, 181–187 CrossRef.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang and R. M. West, Science, 2010, 327, 1110–1114 CrossRef PubMed.
- J. Q. Bond, D. Martin Alonso, R. M. West and J. A. Dumesic, Langmuir, 2010, 26, 16291–16298 CrossRef PubMed.
- V. Fábos, M. Y. Lui, Y. F. Mui, Y. Y. Wong, L. T. Mika, L. Qi, E. Cséfalvay, V. Kovács, T. Szűcs and I. T. Horváth, ACS Sustainable Chem. Eng., 2015, 3, 1899–1904 CrossRef.
- D. Fegyverneki, L. Orha, G. LAng and I. T. Horváth, Tetrahedron, 2010, 66, 1078–1081 CrossRef.
- A. Strádi, M. Molnár, M. Óvári, G. Dibó and F. U. Richter, Green Chem., 2013, 15, 1857 RSC.
- A. Strádi, M. Molnár, P. Szakál, G. Dibó, D. Gáspár and L. T. Mika, RSC Adv., 2015, 5, 72529–72535 RSC.
- L. Orha, J. M. Tukacs, B. Gyarmati, A. Szilágyi, L. Kollár and L. T. Mika, ACS Sustainable Chem. Eng., 2018, 6, 5097–5104 CrossRef.
- M. Chalid, H. J. Heeres and A. A. Broekhuis, J. Appl. Polym. Sci., 2011, 123, 3556–3564 CrossRef.
-
P. K. Wong, C. Li and L. Stubbs, Synthesis of Diacids, Patent Appl., WO2012/134397A1, 2012 Search PubMed.
- J. M. Tukacs, B. Fridrich, G. Dibó, E. Székely and L. T. Mika, Green Chem., 2015, 17, 5189–5195 RSC.
-
Sax's Dangerous Properties of Industrial Materials, ed. R. J. Lewis Sr., Wiley-Interscience, Wiley & Sons, Inc., Hoboken, NJ, 11th edn, 2004, p. 2407 Search PubMed.
- S. Csihony, L. T. Mika, G. Vlád, K. Barta, C. P. Mehnert and I. T. Horváth, Collect. Czech. Chem. Commun., 2007, 72, 1094–1106 CrossRef.
- F. Aricò and P. Tundo, Russ. Chem. Rev., 2010, 79, 479–489 CrossRef.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef PubMed.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef PubMed.
- M. Selva and A. Perosa, Green Chem., 2008, 10, 457–464 RSC.
- A. Caretto and A. Perosa, ACS Sustainable Chem. Eng., 2013, 1, 989–994 CrossRef.
- J. N. G. Stanley, M. Selva, A. F. Masters, T. Maschmeyer and A. Perosa, Green Chem., 2013, 15, 3195–3204 RSC.
- M. Y. Lui, K. S. Lokare, E. Hemming, J. N. G. Stanley, A. Perosa, M. Selva, A. F. Masters and T. Maschmeyer, RSC Adv., 2016, 6, 58443–58451 RSC.
-
http://ec.europa.eu/eurostat/statistics-explained/index.php/Air_pollution_statistics
(accessed on September 10, 2017).
- B. Cornils and E. G. Kuntz, J. Organomet. Chem., 1995, 502, 177–186 CrossRef.
- I. T. Horváth, R. V. Kastrup, A. A. Oswald and E. J. Mozeleski, Catal. Lett., 1989, 2, 85–90 CrossRef.
- Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635 CrossRef PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef PubMed.
- V. Fábos, D. Lantos, A. Bodor, A.-M. Bálint, L. T. Mika, O. E. Sielcken, A. Cuiper and I. T. Horváth, ChemSusChem, 2008, 1, 189–192 CrossRef PubMed.
- S. Csihony, C. Fischmeister, C. Bruneau, I. T. Horváth and P. H. Dixneuf, New J. Chem., 2002, 26, 1667–1670 RSC.
- H. Mehdi, A. Bodor, D. Lantos, I. T. Horváth, A. Dirk, E. De Vos and K. Binnemans, J. Org. Chem., 2006, 72, 517–524 CrossRef PubMed.
- I. T. Horváth, Green Chem., 2008, 10, 1024 RSC.
- P. Pongrácz, L. Kollár and L. T. Mika, Green Chem., 2016, 18, 842–847 RSC.
- P. Pongrácz, B. Bartal, L. Kollár and L. T. Mika, J. Organomet. Chem., 2017, 847, 140–145 CrossRef.
- D. Marosvölgyi-Haskó, B. Lengyel, J. M. Tukacs, L. Kollár and L. T. Mika, ChemPlusChem, 2016, 81, 1224–1229 CrossRef.
- G. Strappaveccia, E. Ismalaj, C. Petrucci, D. Lanari, A. Marrocchi, M. Drees, A. Facchetti and L. Vaccaro, Green Chem., 2015, 17, 365–372 RSC.
- G. Strappaveccia, L. Luciani, E. Bartollini, A. Marrocchi, F. Pizzo and L. Vaccaro, Green Chem., 2015, 17, 1071–1076 RSC.
- E. Ismalaj, G. Strappaveccia, E. Ballerini, F. Elisei, O. Piermatti, D. Gelman and L. Vaccaro, ACS Sustainable Chem. Eng., 2014, 2, 2461–2464 CrossRef.
- D. Rasina, A. Kahler-Quesada, S. Ziarelli, S. Warratz, H. Cao, S. Santoro, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 5025–5030 RSC.
- X. Tian, F. Yang, D. Rasina, M. Bauer, S. Warratz, F. Ferlin, L. Vaccaro and L. Ackermann, Chem. Commun., 2016, 52, 9777–9780 RSC.
-
Sax's Dangerous Properties of Industrial Materials, ed. R. J. Lewis Sr., Wiley-Interscience, Wiley & Sons, Inc., Hoboken, NJ, 11th edn, 2004, p. 285 Search PubMed.
-
Sax's Dangerous Properties of Industrial Materials, ed. R. J. Lewis Sr., Wiley-Interscience, Wiley & Sons, Inc., Hoboken, NJ, 11th edn, 2004, p. 2376 Search PubMed.
-
Sax's Dangerous Properties of Industrial Materials, ed. R. J. Lewis Sr., Wiley-Interscience, Wiley & Sons, Inc., Hoboken, NJ, 11th edn, 2004, p. 1627 Search PubMed.
- Y. Gu, J. Barrault and F. Jérôme, Adv. Synth. Catal., 2008, 350, 2007–2012 CrossRef.
- T. Deligeorgiev, S. Kaloyanova, N. Lesev, R. Alajarín, J. J. Vaquero and J. Álvarez-Builla, Green Sustainable Chem., 2011, 01, 170–175 CrossRef.
- F. Chahdoura, L. Dubrulle, K. Fourmy, J. Durand, D. Madec and M. Gómez, Eur. J. Inorg. Chem., 2013, 2013, 5138–5144 CrossRef.
- E. Farnetti, J. Kašpar and C. Crotti, Green Chem., 2009, 11, 704–709 RSC.
- A. Wolfson, C. Dlugy, Y. Shotland and D. Tavor, Tetrahedron Lett., 2009, 50, 5951–5953 CrossRef.
- A. Azua, J. A. Mata and E. Peris, Organometallics, 2011, 30, 5532–5536 CrossRef.
- A. Azua, J. A. Mata, E. Peris, F. Lamaty, J. Martinez and E. Colacino, Organometallics, 2012, 31, 3911–3919 CrossRef.
- P. Cintas, S. Tagliapietra, E. Calcio Gaudino, G. Palmisano and G. Cravotto, Green Chem., 2014, 16, 1056–1065 RSC.
- S. Tagliapietra, L. Orio, G. Palmisano, A. Penoni and G. Cravotto, Chem. Pap., 2015, 69, 1519–1531 Search PubMed.
- Y. Gu and F. Jérôme, Green Chem., 2010, 12, 1127–1138 RSC.
- J. Yang, J.-N. Tan and Y. Gu, Green Chem., 2012, 14, 3304–3317 RSC.
- J.-P. Wan, C. Wang, R. Zhou and Y. Liu, RSC Adv., 2012, 2, 8789–8792 RSC.
- P. P. Ghosh, S. Paul and A. R. Das, Tetrahedron Lett., 2013, 54, 138–142 CrossRef.
- A. Dandia, A. K. Jain and A. K. Laxkar, Tetrahedron Lett., 2013, 54, 3929–3932 CrossRef.
- W. Keim, Angew. Chem., Int. Ed., 2013, 52, 12492–12496 CrossRef PubMed.
- I. T. Horváth and J. Rábai, Science, 1994, 266, 72–75 Search PubMed.
- I. T. Horváth, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. Rábai and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133–3143 CrossRef.
- J. J. J. Juliette, I. T. Horváth and J. A. Gladysz, Angew. Chem., Int. Ed. Engl., 1997, 36, 1610–1612 CrossRef.
- M. A. Guillevic, M. A. Arif, I. T. Horváth and J. A. Gladysz, Angew. Chem., Int. Ed. Engl., 1997, 36, 1612–1614 CrossRef.
- J. Fawcett, E. G. Hope, R. D. W. Kemmitt, D. R. Paige, D. R. Russel, A. M. Stuart, D. J. Cole-Hamilton and M. J. Pane, Chem. Commun., 1997, 1127–1128 RSC.
- T. J. Malosh, S. R. Wilson and J. R. Shapley, J. Organomet. Chem., 2009, 694, 3331–3337 CrossRef.
- G. Pozzi, F. Montanari and S. Quici, Chem. Commun., 1997, 69–70 RSC , and references therein.
- R. P. Hughes and H. A. Trujillo, Organometallics, 1996, 15, 286–294 CrossRef.
-
J. A. Rossin and S. M. Maurer, Catalytic Processes for the Decomposition of Perfluoroalkanes, US6069291, 2000 Search PubMed.
-
Fluorous Chemistry Top. Curr. Chem, ed. I. T. Horváth, 2012, vol. 308, pp. 1–405 Search PubMed.
- C.-K. E. Law and I. T. Horváth, Org. Chem. Front., 2016, 3, 1048–1062 RSC.
- A. S. W. Lo and I. T. Horváth, Green Chem., 2015, 17, 4701–4714 RSC.
-
Handbook of Fluorous Chemistry, ed. J. A. Gladysz, D. Curran and I. T. Horváth, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- X. Miao, C. Fischmeister, C. Bruneau and P. H. Dixneuf, ChemSusChem, 2008, 1, 813–816 CrossRef PubMed.
- A. Keraani, C. Fischmeister, T. Renouard, M. Le Floch, A. Baudry, C. Bruneau and M. Rabiller-Baudry, J. Mol. Catal. A: Chem., 2012, 357, 73–80 CrossRef.
- P. Arockiam, V. Poirier, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2009, 11, 1871–1875 RSC.
- B. Schäffner, J. Holz, S. P. Verevkin and A. Börner, ChemSusChem, 2008, 1, 249–253 CrossRef PubMed.
- K. Zosel, Angew. Chem., Int. Ed. Engl., 1978, 17, 702–709 CrossRef.
- S. L. Wells and J. Desimone, Angew. Chem., Int. Ed., 2001, 40, 518–527 CrossRef PubMed.
-
Chemical Synthesis Using Supercritical Fluids, ed. P. G. Jessop and W. Leitner, Wiley-VCH, Weinheim-New York, 1999 Search PubMed.
- W. Leitner, Acc. Chem. Res., 2002, 35, 746–756 CrossRef PubMed.
- X. Zhang, S. Heinonen and E. Levänen, RSC Adv., 2014, 4, 61137–61152 RSC.
- E. Fogassy, M. Ács, T. Szili, B. Simándi and J. Sawinsky, Tetrahedron Lett., 1994, 35, 257–260 CrossRef.
- G. Bánsághi, E. Székely, D. M. Sevillano, Z. Juvancz and B. Simándi, J. Supercrit. Fluids, 2012, 69, 113–116 CrossRef.
- L. Lőrincz, G. Bánsághi, M. Zsemberi, S. de Simón Brezmes, I. M. Szilágyi, J. Madarász, T. Sohajda and E. Székely, J. Supercrit. Fluids, 2016, 118, 48–53 CrossRef.
- BP Statistical Review of World Energy June 2017. https://www.bp.com/content/dam/bp/en/corporate/pdf/energy-economics/statistical-review-2017/bp-statistical-review-of-world-energy-2017-full-report.pdf (accessed on November 10, 2017).
- M. A. Delucchi and M. Z. Jacobson, Energy Policy, 2011, 39, 1170–1190 CrossRef.
- M. Z. Jacobson and M. A. Delucchi, Energy Policy, 2011, 39, 1154–1169 CrossRef.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- C.-H. C. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. M. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef PubMed.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef PubMed.
- L. T. Mika, E. Cséfalvay and I. T. Horváth, Catal. Today, 2015, 247, 33–46 CrossRef.
-
T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass, in Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Pacific Northwest National Laboratory (PNNL) and the National Renewable Energy Laboratory (NREL), US Department of Energy, 2004, vol. I Search PubMed.
- E. Cséfalvay, G. R. Akien, L. Qi and I. T. Horváth, Catal. Today, 2015, 239, 50–55 CrossRef.
- I. T. Horváth, E. Cséfalvay, L. T. Mika and M. Debreczeni, ACS Sustainable Chem. Eng., 2017, 5, 2734–2740 CrossRef.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 Search PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef.
- A. Mukherjee, M.-J. Dumont and V. Raghavan, Biomass Bioenergy, 2015, 72, 143–183 CrossRef.
-
W. E. Erner, U.S. Patent, 4364743, 1982 Search PubMed.
- R. H. Leonard, Ind. Eng. Chem., 1956, 48, 1330–1341 CrossRef.
- C. A. Rebeiz, L. J. Gut, K. Lee, J. A. Juvik, C. C. Rebeiz, C. E. Bouton and G. H. N. Towers, Crit. Rev. Plant Sci., 1995, 14, 329–366 CrossRef.
- C. A. Rebeiz, A. Montazer-Zouhoor, H. J. Hopen and S. M. Wu, Enzyme Microb. Technol., 1984, 6, 390–396 CrossRef.
-
DoE
, 10 CFR Part 490: Alternative Fuel Transportation Program; P-Series Fuels; Final Rule. Office of Energy Efficiency and Renewable Energy, Department of Energy (DOE), 1999.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef PubMed.
- A. E. C. Collis and I. T. Horváth, Catal. Sci. Technol., 2011, 1, 912–918 Search PubMed.
- I. T. Horváth, Angew. Chem., Int. Ed. Engl., 1991, 30, 1009–1011 CrossRef.
- D. He and I. T. Horváth, J. Organomet. Chem., 2017, 847, 263–269 CrossRef.
- W. A. Herrmann and C. W. Kohlpaintner, Angew. Chem., Int. Ed. Engl., 1993, 32, 1524–1544 CrossRef.
-
H. Bahrmann and S. Bogdanovich, Hydroformylation of higher olefins in Aqueous phase organometallic catalysis, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, 1998, p. 306 Search PubMed.
- I. T. Horváth, Catal. Lett., 1990, 6, 43–48 CrossRef.
- J. P. Arhancet, M. E. Davis, J. S. Merola and B. E. Hanson, Nature, 1989, 339, 454–455 CrossRef.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef PubMed.
- R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678–2691 RSC.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef PubMed.
- C. C. C. R. de Carvalho, Microb. Biotechnol., 2016, 10, 250–263 CrossRef PubMed.
- J.-M. Choi, S.-S. Han and H.-S. Kim, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef PubMed.
- M. S. Hoekstra, D. M. Sobieray, M. A. Schwindt, T. A. Mulhern, T. M. Grote, B. K. Huckabee, V. S. Hendrickson, L. C. Franklin, E. J. Granger and G. L. Karrick, Org. Process Res. Dev., 1997, 1, 26–38 CrossRef.
- C. A. Martinez, S. Hu, Y. Dumond, J. Tao, P. Kelleher and L. Tully, Org. Process Res. Dev., 2008, 12, 392–398 CrossRef.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef PubMed.
- S. A. B. Dockrey, A. L. Lukowski, M. R. Becker and A. R. H. Narayan, Nat. Chem., 2017, 1–7 Search PubMed.
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Adv. Mater., 2017, 29, 1701139–1701121 CrossRef PubMed.
- Y. Wang, H. Arandiyan, J. Scott, A. Bagheri, H. Dai and R. Amal, J. Mater. Chem. A, 2017, 5, 8825–8846 Search PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
-
K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, Weinheim, 1993 Search PubMed.
-
https://www.nobelprize.org/nobel_prizes/chemistry/laureates/2015/press.html
(Accessed on November 17, 2017).
- P. J. F. De Rege, J. A. Gladysz and I. T. Horváth, Science, 1997, 276, 776–779 CrossRef PubMed.
- P. J. F. De Rege, J. A. Gladysz and I. T. Horváth, Adv. Synth. Catal., 2002, 344, 1059–1062 CrossRef.
- Z. Pusztai, G. Vlád, A. Bodor, I. T. Horváth, H. J. Laas, R. Halpaap and F. U. Richter, Angew. Chem., Int. Ed., 2006, 45, 107–110 CrossRef PubMed.
- S. Csihony, H. Mehdi and I. T. Horváth, Green Chem., 2001, 3, 307–309 RSC.
- L. T. Mika, R. Tuba, I. Tóth, S. Pitter and I. T. Horváth, Organometallics, 2011, 30, 4751–4764 CrossRef.
- G. R. Akien, L. Qi and I. T. Horváth, Chem. Commun., 2012, 48, 5850–5853 RSC.
Footnote |
| † This paper is dedicated to Professor István T. Horváth on the occasion of his 65th birthday. |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b
*b