
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMelt-processing of small molecule organic photovoltaics via bulk heterojunction compatibilization†
Aiman
Rahmanudin
,
Liang
Yao
,
Xavier A.
Jeanbourquin
,
Yongpeng
Liu
 ,
Arvindh
Sekar
,
Emilie
Ripaud
and
Kevin
Sivula
*
,
Arvindh
Sekar
,
Emilie
Ripaud
and
Kevin
Sivula
*
Laboratory for Molecular Engineering of Optoelectronic Nanomaterials, Ecole Polytechnique Fédérale de Lausanne, Station 6, CH-1015, Switzerland. E-mail: kevin.sivula@epfl.ch
First published on 17th April 2018
Abstract
Melt-processing of organic semiconductors (OSCs) is a promising environmentally-friendly technique that can alleviate dependence on toxic chlorinated solvents. While melt-processed single-component OSC devices (e.g. field-effect-transistors) have been demonstrated, multi-component bulk heterojunctions (BHJs) for organic photovoltaics (OPVs) remain a challenge. Herein, we demonstrate a strategy that affords tunable BHJ phase segregation and domain sizes from a single-phase homogeneous melt by employing strongly-crystalline small-molecule OSCs together with a customized molecular compatibilizing (MCP) additive. An optimized photoactive BHJ with 50 wt% MCP achieved a device power conversion efficiency of ca. 1% after melting the active layer at 240 °C (15 min, followed by slow cooling) before deposition of the top electrode. BHJ morphology characterization using atomic force and Kelvin probe microscopy, X-ray diffraction, and photo-luminescence measurements further demonstrate the trade-off between free charge generation and transport with respect to MCP loading in the BHJ. In addition, a functional OPV was also obtained from the melt-processing of dispersed micron-sized solid BHJ particles into a smooth and homogeneous thin-film by using the MCP approach. These results demonstrate that molecular compatibilization is a key prerequisite for further developments towards true solvent-free melt-processed BHJ OPV systems.
Attaining a carbon-neutral, sustainable, and global energy infrastructure based on renewable sources will likely require widespread implementation of photovoltaic cells, thus the development of inexpensive and efficient photovoltaics that can be manufactured by scalable methods is an important goal.1 Amongst various emerging technologies alternative to traditional silicon photovoltaics (including thin film inorganic,2,3 dye sensitized,4 and perovskite5 solar cells), organic photovoltaic (OPV) devices, which rely on a bulk-heterojunction (BHJ) between polymeric or small-molecule carbon-based p-type (electron donor) and n-type (electron acceptor) semiconductors, have attracted significant interest given their promise for achieving high performance while also being low-cost and amenable with scalable roll-to-roll manufacturing.6,7 While the ongoing development of OPVs has led to impressive power conversion efficiency >10%, typical state-of-the-art devices are fabricated using toxic chlorinated or aromatic solvents,8 which undermines their sustainable application at a global scale. Processing in environmentally-benign “green” solvents is a potential alternative,9,10 but requires altering the molecular structure of the organic semiconductors (OSCs) to afford solubility, or dispersing the OSCs in colloidal systems, both of which greatly affect the OSC solid-state self-assembly and the resulting optoelectronic properties.11–14 Alternatively, melt processing, which involves a reversible liquefaction–solidification process and is widely used in industry to produce commodity plastic thin films,15 could be employed to afford solvent-free manufacturing of OPVs. However, while melt-processing techniques have been recently studied and developed for single-component OSC devices (e.g. field effect transistors),16–19 and despite the suggested similarities between solvent quenching and melt quenching techniques from thermodynamic arguments,20–22 the processing of BHJ OPV devices from homogeneous liquid melts into high efficiency OPVs has yet to be demonstrated due to the extreme sensitivity of the BHJ morphology on the device performance. Indeed a percolating network of precisely-sized nanometer donor/acceptor domains in the BHJ is required for high OPV efficiency.23–25 While the optimum BHJ morphology can be readily tuned using solvent processing techniques,26 similar morphology control in melt-processed BHJs remains a challenge.
The use of donor–acceptor block-copolymers27 is a rational route to control the size of donor/acceptor domains under melt-processing conditions given the covalent link between the two components that prevents the macro-scale phase segregation, which is the thermodynamically preferred solid-state arrangement of typical BHJ components. Indeed, Lombeck et al. recently reported a donor–acceptor block-copolymer-based system that afforded promising performance in melt-annealed OPVs.28 However this work also established that the alteration of component molecular structure (through side-chain fluorination) is required with this approach to produce a system wherein the donor and acceptor components remain phase-separated in the melt (to ensure that that they form the required nanometer phase segregated BHJ upon solidification). In addition to this drawback, the processing of the requisite nano-phase separated melts poses additional challenges (e.g. due to shear-induced domain alignment29) toward obtaining the BHJ optimum morphology.
An ideal melt-processed BHJ OPV system would allow a single phase liquid melt to be converted into a nanostructured BHJ with tunable domain sizes in a robust and reproducible manner. We hypothesized that this could be accomplished with strongly-crystalline molecular donor and acceptor components (to drive phase separation in the solid state) together with a molecular compatibilizing additive30 to afford tunability of the BHJ phase domains. Herein we demonstrate this melting/compatibilization approach using a molecular BHJ system with two common OSCs; a diketopyrrolopyrrole-based donor and a perylene diimide acceptor, both of which are known to strongly crystalize in the solid state. We establish that the use of a molecular compatibilizer is essential to tune the BHJ and obtain functional OPVs via melt annealing. Furthermore, we demonstrate a strategy towards the “green” processing of BHJ thin films via a solid dispersion melt-processing technique.
The molecular structures of the electron donor, coded as DPP(TBFu)2,31 and acceptor (coded as EP-PDI), both chosen based on their established high crystallinity in BHJs,32,33 are shown in Fig. 1a along with the corresponding molecular compatibilizer (MCP). A non-conjugated aliphatic linker strategy was chosen for the structure of the MCP as this approach has been established to affect the self-assembly of small-molecule semiconductors without alerting the pi-conjugated semiconducting molecular core.34,35 Detailed synthesis and characterization of the MCP is given in the ESI.† In order to first establish the solid-state phase behavior and melt-processing conditions, differential scanning calorimetry (DSC) was performed on solvent-cast BHJ blends (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 donor:acceptor by weight) with varying wt% of added MCP (see Fig. 1b). For the control BHJ (0 wt% MCP) during the first heating scan, two separate endothermic transitions were observed and are attributed to a liquid-crystal mesophase transition36 in EP-PDI at TLC = 73 °C (the melting temperature of EP-PDI is above 350 °C (ref. 18)) and the melting37 of DPP(TBFu)2 at Tm = 231 °C (see DSC scans for the pure components Fig. S1, ESI†). These transitions were both observed to be reversible upon cooling. A MCP loading of 10 or 25 wt% in the BHJ leads to a decrease in the transition specific enthalpies (see Table S1, ESI†) and a depression of transition temperatures during heating as expected for a compatibilizer.30 Importantly, these results suggest that the donor and acceptor retain the ability to recrystallize from the melt in the presence of significant amounts of the MCP. Notably, no obvious transitions were observed with 50 or 75 wt% MCP, suggesting a disruption of crystallinity in both DPP(TBFu)2 and EP-PDI with sufficiently high MCP loading. The pure compatibilizer (100 wt% MCP) also shows no phase transitions, suggesting no long range crystalline ordering in this material, consistent with similar systems.34
:1 donor:acceptor by weight) with varying wt% of added MCP (see Fig. 1b). For the control BHJ (0 wt% MCP) during the first heating scan, two separate endothermic transitions were observed and are attributed to a liquid-crystal mesophase transition36 in EP-PDI at TLC = 73 °C (the melting temperature of EP-PDI is above 350 °C (ref. 18)) and the melting37 of DPP(TBFu)2 at Tm = 231 °C (see DSC scans for the pure components Fig. S1, ESI†). These transitions were both observed to be reversible upon cooling. A MCP loading of 10 or 25 wt% in the BHJ leads to a decrease in the transition specific enthalpies (see Table S1, ESI†) and a depression of transition temperatures during heating as expected for a compatibilizer.30 Importantly, these results suggest that the donor and acceptor retain the ability to recrystallize from the melt in the presence of significant amounts of the MCP. Notably, no obvious transitions were observed with 50 or 75 wt% MCP, suggesting a disruption of crystallinity in both DPP(TBFu)2 and EP-PDI with sufficiently high MCP loading. The pure compatibilizer (100 wt% MCP) also shows no phase transitions, suggesting no long range crystalline ordering in this material, consistent with similar systems.34
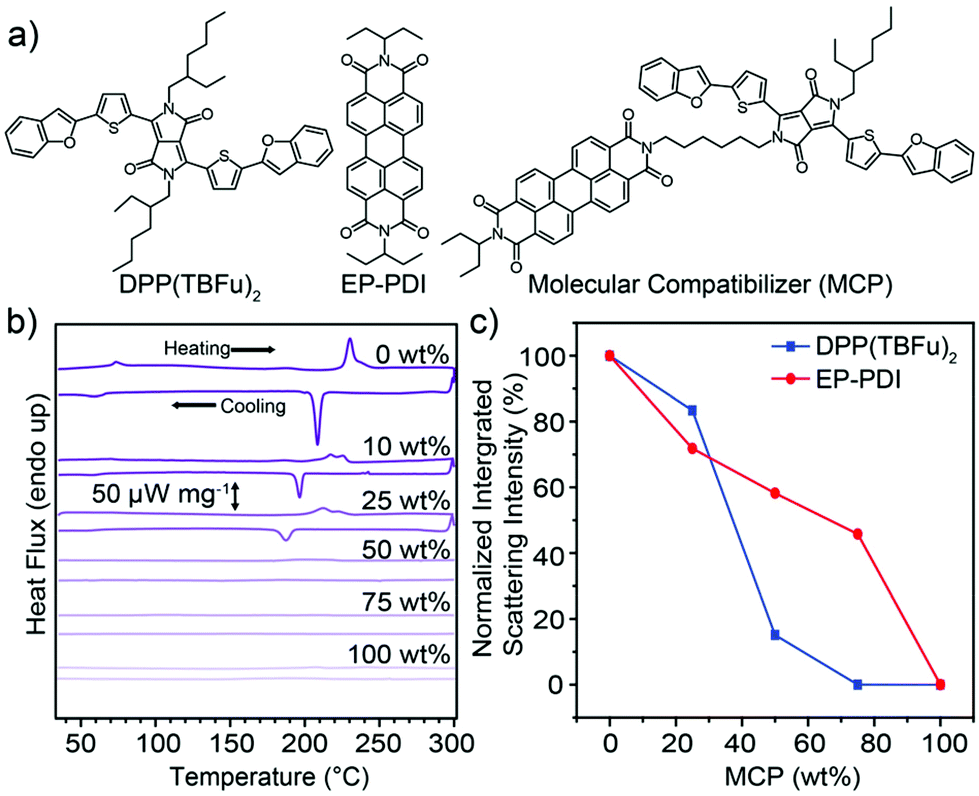 | ||
| Fig. 1 (a) Molecular structures of the organic semiconductors used in this study: DPP(TBFu)2, EP-PDI and the molecular compatibilizer (MCP). (b) DSC of the first heating/cooling scans (10 °C min−1) of 1:1 donor:acceptor BHJs with varying MCP loading (wt%). (c) Normalized integrated XRD scattering intensity of the (020) peak of DPP(TBFu)2 and the (200) peak of EP-PDI in 1:1 BHJ films treated at 240 °C (15 min) and cooled to room temperature. The normalization was performed with respect to the control (0 wt%) BHJ. The XRD data was collected from the grazing incidence geometry in the out-of-plane direction (see Fig. S2a and b, ESI,† for the XRD patterns). | ||
The trend in the molecular ordering in the presence of the MCP as implied by the DSC, was further investigated by X-ray diffraction (XRD) measurements of BHJ films treated at 240 °C (above Tm of the donor) and cooled to ambient temperature. Diffraction peaks for both the DPP(TBFu)2 and EP-PDI (as indexed from previous reports38,39) were observed in the melt-annealed BHJ films (Fig. S2, ESI†), and the relative crystallinity of each component in the BHJs with added MCP was estimated by peak integration. The normalized trends are shown in Fig. 1c. For the DPP(TBFu)2 these results agree qualitatively with the DSC data and confirm that the donor retains some crystallinity even after melting/solidification in the presence of the MCP. In addition, the XRD measurements show that the molecular ordering of the donor exists even after melting with 50 wt% MCP, in contrast to the DSC where no clear transition was detected (likely due to peak broadening). This contrast is greater with the EP-PDI, where the XRD data suggest that, while the crystallinity does decrease with added MCP loading, a significant amount of acceptor phase ordering remains present in films even after melting/solidification with 75 wt% MCP. Given the high melting point of the EP-PDI (>350 °C) this calls into question the nature of the acceptor phase at 240 °C in the presence of the (melted) donor and MCP. To clarify this, we performed in situ XRD measurements of BHJ films with and without the MCP at 240 °C (Fig. S2b, ESI†). These results clearly indicate an absence of crystallinity of the EP-PDI at 240 °C even without the MCP present, consistent with the donor and acceptor forming a single homogeneous liquid phase at a temperature above the melting temperature of the donor phase, similar to other systems.20,21 The formation of a single homogeneous liquid phase at 240 °C was further verified with optical microscopy (see ESI video† that shows the melting and re-solidification of a 1:1 BHJ film without MCP). Thus, overall these results confirm that the DPP(TBFu)2:EP-PDI BHJ blend can be processed from a homogenous melt phase at 240 °C into the solid state, while retaining the crystallinity of both components. Moreover, the degree of solid-state crystallinity of both phases can be tuned by adding the MCP.
To next investigate the effect of the MCP on the BHJ morphology, melt-annealed (240 °C)/solidified thin films were examined by atomic force and Kelvin probe force microscopy (AFM and KPFM, respectively). Relative height and surface potential difference maps are shown in Fig. 2 for 1:1 donor:acceptor BHJs with different MCP loading. The height maps show rod-like crystallite domains hundreds of nanometers in length in the control (0 wt%) BHJ, which are diminished in size with increasing amount of MCP. While some crystalline features are visible in the 50 wt% film, the 75 wt% film appears essentially homogeneous with a morphology similar to the pure MCP (100 wt%) film. The chemical nature of the phase domains was further assessed with the KPFM maps where the absolute values of the surface potential difference (i.e. the difference in potential between the Pt tip and the surface) were related to the pure component phases (as established by measurements on pure component thin films, see Fig. S3, ESI†). Briefly, the donor phase is expected to appear with a lower contact potential difference, VCPD, in the BHJ compared to the acceptor phase. Therefore, the KPFM map of the control BHJ (0 wt%) exhibits a clear phase segregation between the platelet-shaped crystals of DPP(TBFu)2 (yellow) and the rod-like crystals of EP-PDI (purple). The extent of the phase segregation is also indicated by the VCPD histogram (bottom panel Fig. 2), which shows a bimodal distribution. With increasing MCP loading and the corresponding change in topographical morphology, the KPFM data show a decrease in the size of the donor and acceptor domains and an increase in donor:acceptor mixing in the BHJ as implied by the VCPD histograms, which become increasingly unimodal and narrow. Importantly, even with 50 wt% MPC, it appears that some degree of phase segregation (on the <100 nm length scale) still occurs after melt-annealing, while at 75 wt% a surface potential homogeneity similar to the pure MCP is observed (we note that the tip radius convolution prevents resolution of phase domains below ca. 20 nm). Therefore, we conclude that the level of donor:acceptor intermixing and the phase domain sizes in melted BHJs can be modulated with varying MCP loading. To further support this claim, photoluminescence (PL) emission of the respective thin films was characterized (see Fig. S4, ESI†). Upon excitation of the BHJ films at 532 nm, a prominent quenching of two peaks attributed to the PL emission of DPP(TBFu)2 (observed at 816 nm) and EP-PDI (610 nm) with increasing amount of MCP is noted. Since PL quenching corresponds to an increased probability for photogenerated excitons to reach the donor–acceptor interface and undergo charge separation (instead of radiative recombination) this trend corresponds well with the morphological changes exhibited in the KPFM analysis.
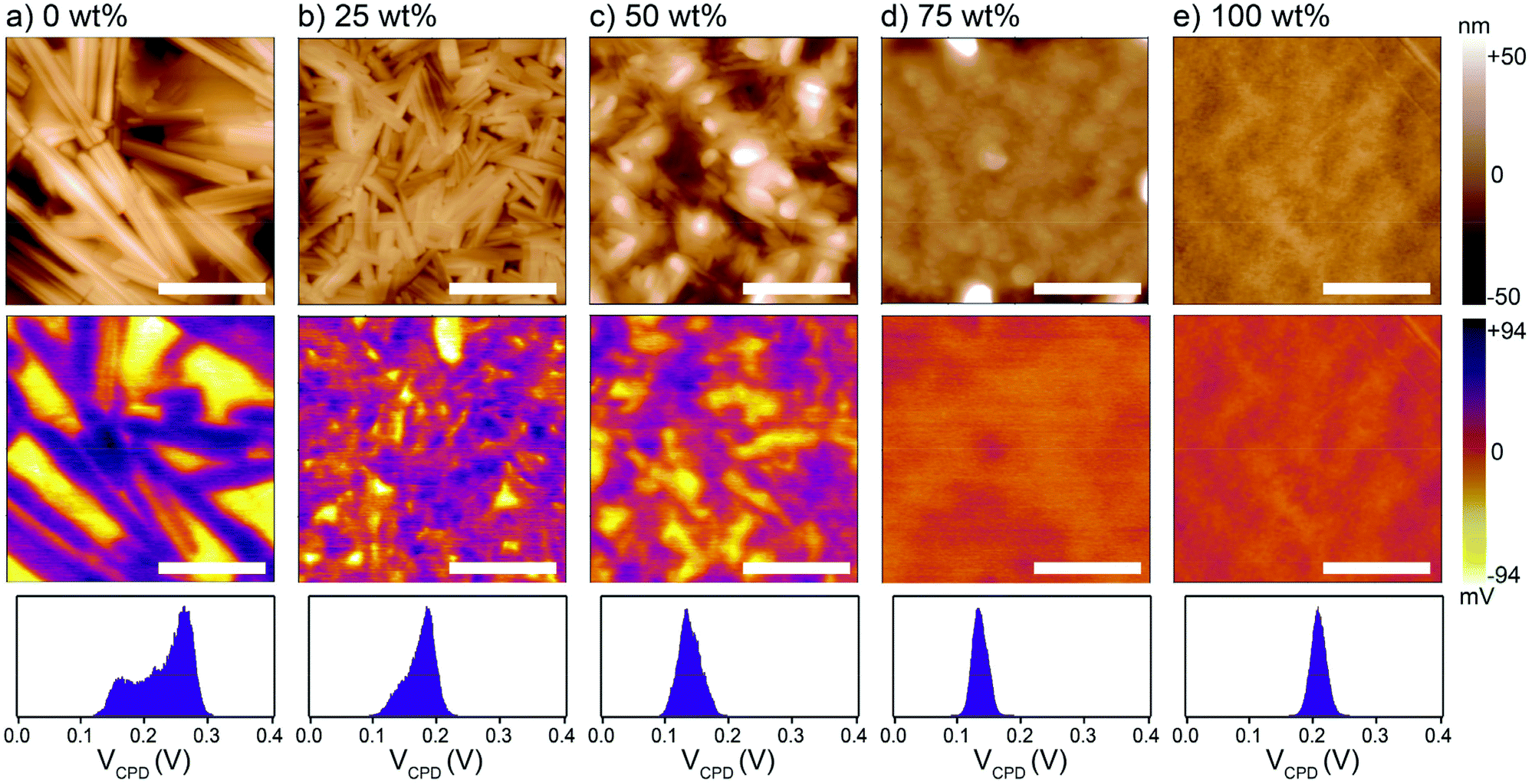 | ||
| Fig. 2 Morphological characterization of melt-annealed (240 °C) BHJ thin films of DPP(TBFu)2:EP-PDI (1:1 by weight) with varying amount of MCP (a–d) and pure MCP (e). Height topography (top panel) and KPFM potential images (middle panel) were measured under dark ambient conditions. The scale bars are 400 nm. The bottom panel shows the contact potential difference, VCPD, histogram for each KPFM image. | ||
To investigate the relation between the BHJ phase segregation after melt-annealing and the photovoltaic performance, devices with an inverted architecture (substrate/ITO/ZnO/BHJ/MO3/Ag) were fabricated and tested under standard 1 Sun illumination. The BHJ was solvent-cast (from chloroform solution) followed by melt-annealing at 240 °C (15 min followed by solidification) before deposition of the top blocking layer and electrode (details of device fabrication and testing are given in the Experimental section). Representative current density–voltage (J–V) curves for devices prepared with different MCP loadings are shown in Fig. 3a while the corresponding performance parameters and external quantum efficiency (EQE) are given in Fig. S5 and Table S2, ESI.† A clear trend in photovoltaic performance was observed with MCP loading. The power conversion efficiency (PCE) increased from 0.01% (control 1:1 BHJ with 0 wt% MCP) to a maximum of 0.83% at 50 wt% MCP. At 75 wt% MCP the PCE decreased to 0.74% and was 0.12% for pure MCP (100 wt%). The change in PCE is attributed primarily to changes in short circuit current density, Jsc, while only slight changes in open-circuit voltage and fill factor were observed with respect to MCP loading. The EQE as a function of the illumination wavelength exhibited a similar trend to the Jsc and did not significantly change shape with respect to the MCP loading (see Fig. S5b, ESI†) suggesting that the change in the device performance can be predominantly attributed to the BHJ morphology.
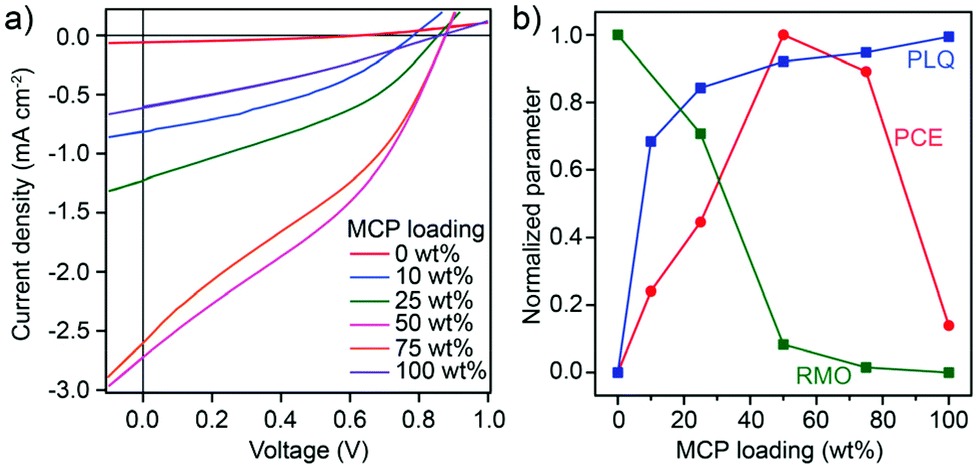 | ||
| Fig. 3 (a) Current–density vs. voltage curves of melt-annealed photovoltaic devices of DPP(TBFu)2:EP-PDI – 1:1 with varying amounts of MCP. (b) Summary on the effect of MCP on the BHJ morphology and the device performance is shown with normalized parameters: the device power conversion efficiency (PCE), the photoluminescence quenching (PLQ, calculated based on the integration of the respective PL emission, see Fig. S4, ESI†) and relative molecular ordering (RMO, estimated from the integration of XRD scattering from both donor and acceptor components, see Fig. S2a†). | ||
Indeed, the trend in the PCE with respect to the MCP loading (as shown normalized as the red curve in Fig. 3b) can be well understood by a trade-off between free charge generation (exciton dissociation) at the donor:acceptor interface and the presence of sufficiently large pure domains to afford the transport of free charges to their respective collecting electrodes. The amount of free charge generation can be estimated by the observed amount of photoluminescence quenching (PLQ) in the BHJ as mentioned before, while the ability to transport free charges in pure domains is related to the relative molecular ordering (RMO) as implied from the previously-discussed XRD results. These two parameters (PLQ and RMO) are shown normalized in Fig. 3b. Thus the optimum melt-annealed BHJ occurs at 50 wt% loading of the MCP, where the trade-off between RMO and PLQ is best balanced. To elucidate this behaviour further, devices processed from chloroform solution and then thermally annealed at temperatures below melting conditions were also fabricated (see Fig. S6†). When the BHJ was annealed for 15 min at 180 °C, a similar trend to devices melt-annealed at 240 °C was observed with an optimum loading of MCP at 50 wt% and a max PCE of 1.53%. However, when thermally annealed for 15 min at 110 °C the optimum PCE occurred at 0 wt% MCP. The observed decrease in PCE with increasing MCP in the 110 °C case is likely due to its role in compatiblizing the BHJ. During solvent quenching, a more homogeneous donor:acceptor blend is formed with the MCP present. The 110 °C, 15 min annealing conditions are evidently insufficient to drive the crystallization and phase segregation of the donor and acceptor to form the necessary percolating network of pure phases. This is supported by the decrease in fill-factor and Jsc in the 110 °C annealed devices with increasing MCP loading. In addition we note that since the results from the 180 °C devices show a similar max PCE compared to the optimum conditions at 110 °C and the 240 °C melt-annealed films, we can conclude that the melt-processing of the film does not lead to a significant decrease in the best possible device performance with the DPP(TBFu)2:EP-PDI system. We note that the slightly lower PCEs observed at 240 °C could be due to degradation of the electrode or blocking layer or their interfaces with the BHJ.40
Given the encouraging photovoltaic behaviour of the chloroform solvent cast, melt-annealed devices in the presence of the MCP, we sought to investigate the feasibility of using melt-processing towards the “green” processing of devices. We dispersed 1:1 BHJ mixtures with or without 50 wt% MCP in isopropanol using a minimal amount of chloroform and sonication to create a colloid of micron-sized particles which were subsequently drop-cast onto ITO/PEDOT:PSS substrates (see Fig. 4 for cross-sectional SEM images of drop-cast films and optical microscope images in Fig. S7, ESI†) to form a rough and non-continuous solid powder coating of BHJ particles. Upon melting at 240 °C for 15 min (and cooling to rt over 30 min) a drastic difference was observed in the resulting thin films: while the control (0 wt%) BHJ exhibited crystal domains microns in length, the BHJ with 50 wt% MCP melted into a remarkably smooth and homogeneous film (Fig. 4). The photovoltaic properties of these melt-processed films were then characterized using Ga–In top contact (to afford comparison with the rough control film) giving the J–V curves shown in Fig. 4c (see Fig. S8† for detailed experimental procedure and Table S3† for individual photovoltaic parameters). As expected the control device (containing 0 wt% MCP) showed a significantly lower performance of 0.002% PCE compared to 0.11% PCE when 50 wt% of MCP was included in the BHJ (ca. 50 times higher than the control). Although the PCE is lower than the chloroform-cast, melt annealed devices discussed above, a relatively thick active layer (ca.1 μm) was used and further optimization to obtain thinner films was not performed for this initial demonstration. Importantly, this validation clearly shows that large solid particles of small-molecule BHJs can be melt-processed into a homogenous and functional active layer through compatibilization with our MCP approach. The further optimization of the processing method and the use of higher-performance molecular donor:acceptor combinations41–43 with their tailored molecular compatibilizers will likely result in melt-processed molecular photovoltaics with high efficiency and excellent thermal stability.
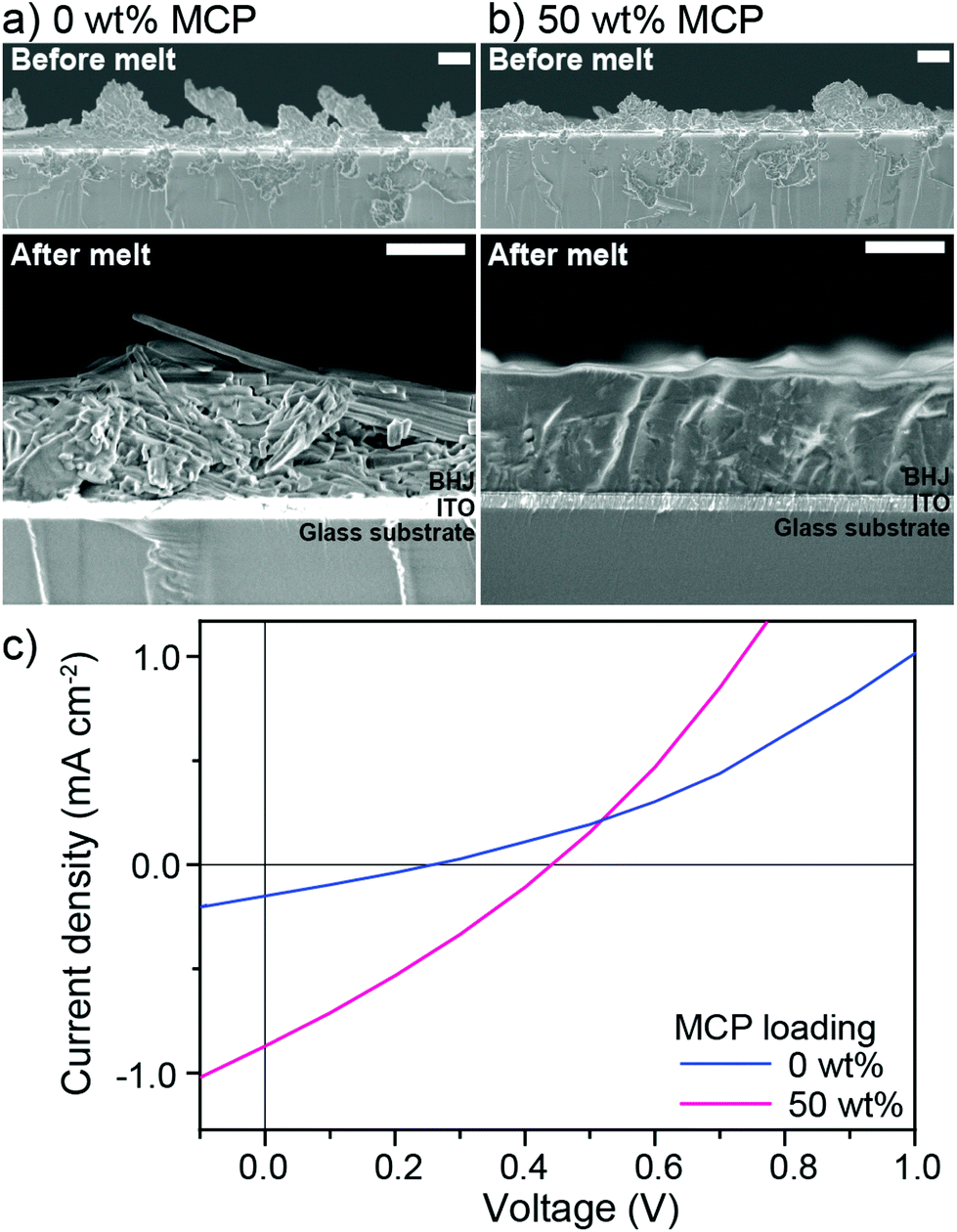 | ||
| Fig. 4 Cross-sectional scanning electron microscopy images of the drop-cast dispersions of 1:1 BHJ of DPP(TBFu)2:EP-PDI with (a) 0 wt% and (b) 50 wt% of MCP before melt-processing (top) and after melting at 240 °C for 15 min (bottom). The scale bars are 1 μm. (c) J–V curves of the corresponding solid-state melt-processed photovoltaic devices. | ||
Conclusions
In conclusion, we have shown that a molecular compatibilizer, which consists of donor and acceptor species covalently-linked with a non-conjugated linker, affords morphology control in melt-processed BHJs. By employing highly crystalline molecular donor and acceptor species we demonstrated the ability to process a BHJ from a homogeneous single phase melt into a photoactive OPV device with tunable domain sizes in the BHJ by modulating the loading of the MCP. Tuning the trade-off between donor:acceptor interfacial area and ordered pure domains using the MCP, as supported by AFM, KPFM, XRD and PL measurements, revealed the optimum device performance with a PCE of ca. 1% at 50 wt% loading of MCP in a 1:1 donor:acceptor BHJ. Furthermore, we demonstrated a functional OPV from the processing of micron-sized solid dispersions of BHJ with 50 wt% MCP into a homogeneous thin films via a melt-processing technique. These results show that molecular compatibilization is the key for further developments toward true solvent-free melt-processed BHJ OPV systems.
Experimental section
Preparation of BHJ (melt-annealed) for morphological characterization and OPV
Stock solutions of DPP(TBFu)2, EP-PDI, and MCP (see ESI† for synthetic procedure and basic characterization) were prepared separately at a concentration of 20 mg ml−1 in chloroform (CF) by stirring for 12 h at 50 °C. Blend solutions were stirred for 2 h before filtering with a 1.0 μm PTFE filter. Pre-patterned tin doped indium oxide (ITO) was cleaned by sequential sonication in water, isopropanol and acetone for 30 min each, and dried by argon. ZnO (20 nm) was used as an electron transport layer. The ZnO precursor solution contained 0.5 M zinc acetate dehydrate and 0.5 M monoethanolamine in 2-methoxyethanol and was stirred at 60 °C for 12 h before deposited on clean ITO substrates via spin-coating (5000 rpm). After removing a portion of the film to expose an electrical contact to the ITO, the substrates were annealed at 200 °C in air for 30 min. The BHJ layer was spin-coated at 3000 rpm for 60 s to obtain a thickness of ca. 140 nm as measured from a Bruker Dektak XT profilometer. The substrates were then annealed at the respective temperatures (110 °C, 180 °C and melting at 240 °C) for 15 min under argon before and allowed to cool down to room temperature in ca. 30 min before further morphological characterization and electrode deposition. For OPV devices 4 nm of MoO3 and 100 nm Ag were thermally deposited at 10−6 mbar through a shadow mask with an active area of 16 mm2. Current density–voltage (J–V) characteristics of the devices were tested under simulated AM1.5G irradiation from a 450 W Xe arc lamp set to 100 mW cm−2 with a calibrated Si photodiode. The external quantum efficiency (EQE) of the devices was characterized by illumination from a Tunable PowerArc illuminator (Optical Building Blocks Corporation). A calibrated Si photodiode was employed to measure the incident photon number at each wavelength.Preparation of BHJ (dispersions) thin films via melt-processing
Patterned and cleaned ITO substrates as described above were used to spin coat a 35 nm layer of PEDOT:PSS (Ossilla M 121 Al 4083). Isopropanol (IPA) was added slowly to a volume of BHJ solution (10 mg mL−1 in CF) to obtain a 8:2 IPA:CF (v:v) biphasic solution (see Fig. S7a, ESI†). This solution was then sonicated in a bath sonicator for 30 min to obtain a homogenous dispersion of BHJ in IPA. The dispersion was sequentially drop-casted (4 × 80 μL) onto the PEDOT:PSS substrate (20 × 20 mm) allowing for the solvent to dry before recoating in order to obtain a relatively homogenous distribution of solid aggregates. The solid powder dispersion films were then melted at 240 °C for 15 min under Argon. A Ga–In eutectic mixture was drop casted on the active layer and used as the top electrode. A micromanipulator was used to form an electrical contact with the respective electrodes to measure the current–voltage (I–V) characteristics of the photovoltaic device under a nitrogen atmosphere using a custom-built probe station and a Keithley 2612A dual-channel source measure unit under simulated AM1.5G irradiation from a 450 W Xe arc lamp set to 100 mW cm−2 with a calibrated Si photodiode.
General BHJ characterization
Atomic force microscopy (AFM) and surface potential measurements (KPFM) were performed using an Asylum Research Cypher using Pt:Ir-coated tips (AC240TM, Olympus) under dark ambient conditions. The work function of the tip was calibrated using reference samples of Pt, Mo, and Cu. Absorption spectra of the BHJ films were acquired with a UV-vis-NIR UV-3600 (Shimadzu) spectrophotometer, and the optical band gap was determined from the absorption edge of the thin film sample. Photoluminescence spectra were acquired with a Fluorolog-3 spectrofluorometer (Horiba) with sample placed at 45° from the lamp to the detector. Differential scanning calorimeter (DSC) thermograms were measured with a PerkinElmer DSC8000 calibrated with indium and zinc, using a scanning rate of 10 °C min−1. Samples were drop-casted onto a SiO2 substrate with a slow evaporation of the solvent (chloroform) at 30 °C under argon atmosphere before transferring the solid powder into an aluminium pan. XRD was measured with a D8 Discovery (Bruker) diffractometer using CuKα radiation and Ni β-filter with a scan rate of 0.05° min−1 and a step width of 0.01°. For the standard measurements, a pseudo-grazing incidence mode was used with an incident angle of 0.2°. In situ thermal X-ray experiments were in Bragg–Brentano geometry at room temperature and in a heated cell at 240 ± 5.0 °C (stabilized for 2 minutes before conducting a 15 min scan). Electrochemical measurements were conducted (to determine the band alignment of the MCP) with a SP-200 potentiostat (Biologic Technologies) with Pt counter and a Ag/Ag+ reference electrode in a 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) acetonitrile solution with a scan rate of 50 mV s−1. OSCs were drop casted on a bare Pt foil from a 2.0 mg mL−1 chloroform solution. Redox energies were calibrated by using Fc/Fc+.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr P. Schouwink is gratefully thanked for XRD data acquisition. We thank the European Research Commission (ERC starting grant “CEMOS” Project 336506) and the Swiss National Science Foundation (200021_169215) for financial support of this work. Y. L. acknowledges support under an Ambizione Energy grant (PZENP2_166871, Dr N. Guijarro).Notes and references
- A. Polman, M. Knight, E. C. Garnett, B. Ehrler and W. C. Sinke, Science, 2016, 352, aad4424 CrossRef PubMed.
- M. P. Suryawanshi, G. L. Agawane, S. M. Bhosale, S. W. Shin, P. S. Patil, J. H. Kim and A. V. Moholkar, Mater. Technol., 2013, 28, 98–109 CrossRef.
- X. Yu and K. Sivula, ACS Energy Lett., 2016, 1, 315–322 CrossRef.
- J. Gong, K. Sumathy, Q. Qiao and Z. Zhou, Renewable Sustainable Energy Rev., 2017, 68, 234–246 CrossRef.
- M. A. Green and A. Ho-Baillie, ACS Energy Lett., 2017, 2, 822–830 CrossRef.
- H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821–7861 CrossRef PubMed.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- I. Burgues-Ceballos, M. Stella, P. Lacharmoise and E. Martinez-Ferrero, J. Mater. Chem. A, 2014, 2, 17711–17722 Search PubMed.
- C. McDowell and G. C. Bazan, Curr. Opin. Green Sustainable Chem., 2017, 5, 49–54 CrossRef.
- I. Burgués-Ceballos, F. Machui, J. Min, T. Ameri, M. M. Voigt, Y. N. Luponosov, S. A. Ponomarenko, P. D. Lacharmoise, M. Campoy-Quiles and C. J. Brabec, Adv. Funct. Mater., 2014, 24, 1449–1457 CrossRef.
- J. Cho, S. Yoon, K. Min Sim, Y. Jin Jeong, C. Eon Park, S.-K. Kwon, Y.-H. Kim and D. S. Chung, Energy Environ. Sci., 2017, 10, 2324–2333 Search PubMed.
- S. Zhang, L. Ye, H. Zhang and J. Hou, Mater. Today, 2016, 19, 533–543 CrossRef.
- J. W. John and M. Dealy, Melt Rheology and its Applications in the Plastics Industry, Springer, Netherlands, 2 edn, 2013 Search PubMed.
- C. Müller, C. P. Radano, P. Smith and N. Stingelin-Stutzmann, Polymer, 2008, 49, 3973–3978 CrossRef.
- J. C. Maunoury, J. R. Howse and M. L. Turner, Adv. Mater., 2007, 19, 805–809 CrossRef.
- M. A. Baklar, F. Koch, A. Kumar, E. B. Domingo, M. Campoy-Quiles, K. Feldman, L. Yu, P. Wobkenberg, J. Ball, R. M. Wilson, I. McCulloch, T. Kreouzis, M. Heeney, T. Anthopoulos, P. Smith and N. Stingelin, Adv. Mater., 2010, 22, 3942–3947 CrossRef PubMed.
- Y. Zhao, X. Zhao, M. Roders, A. Gumyusenge, A. L. Ayzner and J. Mei, Adv. Mater., 2017, 29, 1605056 CrossRef PubMed.
- C. Müller, T. A. M. Ferenczi, M. Campoy-Quiles, J. M. Frost, D. D. C. Bradley, P. Smith, N. Stingelin-Stutzmann and J. Nelson, Adv. Mater., 2008, 20, 3510–3515 CrossRef.
- J. Zhao, A. Swinnen, G. Van Assche, J. Manca, D. Vanderzande and B. V. Mele, J. Phys. Chem. B, 2009, 113, 1587–1591 CrossRef PubMed.
- P. Wolfer, P. E. Schwenn, A. K. Pandey, Y. Fang, N. Stingelin, P. L. Burn and P. Meredith, J. Mater. Chem. A, 2013, 1, 5989–5995 Search PubMed.
- C. J. Brabec, M. Heeney, I. McCulloch and J. Nelson, Chem. Soc. Rev., 2011, 40, 1185–1199 RSC.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef PubMed.
- A. J. Moulé and K. Meerholz, Adv. Funct. Mater., 2009, 19, 3028–3036 CrossRef.
- Y. Lee and E. D. Gomez, Macromolecules, 2015, 48, 7385–7395 CrossRef.
- F. Lombeck, A. Sepe, R. Thomann, R. H. Friend and M. Sommer, ACS Nano, 2016, 10, 8087–8096 CrossRef PubMed.
- M. T. Martello, D. K. Schneiderman and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2014, 2, 2519–2526 CrossRef.
- A. Rahmanudin, X. A. Jeanbourquin, S. Hanni, A. Sekar, E. Ripaud, L. Yao and K. Sivula, J. Mater. Chem. A, 2017, 5, 17517–17524 Search PubMed.
- B. Walker, A. B. Tamayo, X.-D. Dang, P. Zalar, J. H. Seo, A. Garcia, M. Tantiwiwat and T.-Q. Nguyen, Adv. Funct. Mater., 2009, 19, 3063–3069 CrossRef.
- A. Sharenko, M. Kuik, M. F. Toney and T.-Q. Nguyen, Adv. Funct. Mater., 2014, 24, 3543–3550 CrossRef.
- Q. Liang, J. Han, C. Song, Z. Wang, J. Xin, X. Yu, Z. Xie, W. Ma, J. Liu and Y. Han, J. Mater. Chem. C, 2017, 5, 6842–6851 RSC.
- X. A. Jeanbourquin, A. Rahmanudin, A. Gasperini, E. Ripaud, X. Yu, M. Johnson, N. Guijarro and K. Sivula, J. Mater. Chem. A, 2017, 5, 10526–10536 Search PubMed.
- A. Gasperini, X. A. Jeanbourquin, A. Rahmanudin, X. Yu and K. Sivula, Adv. Mater., 2015, 27, 5541–5546 CrossRef PubMed.
- P. E. Keivanidis, I. A. Howard and R. H. Friend, Adv. Funct. Mater., 2008, 18, 3189–3202 CrossRef.
- J. Liu, Y. Zhang, H. Phan, A. Sharenko, P. Moonsin, B. Walker, V. Promarak and T.-Q. Nguyen, Adv. Mater., 2013, 25, 3645–3650 CrossRef PubMed.
- A. Sharenko, D. Gehrig, F. Laquai and T.-Q. Nguyen, Chem. Mater., 2014, 26, 4109–4118 CrossRef.
- A. Viterisi, F. Gispert-Guirado, J. W. Ryan and E. Palomares, J. Mater. Chem., 2012, 22, 15175–15182 RSC.
- W. Greenbank, L. Hirsch, G. Wantz and S. Chambon, Appl. Phys. Lett., 2015, 107, 263301 CrossRef.
- L. Yang, S. Zhang, C. He, J. Zhang, H. Yao, Y. Yang, Y. Zhang, W. Zhao and J. Hou, J. Am. Chem. Soc., 2017, 139, 1958–1966 CrossRef PubMed.
- B. Qiu, L. Xue, Y. Yang, H. Bin, Y. Zhang, C. Zhang, M. Xiao, K. Park, W. Morrison, Z.-G. Zhang and Y. Li, Chem. Mater., 2017, 29, 7543–7553 CrossRef.
- H. Bin, Y. Yang, Z.-G. Zhang, L. Ye, M. Ghasemi, S. Chen, Y. Zhang, C. Zhang, C. Sun, L. Xue, C. Yang, H. Ade and Y. Li, J. Am. Chem. Soc., 2017, 139, 5085–5094 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S8, Tables S1–S3 as described in the main text, full synthetic procedure and characterization of MCP. See DOI: 10.1039/c8gc00335a |
| This journal is © The Royal Society of Chemistry 2018 |