
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Valorization of 2,5-furandicarboxylic acid. Diels–Alder reactions with benzyne†
Eric M.
Serum
 a,
Sermadurai
Selvakumar
b,
Nicolas
Zimmermann
c and
Mukund P.
Sibi
*a
a,
Sermadurai
Selvakumar
b,
Nicolas
Zimmermann
c and
Mukund P.
Sibi
*a
aDepartment of Chemistry and Biochemistry, North Dakota State University, Dept. 2735, PO Box 6050, Fargo, North Dakota 58108, USA. E-mail: mukund.sibi@ndsu.edu
bDepartment of Chemistry, Central University of Haryana, Jant-Pali, Mahendergarh-123031, India
cSigma Informatique, 8 rue Newton, CS 84533-44245 La Chapelle sur Erdre Cedex, France
First published on 20th February 2018
Abstract
Biomass-derived 2,5-furandicarboxylic acid was valorized by conversion to 1,4-naphthalenedicarboxylic acid via benzyne-cycloaddition and reductive aromatization in 66% overall yield (four steps). Two novel bicyclic intermediates were isolated in 80% and 98% yield. These advances diversify the potential end uses of renewable terephalic acid analogs and other furanics available from cellulose biorefinery.
Introduction
Investigation of nonedible biomass as a sustainable alternative to petroleum continues to expand as a field of research. This expansion requires the development of biorefinery technologies along with the diversification and concomitant valorization of platform chemicals. Often, investigators choose a petroleum commodity chemical and attempt to create a sustainable alternative either through production of a facsimile or an analogous structure readily obtained from biorefinery products.One petroleum commodity targeted for replacement by both methods is terephthalic acid 1.1 This has been accomplished by renewable routes2,3 to 1 and by substitution of 1 with renewable analogs (Fig. 1).4 The introduction of furanics derived from hemicellulose and cellulose has resulted in their use as dienes5 in Diels–Alder strategies for renewable 1.6–11
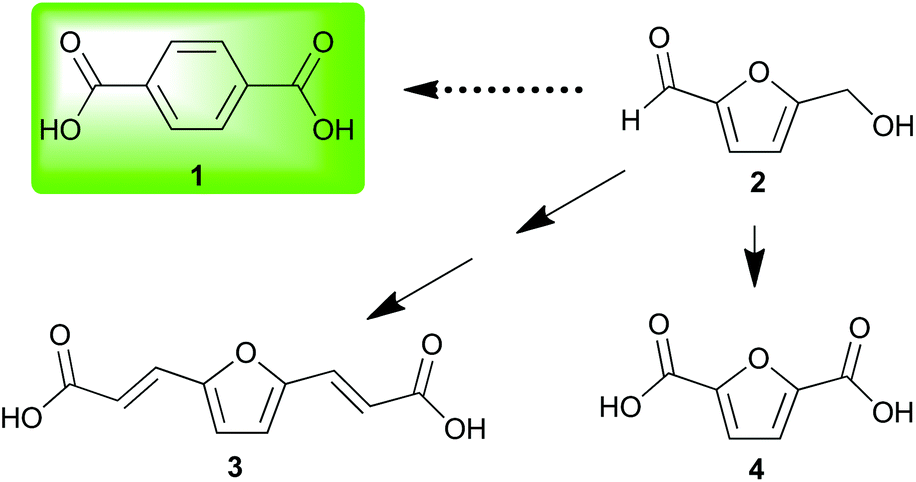 | ||
| Fig. 1 Terephthalic acid and furanic analogs derived from cellulose. | ||
Biomass-derived 5-(hydroxymethyl)furfural 2 along with derivatives—bisacrylic acid 3 and diacid 412—have potential for redox-efficient transformation into benzenoids via cycloaddition followed by aromatization (Scheme 1). However, the electronic demands of the Diels–Alder reaction have led to an impasse which has been met mainly by redox-inefficient use of alkyl-substituted furans and in some cases by use of alkoxyfuranoates prepared via partial oxidation of 2;8,9 no direct synthesis of 1 has been reported from 4 or its esters.
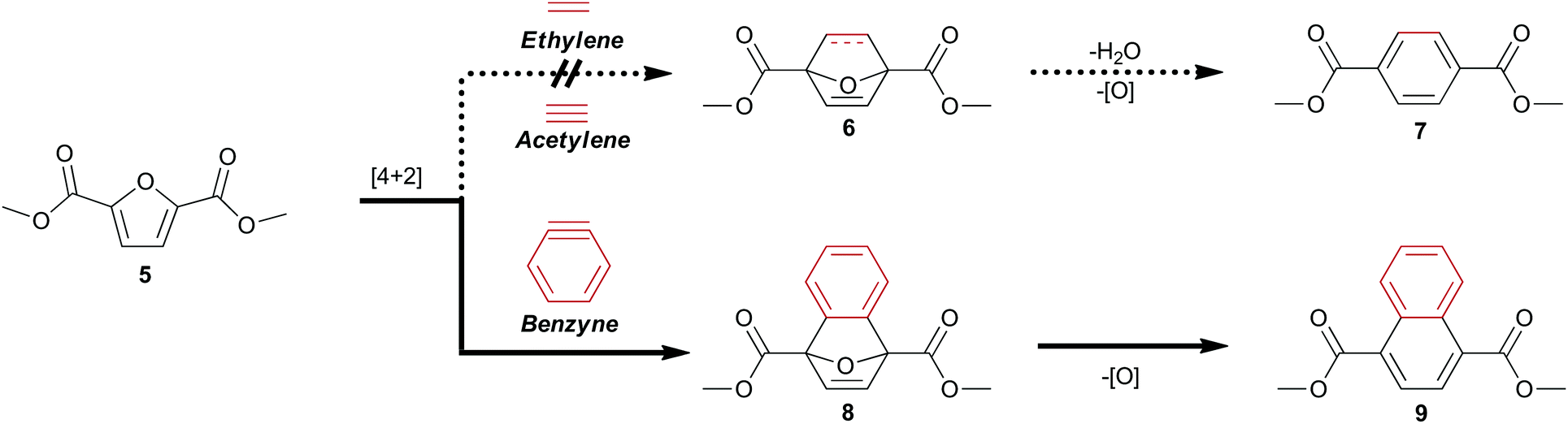 | ||
| Scheme 1 Diels–Alder strategy for direct access to cellulose-derived dimethyl terephthalate 7 and dimethyl 1,4-naphthalenedicarboxylate 9. | ||
Established Diels–Alder protocols involve reaction between derivatives of 2 and pressurized ethylene gas8,9,13 or acrolein.6 A promising approach to 1 under investigation is the thermal rearrangement of phthalic anhydride, which has been prepared from the adducts of furan and maleic anhydride by dehydroaromatization.7,10,11 The 7-oxanorbornene intermediates must be protected from retro–Diels–Alder reaction by immediate dehydration to afford renewable p-xylene13–15 or another drop-in replacement8,9 for industrial precursors of 1.
Having noted the difficulty in obtaining 1 or its ester 7 from biomass-derived furanics by a redox-efficient strategy (Scheme 1), we inferred that the enhanced reactivity16 of benzyne as a dienophile may succeed where the preparation of adduct 6 from ethylene or acetylene would fail. This opportunity to synthesize a naphthalene analog of 1 directly from 4 as well as provide access to novel [2.2.1] oxygen-bridged bicycles (Scheme 1) serves to expand the structural motifs which can be realized from bio-sourced platform chemicals.
This novel valorization strategy relies upon the Diels–Alder reaction between benzyne and electron-deficient furans such as dimethyl 2,5-furandicarboxylate 5. Whereas typical [2.2.1] bicyclic intermediates thusly obtained are unstable, the high energy of benzyne precludes the facile retro–Diels–Alder reaction of adducts such as diester 8 in contrast to traditional adducts such as 6 (Scheme 1).
Diacid substituted norbornenes have been used to prepare amorphous-unsaturated polyester resins which afforded degradable elastomeric thermosets upon treatment with radical initiators.17 The low solids content observed could be remedied by using Diels–Alder adducts such as 7-oxabenzonorbornadienes due to enhanced ring strain.18 Oxabenzonorbornadienes have also been utilized as substrates in ring opening polymerization.19 The sensitivity of those polymers to oxidation has been addressed by blocking the allylic/benzylic bridgeheads with alkyl substitutions;20 presumably carboxy substitutions would also be quite effective while affording additional opportunity for network connectivity. The rich chemistry of 7-oxabenzonorbornadienes led to their use in developing novel polymers as aryne surrogates in the preparation of (o-arylenes).21
While 2,6-naphthalene dicarboxylic acid is considered a high performance alternative to 1,22 comparable polyesters derived from 1,4-naphthalene dicarboxylic acid 10 (Fig. 2) have been classified as amorphous.23 The drastic difference in these two naphthalenedicarboxylic acids is likely due to peri interactions which cause the carboxy moieties to exist in noncoplanar conformations.24 This unique attribute has been used to create integral plasticization of insoluble ridged rod polymers by creating kinks.25 This has the effect of improving their solubility and processability;26 notably, 10 was used to prepare an electrochromic polyamide with good solubility and great potential for use in optoelectronics applications.27
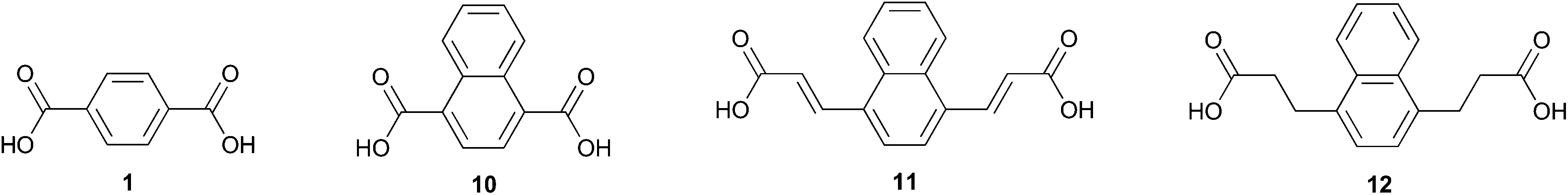 | ||
| Fig. 2 Terephthalic acid and cellulose-derivable naphthalenedicarboxylic acids. | ||
Diacid 10 appears prominently in hybrid organic/inorganic materials such as coordination polymers with diverse applications including chemo sensation of nitro-explosives28 and light-triggered release of adsorbed molecules.29 Often, 10 serves as a spacer in metal organic frameworks (MOFs) and allows for the tuning of structural properties.30–32 Metal–organic gels (MOGs) are important for their application in catalysis,33 gas adsorption, and as chemical sensors. Notably, 10 was used in a MOG to assiduously remove arsenic from aqueous solution; the π–π stacking of the naphthyl ring was credited as driving nanosheet genesis.34
Benzyne reactions with electron-rich furans have been reported in the literature20,35–42 as have their conversion to naphthalenes by various routes. In contrast, there are only a few examples of reactions between benzyne and heavily oxidized furans.43,44 In this work we detail for the first time the Diels–Alder reaction of 5 with benzyne and selective conversion of the adduct to either 1,4-naphthalenedicarboxylic acid 10 (Fig. 2) or its dimethyl ester 9. Diacid 12 was also prepared by application of this strategy while diacid 11 could not be accessed due to side reactions between benzyne and the multiply olefinated oxabenzonorbornadiene.
Results and discussion
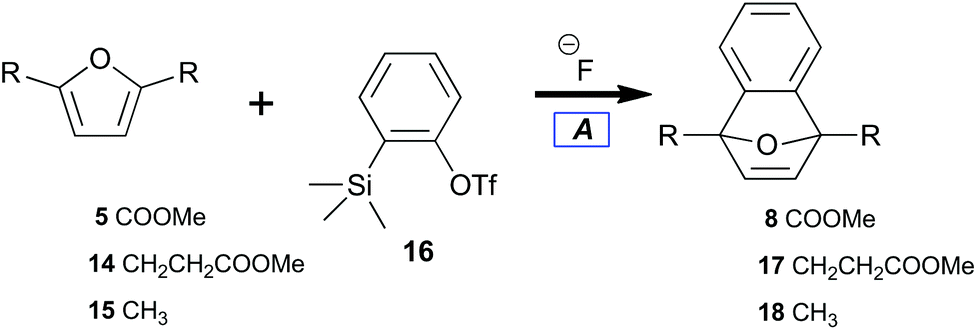
Furan-diesters (5, 13, and 14) with potential for aromatic upgrading to benzenoids were compared with the xylene precursor 15 as targets for conversion to naphthalenes (Fig. 3). The focus of this work was direct access to value added products from furandicarboxylic acids such as 4. However, the extreme reactivity of benzyne makes side reaction with acids45,46 problematic. We therefore required protection of those moieties. Diesters provided an excellent alternative owing to their extensive use in polytransesterification reactions. Diesters are also known for the ease of their purification as compared to corresponding diacids. | ||
| Fig. 3 Cellulose-derived furan-dienes. | ||
Diester 5 was prepared by esterification of crude diacid 4 with methanol; pure 5 was isolated by solid-phase extraction and recrystallization in 93% yield. Diacrylate 13 was prepared from hydroxyaldehyde 2 in three steps with an overall yield of 78%. Dipropionate 14 was prepared by selective reduction of 13 in 93% yield. Furan 15 was commercially available. Complete details of the furan syntheses with discussion of the atom economy47 calculated for each step have been compiled in the ESI.† While atom economy is among the most preliminary of metrics,48 it provides optimal information to guide the initial stage of process development and scale-up.
Benzyne-Diels–Alder reaction
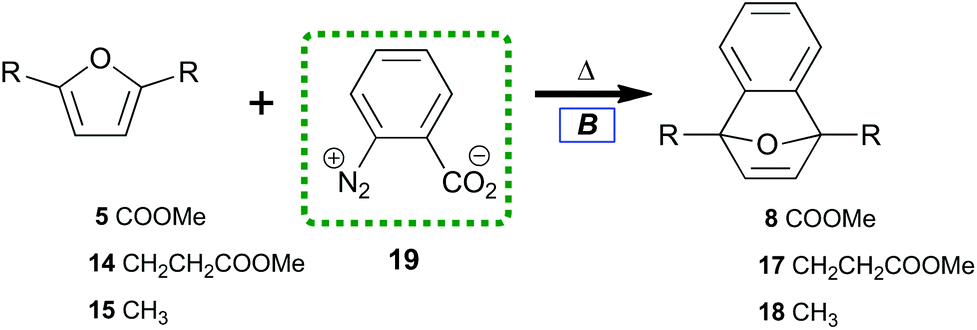
Benzyne can been generated from various synthetic intermediates.49 One of the most useful involves trimethylsilylphenyl-2-triflate50,5116, which undergoes fluoride-induced desilylation followed by elimination of triflate to form benzyne.52 The desilylation protocol tightly controls solution concentrations of benzyne independent of temperature.A preliminary investigation into the feasibility of trapping benzyne with diester 5 (electron deficient) as compared with diester 14 (electron rich) was performed (Table 1); the results indicated the relative benzyne trapping ability of dienes (5, 14, and 15) to afford 7-oxabenzonorbornadienes (8, 17, and 18). While diene 5 required refluxing in acetonitrile to achieve a reasonable conversion (Table 1, entry 1), efficient trapping by alkyl-substituted furans at room temperature was observed. No difference between the sterically more hindered 14versus15 was observed (Table 1, entries 2, 3). Overall, implementation of the desilylation protocol (Table 1) was easy and provided 7-oxabenzonorbornadienes in good yield while indicating a thermal barrier for reaction with diene 5. Unfortunately, there are severe economic limitations to the implementation of such a procedure.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | 20 °C Yielda (%) | 70 °C Yielda (%) |
| A Diene (1 equiv.) was dissolved in MeCN (0.04 M) and CsF (2.2 equiv.) was added to form a suspension. Benzyne precursor 16 (1.3 equiv.) was dissolved in an equal volume of MeCN and added continuously over 16 h at the specified temperature.a Isolated yield. | ||||
| 1 | 5 | 8 | 38 | 84 |
| 2 | 14 | 17 | 97 | — |
| 3 | 15 | 18 | 97 | — |
To contrast the desilylation protocol with the cheapest alternative, we examined the original method for benzyne preparation in non-basic media.53 When first introduced, the thermolysis of benzenediazonium-2-carboxylate 19 (Table 2) provided significant advantages over the organometallic protocols prevalent in the 1960s. The study of diazotized anthranilic acids‡ greatly increased the types of benzyne which could be prepared under neutral conditions. Despite initial excitement for this protocol, recent interest in 19 as a benzyne precursor has lessened due to its established shock sensitivity when isolated.54,55
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yielda (%) |
| B Diene (1 equiv.) and benzyne precursor 19 (1.4 equiv.) were combined and diluted with DCM (0.17 M). The mixture was refluxed for 90 minutes to complete the thermolysis of 19.a Isolated yield.b Diene (98 mmol), 19 (196 mmol), and DCM (0.17 M) were combined and refluxed (90 min). | |||
| 1 | 5 | 8 | 80 |
| 2 | 14 | 17 | 82 |
| 3 | 15 | 18 | 85 |
| 4 | 5 | 8 | 91b |
Importantly, a method of safely employing reactive intermediate 19 has been established on the analytical scale using flow chemistry.56 The advent of flow technology and its proven ability to attenuate dangers associated with harsh reaction conditions/reactive intermediates while providing a linearly scalable platform for organic synthesis may lead to resurgence of aryne research based upon diazotization of anthranilic acids.
Prior to the invention of 16's desilylation,52 procedures had been developed to manage hazards associated with handling of 19. They were addition of an anthranilic acid solution to the reaction mixture at reflux57 or partial isolation of the inner salt without allowing it to dry.58 We chose to modify a partial isolation method59 to juxtapose thermolysis of 19 with the desilylation of 16 for benzyne generation. This decision was motivated by consideration of the excess furan usually required for effective benzyne trapping when 19 is generated in situ.
Under our conditions, each diene trapped benzyne with general uniformity (Table 2). Tetraene 13 did not cleanly transform to 7-oxabenzonorbornadiene-1,4-bisacrylate. Only some adduct could be detected in the reaction along with residual starting material. Many new aromatic peaks along with new aliphatic signals were observed in 1H NMR. This indicated that 13 was an effective diene but that overreaction between benzyne and olefins challenges the efficiency of this process. Furan-dienes (5, 14, and 15) smoothly transformed to 7-oxabenzonorbornadienes (8, 17, and 18). This indicated that 40 °C was sufficient to induce thermolysis while also overcoming the thermal barrier for trapping with electron-deficient dienes.
Further experiments detailed in the ESI† explored the role of initial stoichiometric ratios between 19 and diene; the results showed approximately 60% of benzyne generated by our thermolysis method was trapped by the furans studied. Twofold excess of 19 led to yields above 90% of 7-oxabenzonorbornadienes 8 and 17. Even at that super stoichiometry, some 5 was recovered (6%). The reaction of 5 was safely scaled up to 98 mmol and resulted in 91% isolated yield of 8 as a low-melting crystalline solid (Table 2, entry 4).
Notably, use of excess 19 led to a decreased yield of 18. This result finally differentiated the dialkylfuran substrates by their steric qualities and indicated at least one degradation pathway which disproportionately predated upon sterically vulnerable 7-oxabenzonorbornadienes. Additional degradation of Diels–Alder adducts was observed when reactions were not protected from oxygen and residual nitrites; the nitrogen dioxide–catalyzed epoxidation60 of 7-oxabenzonorbornadienes on the bench has not otherwise been reported.
Diels–Alder reactions have inherent potential for perfect atom economy by incorporation of all atoms from the diene and dienophile into an adduct. However, since benzyne must be generated in situ from a precursor such as 16 or 19, there will be some discarded atoms. These two methods were compared with emphasis on assessing their respective atom economy, nature of side products, and intensity of preparation.
In this preliminary evaluation, we have calculated the atom economy for both Diels–Alder methods as applied to furan 5 since it embodies the direct-access strategy (see ESI† for details). For desilylation, an atom economy of 33% describes the synthesis of 8 as compared with 66% when the thermolysis protocol was employed. There is significantly greater efficiency potential for the thermolysis approach; when the nature of the side products is considered, nitrogen and carbon dioxide would clearly be preferable to trimethylsilyl fluoride and cesium triflate as environmental waste. Preparation of 16 from readily available starting materials is more intensive than preparation of 19. However, the shock sensitivity of 19 requires extreme care and precautions for its safe handling. Halogenated solvents provide a superior reaction medium for the selective conversion of 19 to benzyne.61 This additional environmental limitation to the thermolysis approach was neglected by calculation of atom economy.
Since isolation of the shock-sensitive 19 is diametrically opposed to the principles of green chemistry, the many advantages of this otherwise benign technology are inversely proportional to 19's isolation. For the sake of reducing the process intensity while limiting exposure to hazards associated with 19, we combined the partially-isolated inner salt with furans (5, and 14) and allowed the thermolysis to proceed at 40 °C (Table 2). In attempts to further limit handling, the diazotization of anthranilic acid to afford 19 was kept constant. When the slurry was used without partial isolation, predictably inferior yields were observed. Commensurate yields to the partial isolation procedure were obtained by addition of the raw diazotization mixture—in small aliquots—to the refluxing furan (14) solution! Thus, the hazard associated with 19 can be circumvented by engineering a delivery system which only prepares small quantities of reactive intermediate immediately prior to thermolysis.
Reductive aromatization
With benzyne–Diels–Alder viability established, adducts (8, 17, and 18) were treated with trimethylsilyl iodide to afford naphthalenes (9, 20, and 21) by deoxyaromatization (Table 3).62 Electron-deficient 8 required elevated reaction temperature to undergo deoxygenation (Table 3, entry 1), whereas alkyl-substituted 7-oxabenzonorbornadienes (17 and 18) readily aromatized at ambient temperature (Table 3, entries 2 and 3).|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | 20 °C yielda (%) | 70 °C yielda (%) |
| C 7-Oxabenzonorbornadiene (1 equiv.) was dissolved in MeCN (0.01 M); NaI (5 equiv.) and TMSCl (5 equiv.) were added. The reaction proceeded at the specified temperature for 16 h.a Isolated yield.b Conditions of the aromatization were varied: NaI (3 equiv.), TMSCl (3 equiv.), MeCN (0.15 M). | ||||
| 1 | 8 | 9 | 0 | 25 |
| 2 | 17 | 20 | 98 | — |
| 3 | 18 | 21 | 92b | — |
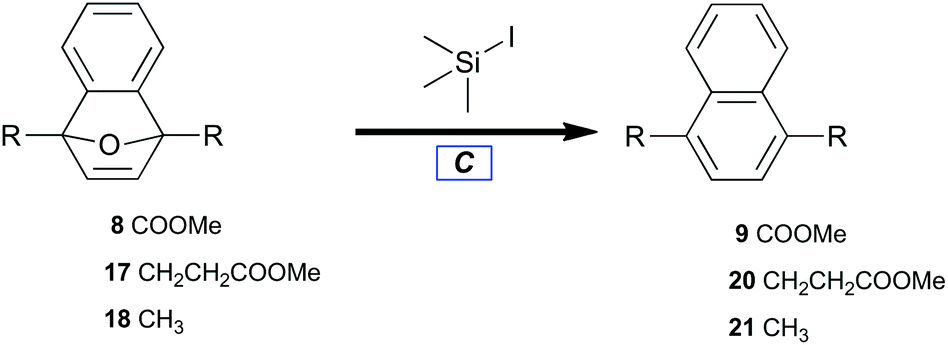
The novelty of bicycle 8 lies not only in its preparation but also in its resistance to ring opening. The stabilizing influence of electron-withdrawing groups at both bridgehead positions may be rationalized by considering the relative stabilities of the tertiary-allylic-benzylic carbenium intermediates developed by spontaneous ring opening. The action of trimethylsilyl iodide likely entails the initial formation of a silyloxonium intermediate. Formation of that intermediate should be irreversible under these conditions due to the comparative bond strengths of Si–O versus Si–I.
To probe the observed stability of bicycle 8, its robust diethyl ester homolog was prepared and subjected to aggressive alkaline hydrolysis. The diester was treated with potassium hydroxide in boiling hot methanol, then neutralized with concentrated hydrochloric acid on ice. The major product was 7-oxabenzonorbornadiene-1,4-dicarboxylic acid (33% yield).
An aromatization strategy that selectively targets reduction apart from the ring-opening reaction offers potential to maximize the efficiency of the overall transformation. The olefin-reactivity of 7-oxabenzonorbornadienes (8, 17, and 18) is independent of substitution at the bridgehead, as evidenced by uniformly efficient catalytic hydrogenations using a flow system (Scheme 2); the hydrogenations also proceeded well under batch conditions and required very little Pd catalyst to convert smoothly (ESI†). Two fringe benefits were observed in the preparation of 7-oxabenzonorbornene diesters (22 and 23): improved processability and stability.
 | ||
| Scheme 2 Flow hydrogenation of 7-oxabenzonorbornadienes. | ||
The diesters were crystalline solids with elevated melting points compared to 7-oxabenzonorbornadienes (8, and 17), and hydrogenation blocked many degradation pathways by removing the reactive olefin. Diesters 22 and 23 were bench stable for over three months with no observable change in appearance or spectrum; liquid 7-oxabenzonorbornene 24 did slowly yellow over time.
A selective method for dehydrating 7-oxabenzonorbornenes (22, 23 and 24) to naphthalenes (9, 20, and 21) was sought (Table 4, Method 1). Amberlyst 15 was chosen as a reusable and highly active acid catalyst for dehydration reactions under nonaqueous conditions. Prevention of ester hydrolysis was a primary concern.
|
|
|||
|---|---|---|---|
| D = Method 1 | |||
| Entry | Substrate | Product | Yielda (%) |
| D 7-Oxabenzonorbornene (1 equiv.) was taken up in DCE (0.3 M), the solution was charged with Amberlyst 15 (15 mol%) and refluxed for 90 minutes. E 7-Oxabenzonorbornene (1 equiv.) was suspended in concentrated HCl (0.17 M), then heated to 100 °C for 2.2 h.a Isolated yield. | |||
| 1 | 22 | 9 | 17 |
| 2 | 23 | 20 | 97 |
| 3 | 24 | 21 | 97 |
| E = Method 2 | |||
| 4 | 22 | 10 | 91 |
| 5 | 23 | 12 | 72 |
| 6 | 24 | 21 | 84 |
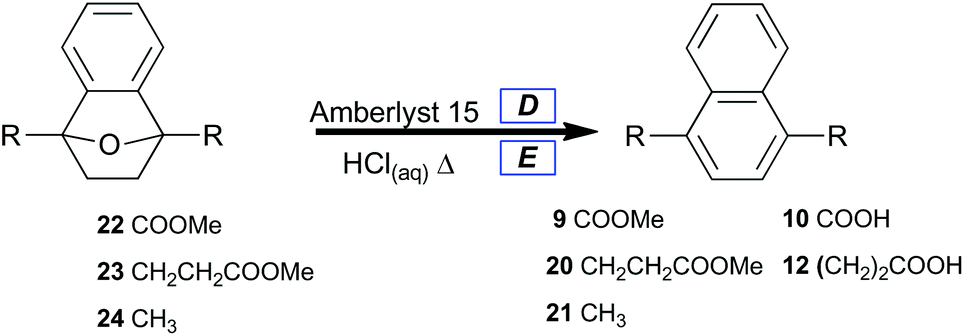
As expected, 7-oxabenzonorbornene 22 converted to dimethyl naphthalene-1,4-dicarboxylate 9 in low yield (Table 4, entry 1) under conditions which completely converted alkyl-substituted 7-oxabenzonorbornenes (23 and 24) to naphthalenes (20 and 21) in high yield (Table 4, entries 2 and 3). This comparison signifies that the enhanced stability of oxanorbornadiene 8 was conserved in oxanorbornene 22, which implies that these novel oxygen-bridged bicycles may lead to the development of new classes of biomass-derived polymers. It also clearly shows that catalytic processes can replace stoichiometric reagents in the aromatization of furan-derived [2.2.1] oxygen-bridged bicycles, which affords significant improvement to the atom economy of the overall approach.
For the deoxyaromatization of 8, an atom economy of only 16% was calculated owing to the use of a super-stoichiometric ring-opening/reducing agent. Even the combination of atom economies from catalytic hydrogenation (100%) and catalytic dehydration (93%) in the case of 8 to 22 to 9 shows more promise for development into an efficient and benign technology. Substituting toluene as the reaction solvent while increasing the reaction time to 12 h afforded diester 9 in 74% isolated yield along with diacid 10 in 15% isolated yield as a side product.
To afford naphthalenedicarboxylic acids directly, 7-oxabenzonorbornenes (22 and 23) were treated with concentrated hydrochloric acid at 100 °C (Table 4, Method 2). This led to the isolation of hydrolyzed naphthalenes (10, and 12) with good yields by suction filtration (Table 4, entries 4, and 5). Naphthalenedicarboxylic acid 12 was isolated in lower yield under the unoptimized conditions, likely due to its greater water solubility. Naphthalene 21 was prepared from 24 and isolated by extraction (Table 4, entry 6).
Since the acid is only required catalytically, atom economy was only slightly lowered to 83% for 10 under these conditions due to the loss of methanol. Owing to the stabilizing influence of the carboxylate moieties at C1 and C4, we surmised that lowering the acid concentration could selectively hydrolyze the ester groups while leaving the strained [2.2.1] bicycle intact.
Obstinate bicycle 22 was treated with 1 M HCl at elevated temperature, and the major component of the isolate was a partial hydrolysis product with an intact oxygen-bridge: 7-oxabenzonorbornyl-1,4-dicarboxylic acid (∼50%). The same conditions afforded dialkylnaphthalene 21 with complete conversion and 82% isolated yield.
Conclusions
Renewable terephthalic acid analog 2,5-furandicarboxylic acid was valorized by efficient diversification (≥80% yield) into several novel bicyclic compounds which may find use in the field of sustainable material science. Those bicycles were further valorized by conversion to naphthalene diacids (≥70%) which are currently derived solely from petroleum and which have been used extensively in the field of coordination polymers.This is the first report describing the challenging direct conversion of dimethyl 2,5-furandicarboxylate into a cycloadduct. Two methods for generating the reactive intermediate, benzyne, were investigated. Anthranilic acid§ was favored as a benzyne precursor to prepare 7-oxabenzonorbornadienes (8 and 17 of Table 2) in ∼80% yield, and catalytic hydrogenation afforded stable 7-oxabenzonorbornenes (22 and 23 of Scheme 2) in >95% yield. Hydrochloric acid mediated hydrolytic-dehydration provided the most atom economical route for preparing naphthalenedicarboxylic acids (10 and 12 of Fig. 2) from oxobridged-bicyclic diesters in good yield (91% and 72% yield respectively).
The atom economy of each step has been compared to assess the potential of a direct-access strategy for developing sustainable chemistries derived from renewable terephthalic acid analogs. Efforts to further quantify and improve the sustainability of this technology while exploring the novel chemistry of these new derivatives of cellulose biorefinery are currently underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science Foundation (grants IIA-1330840 and IIA-1355466) for financial support. EMS thanks ND-EPSCoR for a doctoral dissertation award.Notes and references
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- D. I. Collias, A. M. Harris, V. Nagpal, I. W. Cottrell and M. W. Schultheis, Ind. Biotechnol., 2014, 10, 91–105 CrossRef CAS.
- J. Pang, M. Zheng, R. Sun, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 342–359 RSC.
- D. S. van Es, J. Renewable Mater., 2013, 1, 61–72 CrossRef CAS.
- E. Wenkert, P. D. R. Moeller and S. R. Piettre, J. Am. Chem. Soc., 1988, 110, 7188–7194 CrossRef CAS.
- M. Shiramizu and F. D. Toste, Chem. – Eur. J., 2011, 17, 12452–12457 CrossRef CAS PubMed.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167–175 RSC.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- J. J. Pacheco, J. A. Labinger, A. L. Sessions and M. E. Davis, ACS Catal., 2015, 5, 5904–5913 CrossRef CAS.
- Y. Tachibana, S. Kimura and K.-I. Kasuya, Sci. Rep., 2015, 5, 8249 CrossRef PubMed.
- S. Thiyagarajan, H. C. Genuino, M. Śliwa, J. C. van der Waal, E. de Jong, J. van Haveren, B. M. Weckhuysen, P. C. A. Bruijnincx and D. S. van Es, ChemSusChem, 2015, 8, 3052–3056 CrossRef CAS PubMed.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- Y. P. Wijaya, D. J. Suh and J. Jae, Catal. Commun., 2015, 70, 12–16 CrossRef CAS.
- C. L. Williams, K. P. Vinter, C.-C. Chang, R. Xiong, S. K. Green, S. I. Sandler, D. G. Vlachos, W. Fan and P. J. Dauenhauer, Catal. Sci. Technol., 2016, 6, 178–187 Search PubMed.
- N. G. Rondan, L. N. Domelsmith, K. N. Houk, A. T. Bowne and R. H. Levin, Tetrahedron Lett., 1979, 20, 3237–3240 CrossRef.
- A. H. Brown and V. V. Sheares, Macromolecules, 2007, 40, 4848–4853 CrossRef CAS.
- J. Howell, J. D. Goddard and W. Tam, Tetrahedron, 2009, 65, 4562–4568 CrossRef CAS.
- W. P. Forrest, J. G. Weis, J. M. John, J. C. Axtell, J. H. Simpson, T. M. Swager and R. R. Schrock, J. Am. Chem. Soc., 2014, 136, 10910–10913 CrossRef CAS PubMed.
- J. M. Medina, J. H. Ko, H. D. Maynard and N. K. Garg, Macromolecules, 2017, 50, 580–586 CrossRef CAS PubMed.
- S. Ito, K. Takahashi and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 7547–7550 CrossRef CAS PubMed.
- A. R. Elman, Catal. Ind., 2009, 1, 184 CrossRef.
- C. P. Roupakias, G. Z. Papageorgiou and G. P. Karayannidis, J. Macromol. Sci., Pure Appl. Chem., 2003, 40, 791–805 CrossRef.
- V. Balasubramaniyan, Chem. Rev., 1966, 66, 567–641 CrossRef CAS.
- H. Al-Adwani, A. Bishara and H. Shaban, J. Appl. Polym. Sci., 2003, 89, 1808–1817 CrossRef CAS.
- H.-H. Wang and W.-P. Lin, J. Appl. Polym. Sci., 1997, 65, 1581–1593 CrossRef CAS.
- S.-H. Hsiao, G.-S. Liou, Y.-C. Kung and H.-J. Yen, Macromolecules, 2008, 41, 2800–2808 CrossRef CAS.
- Z.-F. Wu, L.-K. Gong and X.-Y. Huang, Inorg. Chem., 2017, 56, 7397–7403 CrossRef CAS PubMed.
- K. Khaletskaya, J. Reboul, M. Meilikhov, M. Nakahama, S. Diring, M. Tsujimoto, S. Isoda, F. Kim, K.-I. Kamei, R. A. Fischer, S. Kitagawa and S. Furukawa, J. Am. Chem. Soc., 2013, 135, 10998–11005 CrossRef CAS PubMed.
- Y.-L. Liu, P. Liu, K.-B. Li, C.-S. Zhou and K.-F. Yue, J. Mol. Struct., 2017, 1147, 192–196 CrossRef CAS.
- T. Tsuruoka, K. Mantani, A. Miyanaga, T. Matsuyama, T. Ohhashi, Y. Takashima and K. Akamatsu, Langmuir, 2016, 32, 6068–6073 CrossRef CAS PubMed.
- Y. Lou, J. Wang, Y. Tao, J. Chen, A. Mishima and M. Ohba, Dalton Trans., 2014, 43, 8508–8514 RSC.
- M. Tian, X. Cui, M. Yuan, J. Yang, J. Ma and Z. Dong, Green Chem., 2017, 19, 1548–1554 RSC.
- J. Sui, L. Wang, W. Zhao and J. Hao, J. Chem. Soc., Chem. Commun., 2016, 52, 6993–6996 RSC.
- J. Haner, K. Jack, M. L. Menard, J. Howell, J. Nagireddy, M. A. Raheem and W. Tam, Synthesis, 2012, 2713–2722 CAS.
- E. Carlson, J. Haner, M. McKee and W. Tam, Org. Lett., 2014, 16, 1776–1779 CrossRef CAS PubMed.
- M. McKee, J. Haner, E. Carlson and W. Tam, Synthesis, 2014, 1518–1524 Search PubMed.
- A. Tigchelaar, J. Haner, E. Carlson and W. Tam, Synlett, 2014, 2355–2359 CAS.
- W. Yang, R. Luo and D. Yang, Molecules, 2015, 20, 19748 CrossRef PubMed.
- E. Carlson, G. Duret, N. Blanchard and W. Tam, Synth. Commun., 2016, 46, 55–62 CrossRef CAS.
- C. C. J. Loh, M. Schmid, R. Webster, A. Yen, S. K. Yazdi, P. T. Franke and M. Lautens, Angew. Chem., Int. Ed., 2016, 55, 10074–10078 CrossRef CAS PubMed.
- A. Yen, K.-L. Choo, S. K. Yazdi, P. T. Franke, R. Webster, I. Franzoni, C. C. J. Loh, A. I. Poblador-Bahamonde and M. Lautens, Angew. Chem., Int. Ed., 2017, 56, 6307–6311 CrossRef CAS PubMed.
- M. S. Newman and J. A. Cella, J. Org. Chem., 1973, 38, 3482–3484 CrossRef CAS.
- S. Sato, T. Kawada, H. Takikawa and K. Suzuki, Synlett, 2017, 1719–1723 CAS.
- Z. Liu and R. C. Larock, J. Org. Chem., 2006, 71, 3198–3209 CrossRef CAS PubMed.
- M. Stiles, R. G. Miller and U. Burckhardt, J. Am. Chem. Soc., 1963, 85, 1792–1797 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- T. Kitamura, Aust. J. Chem., 2010, 63, 987–1001 CrossRef CAS.
- S. M. Bronner and N. K. Garg, J. Org. Chem., 2009, 74, 8842–8843 CrossRef CAS PubMed.
- D. J. Atkinson, J. Sperry and M. A. Brimble, Synthesis, 2010, 911–913 CAS.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 12, 1211–1214 CrossRef.
- M. Stiles and R. G. Miller, J. Am. Chem. Soc., 1960, 82, 3802–3802 CrossRef CAS.
- T. F. Mich, E. J. Nienhouse, T. E. Farino and J. J. Tufariello, J. Chem. Educ., 1968, 45, 272 CrossRef CAS.
- J. M. Sullivan, J. Chem. Educ., 1971, 48, 419 CrossRef CAS.
- D. L. Browne, S. Wright, B. J. Deadman, S. Dunnage, I. R. Baxendale, R. M. Turner and S. V. Ley, Rapid Commun. Mass Spectrom., 2012, 26, 1999–2010 CrossRef CAS PubMed.
- L. Friedman and F. M. Logullo, J. Org. Chem., 1969, 34, 3089–3092 CrossRef CAS.
- F. M. Logullo, A. H. Seitz and L. Friedman, Org. Synth., 1968, 48, 12 CrossRef CAS.
- L. Friedman and D. F. Lindow, J. Am. Chem. Soc., 1968, 90, 2329–2333 CrossRef CAS.
- E. Bosch and J. K. Kochi, J. Am. Chem. Soc., 1996, 118, 1319–1329 CrossRef CAS.
- C. P. Buxton, M. Fensome, H. Heaney and K. G. Mason, Tetrahedron, 1995, 51, 2959–2968 CrossRef.
- K. Y. Jung and M. Koreeda, J. Org. Chem., 1989, 54, 5667–5675 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc00308d |
| ‡ Anthranilic acid is predominantly prepared industrially from phthalic anhydride. Thus, benzyne prepared from 19 could be made by renewable routes in the future. |
| This journal is © The Royal Society of Chemistry 2018 |