
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatially resolved spectral determination of polysaccharides in hydrothermally carbonized biomass†
Mikko
Mäkelä
 *ab,
Maurizio
Volpe
c,
Roberto
Volpe
d,
Luca
Fiori
c and
Olli
Dahl
a
*ab,
Maurizio
Volpe
c,
Roberto
Volpe
d,
Luca
Fiori
c and
Olli
Dahl
a
aAalto University, School of Chemical Engineering, Department of Bioproducts and Biosystems, PO Box 16300, 00076 Aalto, Finland. E-mail: mikko.makela@aalto.fi; mikko.makela@slu.se
bSwedish University of Agricultural Sciences, Department of Forest Biomaterials and Technology, Skogsmarksgränd, 90183 Umeå, Sweden
cUniversity of Trento, Department of Civil, Environmental and Mechanical Engineering, via Mesiano 77, 38123 Trento, Italy
dQueen Mary University of London, School of Engineering and Materials Science, Mile End Road, London E1 4NS, UK
First published on 7th February 2018
Abstract
Reliable information on the decomposition of polysaccharides is important to evaluate the evolution and properties of hydrothermally carbonized chars. Hyperspectral imaging offers a quick and robust alternative to expensive and time-consuming laboratory methods to determine the polysaccharide contents of biomass and biomass-derived chars. Here, we show that the decomposition of hemicellulose and cellulose were visible in the acquired hyperspectral images even without image calibration. Image regression based on sample holocellulose, glucan and the sum of xylan, galactan, arabinan and mannan provided good calibration models and enabled visualizing the decomposition of polysaccharides based on carbonization temperature. Hyperspectral imaging thus provides a non-destructive alternative to traditional polysaccharide analyses of hydrochars for laboratory and potential future industrial applications.
1. Introduction
The need to develop sustainable and cost-effective bioenergy has increased the innovation and technology development for sustainable energy application of biomass within the European Union. Thermochemical conversion of agricultural and agro-industrial residues into biofuels is particularly attractive, as they do not compete with food crops and are often seen as wastes generated during the primary production of other products. As an example, the olive oil industry generates significant quantities of wet industrial residues, which are currently not utilized to their full potential. A global production of over 2500 kilotons of olive oil per year is associated with the generation of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kilotons of effluents and 20000 kilotons of tree prunings and trimmings, which could potentially be converted into low-cost solid biofuels.1
000 kilotons of effluents and 20000 kilotons of tree prunings and trimmings, which could potentially be converted into low-cost solid biofuels.1
Among different thermochemical conversion technologies, hydrothermal carbonization (HTC) is particularly suitable for wet residual biomass and heterogeneous organic residues.2,3 HTC is performed in subcritical water under relatively low temperatures and self-generated pressure and does not require prior drying of wet feedstocks. During HTC, the hydrolysis of biomass hemicellulose and cellulose to soluble sugars leads to the formation of an increasingly aromatic solid hydrochar.4 Further degradation of soluble sugar monomers into furfurals and organic acids lowers liquid pH and can further catalyze the dehydration of remaining polysaccharides.5 The lack of repeated glycosidic bonds and a more heterogeneous structure makes hemicellulose more vulnerable to hydrothermal degradation than cellulose.6,7 Several research groups have studied the behavior of polysaccharides under hydrothermal conditions and suggested reaction pathways using model compounds, such as cellulose,4,8–10 glucose,4,11 xylan10,12 and xylose.11 Information on the decomposition of polysaccharides is valuable to evaluate the evolution and properties of hydrochars for energy or material applications.
The decomposition of polysaccharides can also be studied using biomass or biomass-derived chars to acquire information on specific feedstocks. Several methods exist to determine the polysaccharide contents of lignocellulosic samples, based mainly on wet chemical analysis13,14 or thermogravimetry.15,16 However, these methods are either expensive, time-consuming and require the use of chemicals, or only provide information on pseudo-components. Spectroscopic methods provide an alternative for quantitative determination of polysaccharides. As an example, near infrared (NIR) spectroscopy measures the overtones and combinations of fundamental vibrational modes of polar molecular bonds and can be used to determine the polysaccharide content of biomass and biomass-derived chars.17–20 Lower molecular absorptivity in the NIR requires longer path lengths than in mid-infrared (IR), but eliminates the need for laborious sample preparation. This opens up possibilities to perform quick and non-destructive spectral measurements on-line.
Spatially resolved NIR spectra can be acquired through hyperspectral imaging, where the chemical information of spectrum is combined with the spatial information of an image.21 Instead of recording a spectrum from an arbitrary point of a sample, entire images are acquired using a continuous range of several hundred different wavelengths. Each pixel then contains a spectrum, which allows evaluating the chemical properties of a sample surface in spatial dimensions. This is an important advantage with heterogeneous samples. One possibility to perform quantitative analysis on hyperspectral images is multivariate image regression, where one or several average spectra are collected from an image and regressed against external reference measurements. A calibration model is determined, which can then be used to predict analyte concentrations in individual image pixels. A useful property of hyperspectral images is that they contain many more pixel objects than spectral variables, which enhances signal to noise ratio and provides distributions of pixel values rather than just one average value.22 This together with predicted concentration images provides robust information on the properties of biomass and biomass-derived chars.
Here we show that hyperspectral NIR imaging and multivariate image regression can be used to determine the polysaccharide contents hydrothermally carbonized agro-industrial residues from olive mills. This method provides a fast and non-destructive alternative to traditional laboratory methods. Our results also illustrate the hydrothermal decomposition of polysaccharides from olive mill residues, which has not yet been clearly shown in the HTC literature. In the future, the decomposition of polysaccharides could possibly be determined on-line at industrial installations that deal with HTC of agro-industrial wastes or other biomass streams for energy or material applications. This would provide rapid information on hydrothermal carbonization and reduce dependency on time-consuming and chemical-intensive laboratory analyses.
2. Materials and methods
2.1. Carbon materials
Hydrochars were produced by HTC of olive mill trimmings and olive mill pulp. A total of 16 samples including untreated raw materials were produced through carbonization at 120, 150, 180, 200, 220, 235 and 250 °C for 0.5 hours with a reactor solid load of 25%. In addition, hydrochars from HTC of Opuntia ficus indica at 180 °C for 0.5 hours and 250 °C for 3 hours and pyrochars from the pyrolysis of olive mill trimmings at 250, 300, 500 and 650 °C for 0.5 hours were prepared. The hydrochars from olive mill pulp and Opuntia ficus indica and the pyrochars from olive mill trimmings were included for calibration purposes. This provided a population of 22 calibration samples. Details on the preparation of the samples have been given elsewhere.23–252.2 Reference analyses
The carbon contents of dried samples were determined with a LECO 628 CHN analyser (LECO Corp.) according to ASTM D-5373. Sample polymer and holocellulose contents were estimated based on respective glucose, xylose, galactose, arabinose, mannose and rhamnose contents determined through dilute acid hydrolysis. The samples were first extracted with acetone according to the guidelines of SCAN-CM 49:03. 250 mL acetone was used in a Soxhlet apparatus with approximately 0.5 g of sample for 2 hours to guarantee removal of extractives and potential oils remaining on char particles. The monosaccharide contents of the extractive-free samples were determined based on the National Renewable Energy Laboratory (NREL) procedure for determination of structural carbohydrates in biomass.26 Sugar recovery standards were prepared from analytical grade D-(+)-glucose, D-(+)-xylose, D-(+)-galactose, D-(+)-mannose, L-(+)-arabinose and L-(+)-rhamnose. Hydrolysed monomers were quantified after filtration based on respective peak areas using a Dionex ICS-3000 ion chromatograph (Dionex Corp.) and corrected to respective polymeric forms on a dried, as-received basis.26 Replicate analyses were performed where sufficient sample was available. The root mean squared errors (RMSE) of replicate analyses were calculated and were 0.70% for the sum of xylan, galactan, arabinan and mannan including anhydro rhamnose, 0.39% for glucan and 0.62% for holocellulose calculated as the sum of all oligomers (Table 1). Performed replicates were not evenly distributed within the sample population, but provided a rough estimate of the repeatability of the method.| Parameter | No of replicates | Range (%) | RMSE (%) |
|---|---|---|---|
| RMSE = root mean squared error. | |||
| Xylan, galactan, arabinan and mannan | 14 | 0–16.5 | 0.70 |
| Glucan | 14 | 0–18.0 | 0.39 |
| Holocellulose | 14 | 0–29.5 | 0.62 |
2.3. Hyperspectral imaging
Hyperspectral images of the samples were taken with a Specim SWIR 3 (Specim, Spectral Imaging, Ltd) camera equipped with a 105 mm OLES macro lens provided by Specim. In the camera setup, two rows of quartz halogen lamps generated polychromatic light and the reflected wavelengths were separated by a grating-prism monochromator followed by a HgCdTe detector array. The camera was operated in line-scanning mode where a line of 384 pixels was continuously recorded on different wavelengths. The spectral range was limited to 1000–2550 nm with a nominal spectral resolution of 5.6 nm, which provided 276 spectral variables. The field of view provided by the lens was 10 mm, which resulted in a pixel size of 26 μm × 26 μm. A small pixel size provided a shallower depth of field, and the samples were pressed in spectrograph sample holders and imaged through quartz glass to minimize variations in surface depth and random light scattering, Fig. A.1 (ESI†). The acquisition time was 10 ms per line, resulting in approximately 30 s per an image that contained two samples. The absorbance in each pixel was calculated based on reflectance and measured Spectralon white reference and dark current intensities.2.4. Image and data analysis
After the imaging procedure, a 576 × 384 pixel hypercube (spatial dimensions 15 × 10 mm) was cropped from the center of each sample image. The individual images of the olive mill trimming samples were then combined to one larger 1152 × 1576 pixel hypercube, which included all eight samples. Principal component analysis (PCA)27 was performed on the combined image after unfolding and spectral preprocessing by standard normal variate (SNV) transformation and mean-centering. Spectral calibration on measured reference values was performed by partial least squares (PLS) regression28,29 on each reference variable separately. The calibration spectra were obtained by vertically splitting each sample image in six parts and calculating the median absorbance on each wavelength. The obtained median spectra were further divided into separate calibration and validation sets. This resulted in a total of 88 calibration and 44 validation objects from 22 samples. Spectral preprocessing was performed by SNV transformation and mean-centering.The PLS calibration models were validated by comparing the measured and predicted values of the validation set. The RMSE of prediction (RMSEP) were determined as:
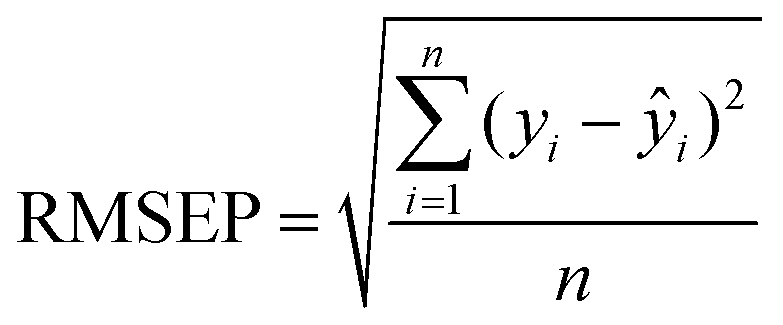 | (1) |
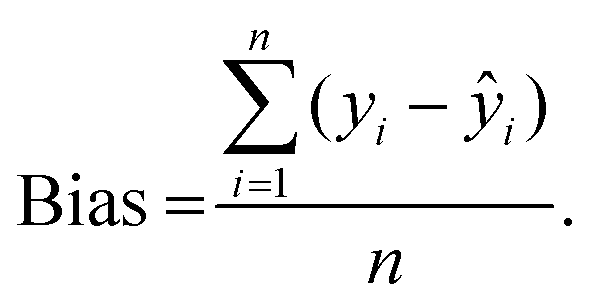 | (2) |
Determined RMSEPs were used to describe model range error ratio (RER) and residual prediction deviation (RPD) parameters based on the validation set through:
 | (3) |
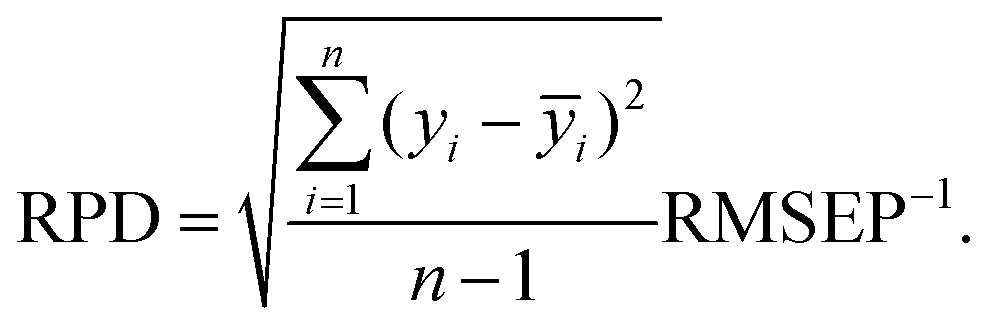 | (4) |
Data analysis and plotting were performed with the Matlab (The Mathworks, Inc.), Matlab PLS Toolbox (Eigenvector Research, Inc.) and OriginPro (Originlab Corp.) software packages.
3. Results
Spectral calibration models for sample carbon, the sum of xylan, galactan, arabinan and mannan, and glucan and holocellulose contents were successfully determined. The final PLS models were composed of 4–5 latent variables and explained 97–99% of the determined reference values during model validation (Table 2). Determined RMSEP values indicated the mean deviations of observed and predicted values during model validation and are given in the original reference units. The RMSEPs for the sum of xylan, galactan, arabinan and mannan, and glucan and holocellulose (0.90–1.7%) were higher, but in the same range as the reference RMSE values (0.35–0.70%) calculated from the performed replicate analyses. The models also showed low prediction bias. The RER and RPD parameters took into account the prediction error and the original range and variation of the determined reference values. The parameters values indicated that the models performed very well and were suitable for quality control.21 As shown in Table 2, the holocellulose model was the most reliable one from the three polysaccharide models. Examples of the used calibration spectra, spectral preprocessing and model predictions for holocellulose are given in Fig. 1.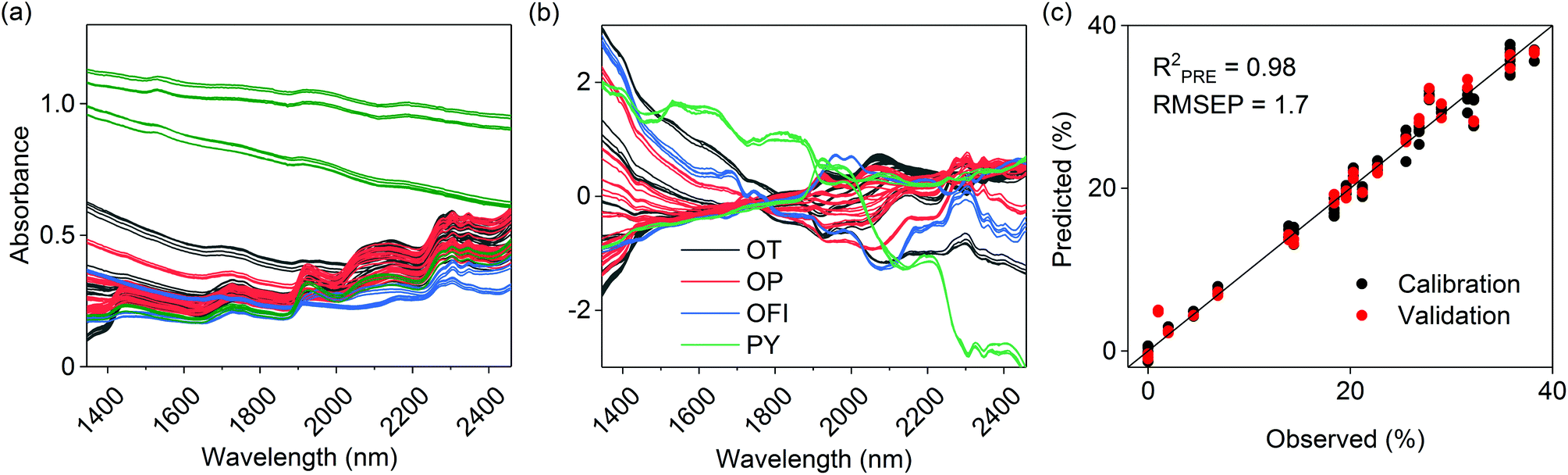 | ||
| Fig. 1 (a) Original and (b) preprocessed calibration spectra for holocellulose calculated as the sum of xylan, galactan, arabinan, mannan and glucan. Observed vs. predicted holocellulose values based on model validation are illustrated in (c). Different colors in (a) and (b) illustrate the classes of included calibration samples; olive mill trimmings (OT), olive mill pulp (OP), Opuntia ficus indica (OFI) and pyrochars (PY). | ||
| Parameter | Calibration/validation objects | Range (%) | Spectral variables (nm) | Model components | R 2 CAL | RMSECAL (%) | R 2 PRE | RMSEP (%) | Prediction bias | RER | RPD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CAL = calibration. PRE = prediction. RMSE = root mean squared error. RMSEP = root mean squared error of prediction. RER = range error ratio. RPD = residual prediction deviation. | |||||||||||
| C (%) | 88/44 | 41.8–73.1 | 1005–2548 | 6 | 0.98 | 1.1 | 0.99 | 0.95 | 0.17 | 32.6 | 8.0 |
| Xylan and others (%) | 86/44 | 0–16.6 | 1005–2236 | 5 | 0.99 | 0.71 | 0.98 | 0.90 | −1.4 × 10−2 | 18.5 | 7.2 |
| Glucan (%) | 84/44 | 0–24.9 | 1345–1900; 2018–2236; 2354–2459 | 5 | 0.98 | 1.25 | 0.97 | 1.2 | −0.31 | 21.2 | 6.9 |
| Holocellulose (%) | 84/44 | 0–38.2 | 1345–2459 | 4 | 0.99 | 1.4 | 0.98 | 1.7 | 0.15 | 22.6 | 7.3 |
4. Discussion
A false color image of untreated olive mill trimmings and the resulting hydrochars is given in Fig. A.2 (ESI†). PCA was first performed on the combined image to determine differences between the samples based on spectral information alone. This enabled to separate measurement noise and summarize relevant information in only a few dimensions before image calibration. The principal component model is essentially a series of linear combinations of pixel scores and the respective loadings, which indicate changes in absorbance on specific wavelengths. Individual principal components describe non-correlated variation in the image data, which enables to obtain information on the physical or chemical properties of the samples. With images, the PCA scores can also be refolded back to the original image dimensions which is helpful for visual interpretation. The first principal component explained 88% of data variation and mainly separated the images based on sample color (Fig. A.3†). This was expected as short wave NIR approaches the visible range, where darker colors manifest as increased absorbance.The second principal component explained 10% of the remaining data variation and provided more detailed information on the chemical properties of the samples. As illustrated in Fig. 2a, clear pixel groupings were observed based on the scores. Hydrochars prepared within 180–235 °C were separated from all the other samples based on positive score values, Fig. A.4.† This was an initial indication that polysaccharides were visible in the images, as the temperature range generally corresponds with the hydrothermal degradation of cellulose.4,20 The positive loading peaks at 2281, 2348 and 2487 nm in Fig. 2b also indicated increasing absorbance of the O–H, C–H and C–C bonds of cellulose30 likely due to the lack of hemicellulose. The peak at 2136 nm suggested that some extractives, acetyl groups of hemicellulose, or lignin was still present.30 Modes of aromatic C–H vibrations have been reported at 1417 nm,30 which together with the decreasing score values of the sample prepared at 250 °C suggested an increasingly aromatic character of the char from higher carbonization temperature. The third and fourth principal components respectively described 0.6 and 0.4% of data variation and mainly seemed to separate differences between polysaccharides and lignin based on the loadings (not shown). However, as shown by the score images in Fig. A.5 and A.6,† they no longer described differences between but rather within the individual samples.
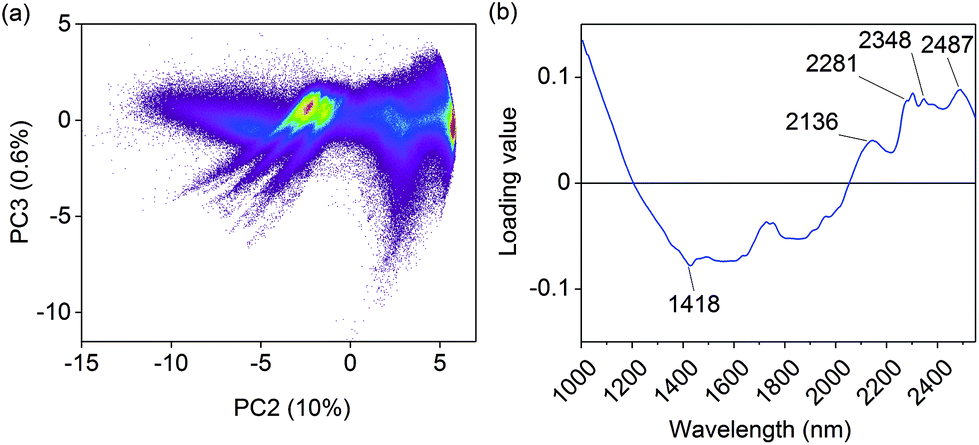 | ||
| Fig. 2 (a) Pixel score values on the second and the third principal component (PC) and (b) the second loading vector. The colors in (a) indicate pixel count density, calculated based on 1.8 million pixels on a grid with square grid bin size. | ||
Calibration based on latent variables is especially suitable for spectroscopic applications as there are often more spectral variables than available samples. In addition, there is often serious collinearity in spectral data as the spectral variables are linearly dependent based on the absorbing constituents. This makes least squares regression based on the original spectral variables either impossible, or highly unstable. PLS regression avoids these problems by reducing the number of variables to a few latent variables that describe the internal dependencies of the data. Where a PCA model describes variation in the spectral data, a PLS model is determined based on covariance between the spectral data and the reference measurements. Thus, not all spectral variables are equally important for predicting the reference values.
We chose the NREL method based on dilute acid hydrolysis26 to determine the monosaccharide contents of the samples and corrected the results to respective polymeric forms for use as reference values for image calibration. Although the polymerization of sugars can bias the estimation of solid residual lignin during acid hydrolysis,31 the method has been reported to provide reliable estimates of soluble polysaccharides if sugar recovery standards are used.32 In our case the total mass yields including extractives, polysaccharides, soluble and Klason lignin, and ash were incomplete and ranged from 62% to 95% in the calibration samples. Of the determined monomers, glucose (≤28%) and xylose (≤15%) were the major species followed by arabinose (≤4%), galactose (≤2%) and mannose (≤1%) on an extractive-free basis. Rhamnose was only found in minute concentrations (<1%) in all samples. The resulting concentrations for glucan and the sum of xylan, arabinan, galactan and mannan could not be used as an absolute measure of cellulose and hemicellulose without further information, but provided reliable indirect estimates on the decomposition of polysaccharides during hydrothermal carbonization. Rhamnose was included in the calculation of mannan. The sum of all corrected polymers however included both hemicellulose and cellulose derived oligomers and could be used as a direct measure of holocellulose in the samples.
The selection of spectral variables for the final polysaccharide models was performed after outlier removal using a combination of genetic algorithms and interval PLS using a window width of 20 variables. Variables were selected based on internal cross-validation, a method to test calibration models without a separate validation set.33 Due to the random nature of genetic algorithms, the final decision on inclusion was based on interval PLS except for the holocellulose model where the algorithm suggested a continuous spectral range (Table 2). The final number of PLS model components was chosen based on RMSEP and visual inspection of the predicted concentration images. For all three polysaccharide models, internal cross-validation overestimated the required number of components based on image inspection. This is a known property of cross-validation.33 A higher number of variables increased image noise, an indication of over-fitting. Future work will be performed with genuine validation samples to verify the performance of the prediction models.
A predicted concentration image of the holocellulose contents of the olive mill trimmings residue and the respective hydrochars is illustrated in Fig. 3a. As shown in the image, a decrease in holocellulose content was already observed in the char prepared at 150 °C followed by complete decomposition in the char from 250 °C. The exact spatial properties of the subimages cannot be compared as they represent different samples, but the images provide an important visualization of the gradual decomposition of holocellulose. Each sample image contained approximately 220000 pixels providing a total of 1.8 million pixels for the whole image. This provides robust information on the variation of holocellulose contents within the samples (Fig. 3b).
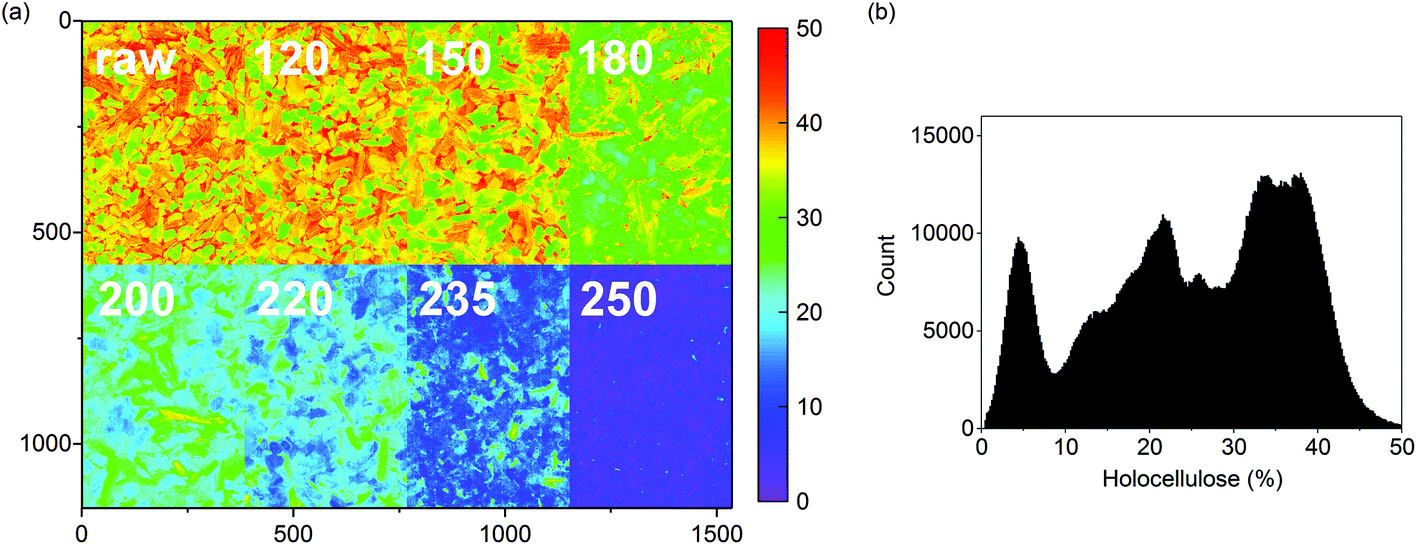 | ||
| Fig. 3 (a) Predicted holocellulose concentrations (%) for the olive mill trimmings samples and (b) the respective image histogram. The color scale in (a) was based on 256 levels within 0–50%. Outlier pixels were removed from (b) by excluding values that situated >|3| standard deviations from the mean based on a normal distribution. This still enabled plotting 99.7% of the predicted pixel values. | ||
Models for the decomposition of glucan and the sum of xylan, galactan, arabinan and mannan were also determined individually. Part of the glucan could have derived from galactoglucomannans or glucomannan commonly found in the hemicellulose fractions of wood, but the low mannose contents suggest that the glucan was mainly cellulose-based. As illustrated in Fig. 4, glucan and the sum of xylan, galactan, arabinan and mannan also showed very different degradation behavior based on carbonization temperature. As also seen in Fig. 3, decrease in the content of xylan and the other oligomers was observed already at 150 °C whereas a significant change in glucan was not observed until 200 °C. Most of the former were also decomposed in the chars prepared at 200 °C, as the char from 235 °C still showed considerable contents of glucan.
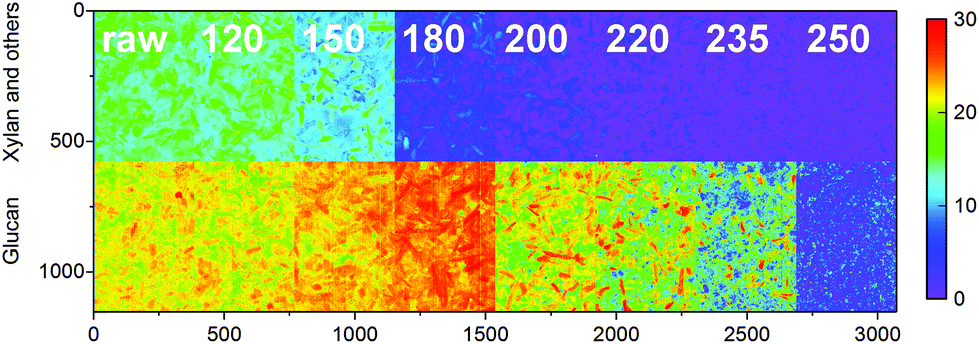 | ||
| Fig. 4 Predicted xylan, galactan, arabinan and mannan, and glucan contents (%) of the olive mill trimmings samples. The color scale in was based on 256 levels within 0–30%. | ||
The decomposition of biomass polysaccharides is often discussed but rarely reported in the HTC literature. The hemicellulose and cellulose contents of loblolly pine and respective hydrochar prepared at 200–260 °C have been determined based on the Van Soest method.34 This method provided direct estimates of the hemicellulose and cellulose content of biomass, but its selectivity for biomass-derived chars has been questioned.35,36 The authors reported complete degradation of hemicellulose by 200 °C, while the char from 260 °C still contained 34% cellulose. Hemicellulose and cellulose have also been determined from sawdust and sawdust-derived chars prepared by HTC at 200–260 °C without detailed specification of the used method.37 Hemicellulose was also almost entirely decomposed by 200 °C, as the char prepared at 260 °C still contained 11% cellulose. Our results on holocellulose and glucan are in closer agreement with the cellulose content of HTC chars from maize silage, another agro-industrial residue.38 Although maize silage chars from 230 °C showed higher cellulose content than suggested by Fig. 3 and 4 within 220–235 °C, the results from 250 °C are comparable.
5. Conclusions
The properties of biomass-derived HTC chars are governed by the hydrolysis of hemicellulose and cellulose. Although their hydrothermal degradation is often discussed in the HTC literature, laboratory methods to determine the polysaccharide contents of biomass and biomass-derived chars are expensive, time-consuming and require the use of chemicals. Hyperspectral imaging provides a robust and reliable alternative for quantitative determination of polysaccharides in biomass and biomass-derived chars. As illustrated by our results, spectral PCA alone provided clear evidence on the decomposition of hemicellulose and cellulose based on carbonization temperature. Further calibration models for sample holocellulose, glucan and the sum xylan, galactan, arabinan and mannan showed good prediction performance and enabled visualizing the decomposition of polysaccharides as a result of HTC. Although the model results for glucan and the sum of xylan, galactan, arabinan and mannan can only be used as indirect estimates for cellulose and hemicellulose, the holocellulose parameter included all cellulose and hemicellulose derived oligomers and also showed the best prediction performance. In the future the decomposition of polysaccharides could possibly be determined and visualized on-line at industrial installations that deal with HTC of agro-industrial residues or other biomass streams for energy and material applications.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank Prof. Paul Geladi from the Swedish University of Agricultural Sciences, Department of Forest Biomaterials and Technology for advice on hyperspectral imaging and Marja Kärkkäinen and Rita Hatakka from Aalto University, Department of Bioproducts and Biosystems for guidance on reference analyses.References
- A. Messineo, R. Volpe and F. Asdrubali, Energies, 2012, 5, 1384–1397, DOI:10.3390/en5051384.
- P. Burguete, A. Corma, M. Hitzl, R. Modrego, E. Ponce and M. Renz, Green Chem., 2016, 18, 1051–1060, 10.1039/c5gc02296g.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290, 10.1039/c4cs00232f.
- C. Falco, N. Baccile and M.-M. Titirici, Green Chem., 2011, 13, 3273–3281, 10.1039/c1gc15742f.
- J. Deng, M. Li and Y. Wang, Green Chem., 2016, 18, 4824–4854, 10.1039/c6gc01172a.
- O. Bobleter, Prog. Polym. Sci., 1994, 19, 797–841, DOI:10.1016/0079-6700(94)90033-7.
- A. Peterson, F. Vogel, R. P. Lachange, M. Fröling, M. J. Antal Jr. and J. W. Tester, Energy Environ. Sci., 2008, 1, 32–65, 10.1039/b810100k.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289, DOI:20.1016/j.carbon.2009.04.026.
- X. Lu, J. R. V. Flora and N. D. Berge, Bioresour. Technol., 2014, 154, 229–239, DOI:10.1016/j.biortech.2013.11.069.
- D. Kim, K. Lee and K. Y. Park, J. Ind. Eng. Chem., 2016, 42, 95–100, DOI:10.1016/j.jiec.2016.07.037.
- M.-M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204–1212, 10.1039/b807009a.
- H. Pińkowska, P. Wolak and A. Złocińska, Biomass Bioenergy, 2011, 35, 3902–3912, DOI:j.biombioe.2011.06.015.
- T. Yokoyama, J. F. Kadla and H.-M. Chang, J. Agric. Food Chem., 2002, 50, 1040–1044, DOI:10.1021/jfo11173q.
- S.-J. Jung, S.-H. Kim and I.-M. Chung, Biomass Bioenergy, 2015, 83, 322–327, DOI:10.1016/j.biombioe.2015.10.007.
- Q.-V. Bach, K.-Q. Tran and Ø. Skreiberg, Appl. Energy, 2016, 185, 1059–1066, DOI:10.1016/j.apenergy.2016.02.056.
- A. Strandberg, P. Holmgren and M. Broström, Fuel Process. Technol., 2017, 156, 107–112, DOI:10.1016/j.fuproc.2016.10.021.
- J. Skravil, K. G. Kyprianidis and E. Dahlquist, Appl. Spectrosc. Rev., 2017, 52, 675–728, DOI:10.1080/05704928.2017.1289471.
- J. Xue, Z. Yang, L. Han, Y. Liu, Y. Liu and C. Zhou, Appl. Energy, 2015, 137, 18–25, DOI:10.1016-j.apenergy.2014.09.089.
- K. Zhang, Y. Xu, L. Johson, W. Yuan, Z. Pei and D. Wang, Renewable Energy, 2017, 109, 101–109, DOI:10.1016/j.renene.2017.03.020.
- M. T. Reza, W. Becker, K. Sachsenheimer and J. Mumme, Bioresour. Technol., 2014, 161, 91–101, DOI:10.1016/j.biortech.2014.03.008.
- M. Manley, Chem. Soc. Rev., 2014, 43, 8200–8214, 10.1039/c4cs00062e.
- M. Mäkelä and P. Geladi, ChemSusChem, 2017, 10, 2751–2757 CrossRef PubMed.
- M. Volpe and L. Fiori, J. Anal. Appl. Pyrolysis, 2017, 124, 63–72, DOI:10.1016/j.jaap.2017.02.022.
- M. Volpe, J. L. Goldfarb and L. Fiori, Bioresour. Technol., 2018, 247, 310–318, DOI:10.1016/j.biortech.2017.09.072.
- R. Volpe, A. Messineo, M. Millan, M. Volpe and R. Kandiyoti, Energy, 2015, 82, 119–127, DOI:10.1016/j.energy.2015.01.011.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, Report TP-510-42618, National Renewable Energy Laboratory, Golden, CO, 2011 Search PubMed.
- S. Wold, K. H. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37–52, DOI:10.1016/0169-7439(87)80084-9.
- P. Geladi and B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1–17, DOI:10.1016/0003-2670(86)80028-9.
- S. Wold, M. Sjöström and L. Eriksson, Chemom. Intell. Lab. Syst., 2001, 58, 109–130, DOI:10.1016/S0169-7439(01)00155-1.
- M. Schwanninger, J. C. Rodrigues and K. Fackler, J. Near Infrared Spectrosc., 2011, 19, 287–308, DOI:10.1255/jnirs.955.
- R. Katahira, J. B. Sluiter, D. J. Schell and M. F. Davis, J. Agric. Food Chem., 2013, 61, 3286–3292, DOI:10.1021/jf303727t.
- S. Zhou and T. M. Runge, Carbohydr. Polym., 2014, 112, 179–185, DOI:10.1016/j.carbpol.2014.05.088.
- K. H. Esbensen and P. Geladi, J. Chemom., 2010, 24, 168–187, DOI:10.1002/cem.1310.
- M. T. Reza, M. H. Uddin, J. G. Lynam, S. K. Hoekman and C. J. Coronella, Biomass Convers. Biorefin., 2014, 4, 311–321, DOI:10.1007/s13399-014-0115-9.
- H. K. Goering and P. J. van-Soest, Forage fiber analysis, Agricultural Handbook No. 379, United States Department of Agriculture, Washington DC, 1970, pp. 1–9 Search PubMed.
- J. G. Lynam, M. T. Reza, W. Yan, V. R. Vásquez and C. J. Coronella, Biomass Convers. Biorefin., 2015, 5, 173–181, DOI:10.1007/s13399-014-0137-3.
- H. Li, S. Wang, X. Yuan, Y. Xi, Z. Huang, M. Tan and C. Li, Bioresour. Technol., 2018, 249, 574–581, DOI:10.1016/j.biortech.2017.10.046.
- M. T. Reza, A. G. Borrego and B. Wirth, Int. J. Coal Geol., 2014, 134–135, 74–79, DOI:10.1016/j.coal.2014.09.015.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc03676k |
| This journal is © The Royal Society of Chemistry 2018 |