
Closed cycle production of concentrated and dry redispersible colloidal lignin particles with a three solvent polarity exchange method†

Abstract
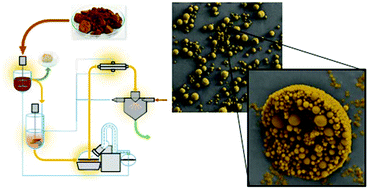
Lignin, an aromatic biopolymer, is the main by-product of pulp manufacture, and has been under intense study, as it offers great promise as an alternative for petrochemical polymers. However due to its heterogeneity, the applications where lignin can be used have been limited, leading to the vast majority of it being burned for fuel. Colloidal lignin particles (CLPs) offer a means to disperse lignin homogenously into both water and other media, such as polymers. However, no means thus far have been presented that would allow for a large-scale production of CLPs. Herein we show an industrially scalable closed cycle process of CLP production. In the process, a concentrated solution of lignin in tetrahydrofuran (THF) and ethanol (EtOH) is added into the non-solvent water, instantaneously forming CLPs through self-assembly. The organic solvents are recovered and reused in the process. The aqueous CLPs are concentrated by ultrafiltration and the concentrated particles are spray dried, leading to redispersible microclusters. CLPs can be used in multiple applications, such as Pickering emulsions and composite materials. A significant portion of the 50 million tons of lignin produced by the pulp industry could be made into CLPs with this low cost process, which would open a whole new class of materials for industrial applications.
 Please wait while we load your content...
Please wait while we load your content...

