
Catalyst- and solvent-free bisphosphinylation of isothiocyanates: a practical method for the synthesis of bisphosphinoylaminomethanes†
Li-Rong
Wen
 ab,
Yong-Xu
Sun
a,
Jin-Wei
Zhang
b,
Wei-Si
Guo
*ab and
Ming
Li
*a
ab,
Yong-Xu
Sun
a,
Jin-Wei
Zhang
b,
Wei-Si
Guo
*ab and
Ming
Li
*a
aState Key Laboratory Base of Eco-Chemical Engineering, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: nick8110@163.com; liming928@qust.edu.cn
bState Key Laboratory for Marine Corrosion and Protection, Luoyang Ship Material Research Institute (LSMRI), Qingdao 266101, P. R. China
First published on 27th November 2017
Abstract
A general and convenient double addition of phosphine oxides to isothiocyanates is described. It is a practically useful protocol for the construction of bisphosphinoylaminomethanes. The reaction can be carried out smoothly under metal- and solvent-free conditions. It also features a broad substrate scope, simple operation and purification. A possible mechanism involving a tandem double nucleophilic addition/H2S elimination/in situ imine reduction process is proposed.
Introduction
Organophosphorus reagents are an important class of compounds in organic synthesis, pharmaceutical chemistry, coordination chemistry, and materials science.1 Recently, the phosphinoyl group as a directing group for C–H activation has also been reported.2 Therefore, synthetic methods of constructing organophosphorus compounds have received great attention in the past decade.3 Among them, bisphosphorous aminomethane derivatives have gained significant attention. Several clinical drugs containing the bisphosphorous aminomethane framework have been used to treat a variety of diseases such as hypercalcemia and osteoporosis.4 Some derivatives are reported to possess herbicidal,5 antibacterial,6 and antiparasitic activities.7 However, synthetic methods for these bisphosphorus compounds have been limited,8 especially for bisphosphinoyl derivatives. As shown in Scheme 1a, Han et al. demonstrated that bisphosphinoylaminomethanes were obtained by rhodium catalysed double addition of diarylphosphine oxide to isocyanide.9 Very recently, Schmidt et al. developed a lanthanum-catalyzed reaction through the bisphosphinylation of nitriles (Scheme 1b).10 Although these protocols are efficient, they all used expensive catalysts, such as Rh and La reagents. They also have other drawbacks including a limited substrate scope and a long reaction time. Therefore, from a sustainability perspective, developing a general and environmentally benign approach for the construction of bisphosphinoylaminomethanes would be highly desirable.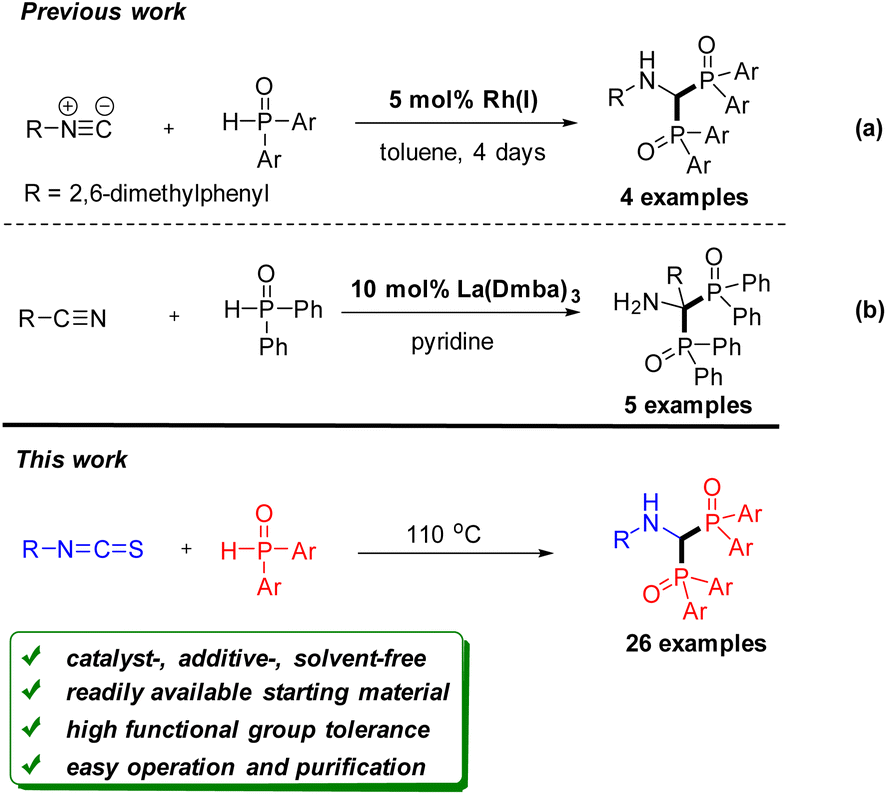 | ||
| Scheme 1 The synthesis of bisphosphinoylaminomethanes. | ||
Isothiocyanates are versatile intermediates in organic synthesis. Because of their high electrophilicity, isothiocyanates could be easily trapped by many nucleophiles for constructing synthetically useful structures through the carbon–carbon and carbon–heteroatom bond formation.11 In addition, they could also be used as nucleophiles12 and radical receptors.13 Recently, our group has introduced a series of convenient methods with isothiocyanates to provide sulphur-containing heterocyclic compounds.14 As an ongoing endeavour to demonstrate the wide-ranging applications of isothiocyanates, we describe herein a new convenient protocol to synthesize bisphosphinoylaminomethanes from isothiocyanates and phosphine oxides under solvent-free conditions. The bis-addition of phosphine oxides to isothiocyanates could be an attractive method for the construction of the C–P bond, due to the following advantages: (1) The reaction was carried out under catalyst-, additive-, and solvent-free conditions. (2) The procedure is easy to operate, and pure products can be readily afforded by washing the crude adducts with a small amount of solvent. (3) An interesting mechanism involving a double nucleophilic addition/H2S elimination/in situ imine reduction cascade is proposed.
Results and discussion
Initially, phenyl isothiocyanate 1a and diphenylphosphine oxide 2a were employed as model substrates for reaction condition optimization. To our delight, the reaction proceeded smoothly to afford the desired product 3a in 45% yield in the presence of Cu(OAc)2 in 1,4-dioxane at 110 °C for 6 h (Table 1, entry 1). However, no improvement was observed when other low-cost Lewis acids such as Fe(OAc)2 and Mn(OAc)2·4H2O were examined (Table 1, entries 2 and 3). Interestingly, the yield of 3a was increased to 53% in the absence of Lewis acid, indicating that the acid was not essential in the transformation (Table 1, entry 4). Comparable yields were obtained in other solvents such as acetonitrile, toluene, and DCE (Table 1, entries 5–7). To our surprise, the yield of 3a was increased to 70% under solvent-free conditions (Table 1, entry 8).15 Furthermore, the reaction also proceeded well under air (Table 1, entry 9), which makes the operation more convenient. A diminished yield was obtained when the reaction was carried out at a lower temperature (Table 1, entry 10), while a comparative yield was achieved at a higher temperature (Table 1, entry 11). Finally, the optimized procedure involved stirring 1a and 2a under catalyst- and solvent-free conditions at 110 °C for 6 h.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (equiv.) | Solvent | T [°C] | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (0.8 mmol), 6 h, under a N2 atmosphere. b Isolated yield based on 1a. c Under air. | ||||
| 1 | Cu(OAc)2 (1.0) | 1,4-Dioxane | 110 | 45 |
| 2 | Fe(OAc)2 (1.0) | 1,4-Dioxane | 110 | 30 |
| 3 | Mn(OAc)2·4H2O (1.0) | 1,4-Dioxane | 110 | 43 |
| 4 | 1,4-Dioxane | 110 | 53 | |
| 5 | CH3CN | 110 | 49 | |
| 6 | Toluene | 110 | 55 | |
| 7 | DCE | 110 | 47 | |
| 8 | 110 | 70 | ||
| 9 | 110 | 70 | ||
| 10c | 100 | 52 | ||
| 11c | 120 | 68 | ||
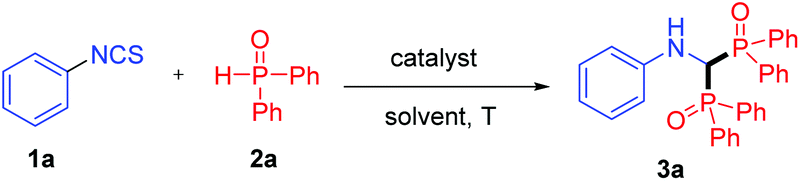
With the optimized conditions being established, the substrate scope of isothiocyanates 1 was explored (Table 2). Isothiocyanates bearing electron-withdrawing substituents (–COOMe, –COMe, –CN, –CF3, –NO2) at the para position of the phenyl ring all served as good substrates, and the corresponding products 3b–f were obtained in good yields (76–87%). Substrates containing halogen groups (–F, –Cl, –Br) also transformed smoothly to products 3g–i in good yields (74–77%), facilitating further functionalization. Whereas when isothiocyanates bearing an electron-donating group such as the methyl group and the methoxy group were employed, the products 3j and 3k were generated in moderate yields. These results indicate that the electronic properties of isothiocyanates play an important role in the reactions. In addition, isothiocyanates with a chloro substituent at the ortho- or meta-position of the phenyl ring could react with 2a to afford the products 3l and 3m in 66% and 71% yields, respectively. It is noteworthy that the reaction also proceeded with sterically hindered ortho substituents, albeit with relatively lower yields (3n and 3o). Moreover, the reaction was compatible with disubstituted substrates, providing the corresponding products 3p and 3q in good yields. A heterocycle such as pyridine was also tolerated in the reaction, and the product 3r was isolated in 57% yield. The structures of compounds 3 were undoubtedly confirmed by the X-ray crystallographic analysis of 3o (see the ESI, Fig. S1†). Aliphatic (methyl or cyclohexyl) isothiocyanates were also employed under the optimized reaction conditions, however, instead of the bisphosphinylation, the mono-addition adducts were obtained in good yields. In addition, a similar mono-addition product was generated when phenyl isocyanate was used to react with 2a.16
It is worth mentioning that this solvent-free protocol is operationally simple. Firstly, isothiocyanates and diphenylphosphine oxide were mixed in a 15 mL sealed tube. Then, a homogeneous solution was generated when heating the mixture to 110 °C. After completion, ethyl acetate was added to the mixture. The pure product 3 was obtained as a solid by simple filtration (see the ESI, Fig. S2†).
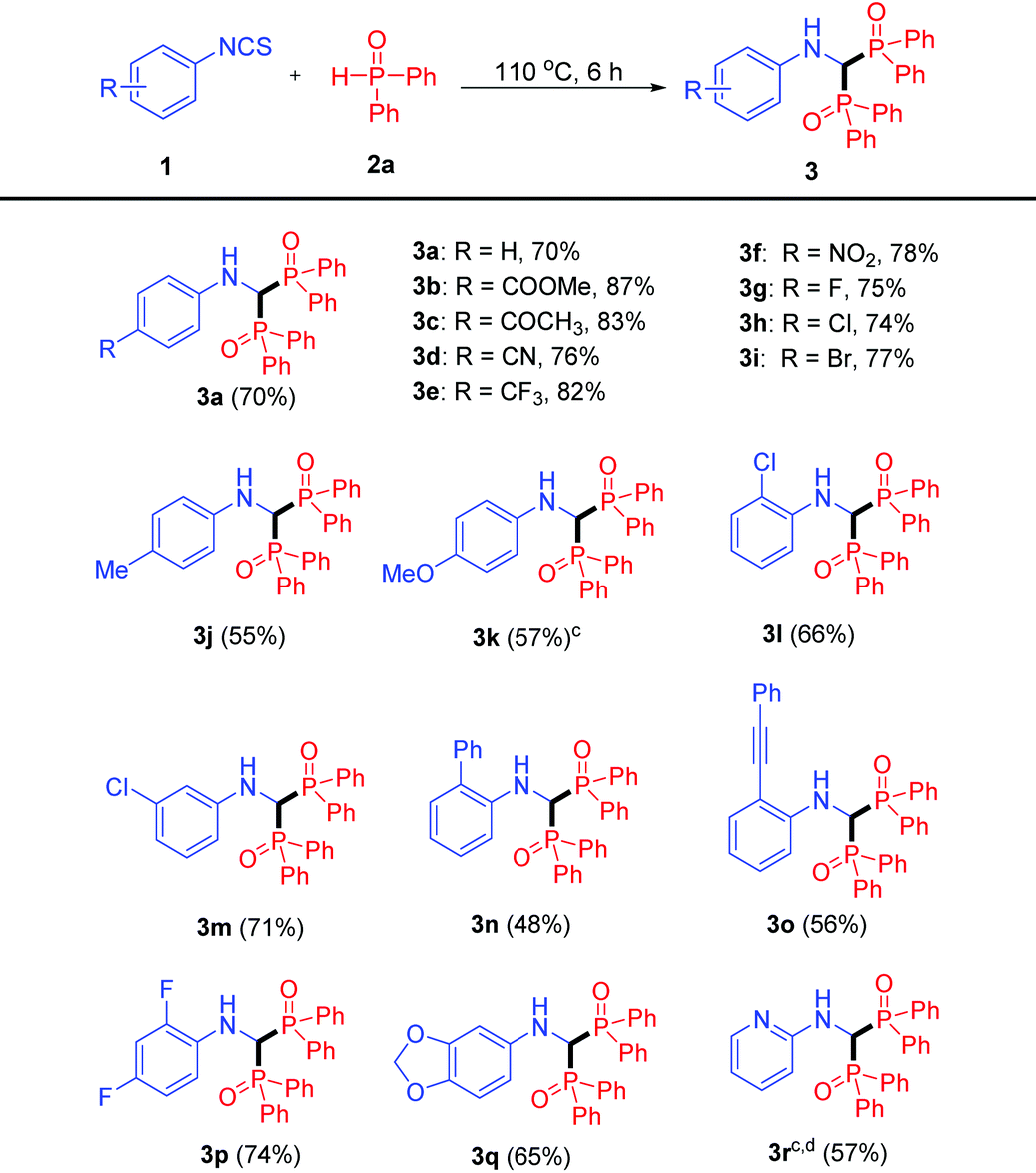
Having established the scope of isothiocyanates 1, we moved on to screen the scope of P-reagents 2 (Table 3). Regardless of their electronic properties, the desired products 3s and 3t were afforded with good yields. The reaction also proceeded smoothly with the ortho methyl group on P-reagents, albeit with a lower yield (35%). In addition, the 3,5-dimethyl substrate still achieved 3v in 65% yield. Unfortunately, no desired product 3w was generated when dimethyl phosphonate was used.
To demonstrate the practicality of this transformation, a scaled-up synthesis was performed under the standard conditions (Scheme 2, eqn (1)). The reaction of 4 mmol isothiocyanate 1a with 2a produced the corresponding product 3a in 65% yield (1.32 g).
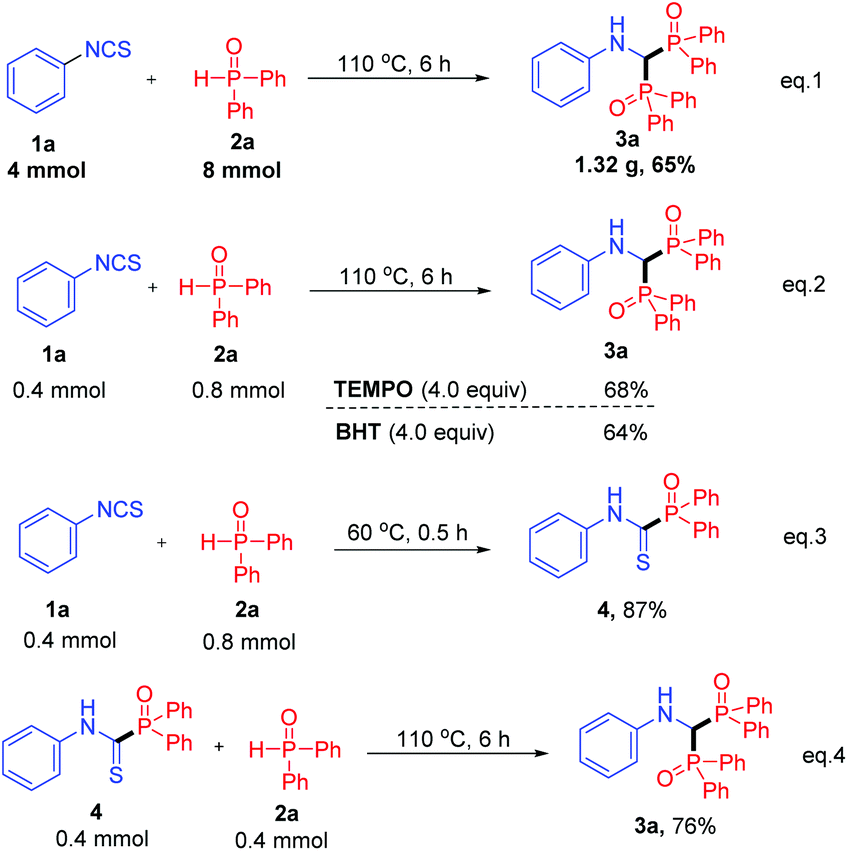 | ||
| Scheme 2 Gram-scale reactions and control experiments. | ||
Some control experiments were conducted to elucidate the reaction mechanism. Firstly, considering that the reactions with Ph2P(O)H usually proceeded in a radical pathway, 4 equiv. of 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) were added to a mixture of 1a and 2a. The reaction was not influenced and product 3a was isolated in 68% or 64% yield, respectively (Scheme 2, eqn (2)). These results indicated that the reaction might not proceed in a radical pathway. Remarkably, an intermediate thioamide 4 was separated in 87% yield when the reaction temperature was decreased to 60 °C (Scheme 2, eqn (3)), which smoothly converted to product 3a under the standard conditions, confirming the hypothesis that compound 4 was an important intermediate (Scheme 2, eqn (4)).
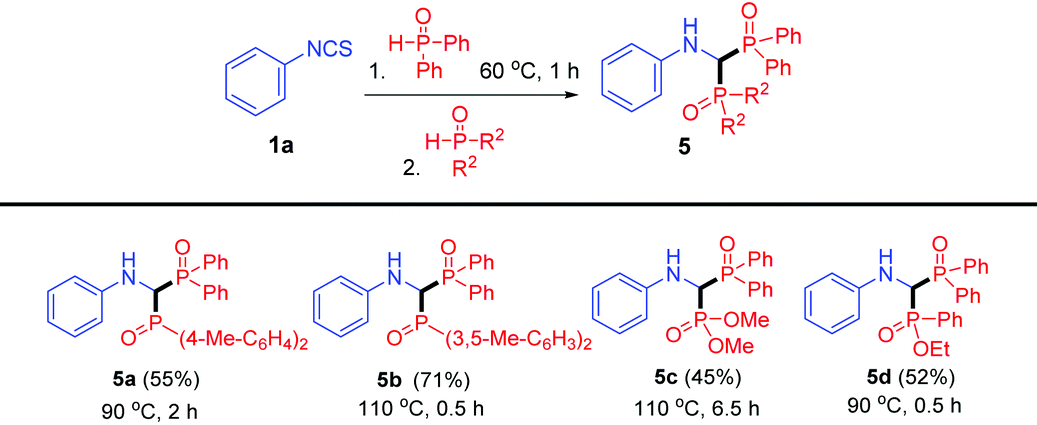
Inspired by the isolation of thioamide 4 (Scheme 2, eqn (3)), we proposed that bisphosphinoylaminomethanes 5 would be obtained with two different phosphorus reagents (Table 4). Compared to products 3, the synthesis of bisphosphinoylaminomethane derivatives with two different P-substituents is still undeveloped. To our delight, the reactions of 1a with two different diarylphosphine oxides were carried out smoothly, and the desired products 5a and 5b were generated in 55% and 71% yields, respectively. It is worth mentioning that dimethyl phosphonate and ethyl phenylphosphinate were also tolerated, and the unsymmetrical products 5c and 5d were generated in moderate yields.
Based on the above experimental results, a possible mechanism is proposed for this transformation. As described in Scheme 3, initially, the diarylphosphine oxide 2 can tautomerize to the P–OH form 2′.17 Then, thioamide 4 is formed by the nucleophilic addition of 2′ to the NCS group in 1. Next, a second nucleophilic addition18 occurs on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond of thioamide 4 to generate intermediate A. Consequently, the 1,1-bisphosphorylimine B is produced with H2S elimination.19 Finally, reduction of the imine with the in situ generated H2S results in the formation of bisphosphinoylaminomethane derivatives 3 with loss of sulfur element (see Fig. S5†). To the best of our knowledge, the efficient imine reduction by the in situ generated H2S is unprecedented.
S bond of thioamide 4 to generate intermediate A. Consequently, the 1,1-bisphosphorylimine B is produced with H2S elimination.19 Finally, reduction of the imine with the in situ generated H2S results in the formation of bisphosphinoylaminomethane derivatives 3 with loss of sulfur element (see Fig. S5†). To the best of our knowledge, the efficient imine reduction by the in situ generated H2S is unprecedented.
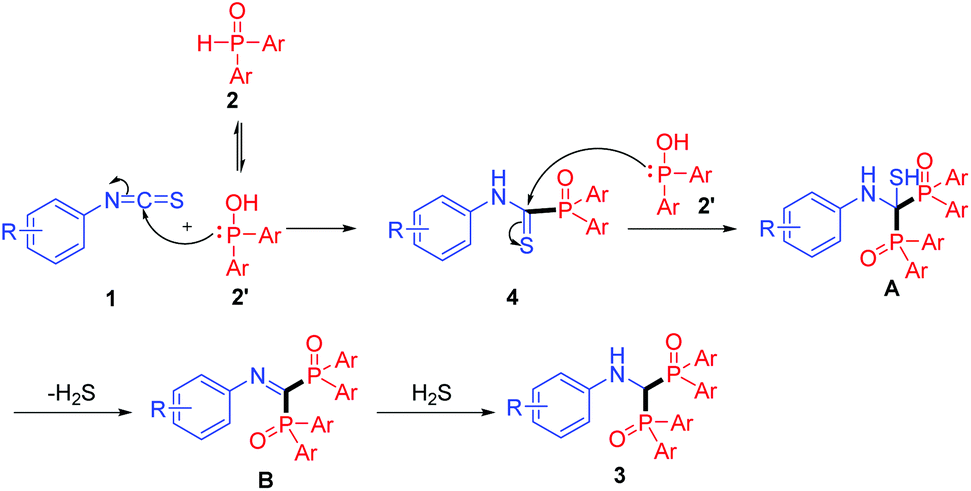 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed the first bisphosphorylation reaction of isothiocyanates. The environmentally friendly reaction proceeded smoothly in a solvent-free manner, and no extra catalysts and additives are required. A variety of bisphosphinoylaminomethanes can be readily isolated in pure form by simply washing the crude products with ethyl acetate. This protocol is very practical and easily scaled-up, which provides a direct way for the preparation of bisphosphoryl derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21372137 and 21572110), the Key Research and Development project of Shandong province (2017GHY15125), the Science and Technology Development Project in the University of Shandong Province (J16LC12), the Applied Basic Research Project (Youth Special) of Qingdao (16-5-1-98-jch), and the Research Fund of State Key Laboratory for Marine Corrosion and Protection of Luoyang Ship Material Research Institute (No. KF160404). Zhen-Jiang Liu and Yue-Xiang Chen (QUST) are acknowledged for their contributions in the synthesis of isothiocyanates.Notes and references
- (a) Phosphorus Chemistry, Biochemistry and Technology, ed. D. E. C. Corbridge, Taylor and Francis Group, London, 2012 Search PubMed; (b) Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological, ed. V. P. Kukhar and H. R. Hudson, Wiley and Sons, Chichester, New York, 2000, pp. 1–634 Search PubMed; (c) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC; (d) P. Sutra and A. Igau, Coord. Chem. Rev., 2016, 308, 97–116 CrossRef CAS; (e) A. Nordheider, J. D. Woollins and T. Chivers, Chem. Rev., 2015, 115, 10378–10406 CrossRef CAS PubMed; (f) J. Crassous and R. Réau, Dalton Trans., 2008, 6865–6876 RSC; (g) A. A. Reszka and G. A. Rodan, Mini–Rev. Med. Chem., 2004, 4, 711–719 CAS.
- (a) X. Qiu, M. Wang, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 7233–7237 CrossRef CAS PubMed; (b) Y. Yang, R. Li, Y. Zhao, D. Zhao and Z. Shi, J. Am. Chem. Soc., 2016, 138, 8734–8737 CrossRef CAS PubMed.
- (a) X. Gong, J. Chen, J. Liu and J. Wu, Org. Chem. Front., 2017, 4, 2221–2225 RSC; (b) J.-Q. Zhang, T. Chen, J.-S. Zhang and L.-B. Han, Org. Lett., 2017, 19, 4692–4695 CrossRef CAS PubMed; (c) J. Li, W.-W. Zhang, X.-J. Wei, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Org. Lett., 2017, 19, 4512–4515 CrossRef CAS PubMed; (d) H. Guo, A. Yoshimura, T. Chen, Y. Saga and L.-B. Han, Green Chem., 2017, 19, 1502–1506 RSC; (e) P. Xie, J. Wang, J. Fan, Y. Liu, X. Wo and T.-P. Loh, Green Chem., 2017, 19, 2135–2139 RSC; (f) F. Su, W. Lin, P. Zhu, D. He, J. Lin, H.-J. Zhang and T.-B. Wen, Adv. Synth. Catal., 2017, 359, 947–951 CrossRef CAS; (g) J. Sun, J.-K. Qiu, Y.-N. Wu, W.-J. Hao, C. Guo, G. Li, S.-J. Tu and B. Jiang, Org. Lett., 2017, 19, 754–757 CrossRef CAS PubMed; (h) X.-T. Zhu, Q. Zhao, F. Liu, A.-F. Wang, P.-J. Cai, W.-J. Hao, S.-J. Tu and B. Jiang, Chem. Commun., 2017, 53, 6828–6831 RSC; (i) P. Zhang, Y. Gao, L. Zhang, Z. Li, Y. Liu, G. Tang and Y. Zhao, Adv. Synth. Catal., 2016, 358, 138–142 CrossRef CAS; (j) P. Xu, Z. Wu, N. Zhou and C. Zhu, Org. Lett., 2016, 18, 1143–1145 CrossRef CAS PubMed; (k) Y. Zhu, T. Chen, S. Li, S. Shimada and L.-B. Han, J. Am. Chem. Soc., 2016, 138, 5825–5828 CrossRef CAS PubMed; (l) J. Ke, Y. Tang, H. Yi, Y. Li, Y. Cheng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 6604–6607 CrossRef CAS PubMed; (m) J. Xu, X. Li, Y. Gao, L. Zhang, W. Chen, H. Fang, G. Tang and Y. Zhao, Chem. Commun., 2015, 51, 11240–11243 RSC; (n) N. Liu, L.-L. Mao, B. Yang and S.-D. Yang, Chem. Commun., 2014, 50, 10879–10882 RSC.
- (a) L. Widler, K. A. Jaeggi, M. Glatt, K. Müller, R. Bachmann, M. Bisping, A.-R. Born, R. Cortesi, G. Guiglia, H. Jeker, R. Klein, U. Ramseier, J. Schmid, G. Schreiber, Y. Seltenmeyer and J. R. Green, J. Med. Chem., 2002, 45, 3721–3738 CrossRef CAS PubMed; (b) R. G. G. Russell, P. I. Croucher and M. J. Rogers, Osteoporosis Int., 1999, 2, 66–80 CrossRef.
- P. Kafarski, B. Lejczak and G. Forlani, Heteroat. Chem., 2000, 11, 449–453 CrossRef CAS.
- A. Leon, L. Liu, Y. Yang, M. P. Hudock, P. Hall, F. Yin, D. Studer, K.-J. Puan, C. T. Morita and E. Oldfield, J. Med. Chem., 2006, 49, 7331–7341 CrossRef CAS PubMed.
- E. Kotsikorou, Y. Song, J. M. W. Chan, S. Faelens, Z. Tovian, E. Broderick, N. Bakalara, R. Docampo and E. Oldfield, J. Med. Chem., 2005, 48, 6128–6139 CrossRef CAS PubMed.
- (a) E. Bálint, Á. Tajti, A. Ádám, I. Csontos, K. Karaghiosoff, M. Czugler, P. Ábrányi-Balogh and G. Keglevich, Beilstein J. Org. Chem., 2017, 13, 76–86 CrossRef PubMed; (b) A.-E. Wang, Z. Chang, W.-T. Sun and P.-Q. Huang, Org. Lett., 2015, 17, 732–735 CrossRef CAS PubMed; (c) J. M. Sanders, A. O. Gómez, J. Mao, G. A. Meints, E. M. V. Brussel, A. Burzynska, P. Kafarski, D. González-Pacanowska and E. Oldfield, J. Med. Chem., 2003, 46, 5171–5183 CrossRef CAS PubMed.
- T. Hirai and L.-B. Han, J. Am. Chem. Soc., 2006, 128, 7422–7423 CrossRef CAS PubMed.
- M. M. I. Basiouny and J. A. R. Schmidt, Organometallics, 2017, 36, 721–729 CrossRef CAS.
- (a) A. K. Mukerjee and R. Ashare, Chem. Rev., 1991, 91, 1–24 CrossRef CAS; (b) Z.-W. Zhou, F.-C. Jia, C. Xu, S.-F. Jiang, Y.-D. Wu and A.-X. Wu, Chem. Commun., 2017, 53, 1056–1059 RSC; (c) P. Wang, S. Tang and A. Lei, Green Chem., 2017, 19, 2092–2095 RSC; (d) T. Xie, Y. Xiao, S. Zhao, X.-Q. Hu and P.-F. Xu, J. Org. Chem., 2016, 81, 10499–10505 CrossRef CAS PubMed.
- (a) S. Khan and C. M. R. Volla, Chem. – Eur. J., 2017, 23, 1–6 CrossRef PubMed; (b) W.-S. Guo, Y.-C. Wang, Q. Dou, L.-R. Wen and M. Li, Org. Chem. Front., 2017, 4, 510–513 RSC; (c) P. Zhao, Y. Liu and C. Xi, Org. Lett., 2015, 17, 4388–4391 CrossRef CAS PubMed; (d) P. Zhao, X. Yan, H. Yin and C. Xi, Org. Lett., 2014, 16, 1120–1123 CrossRef CAS PubMed; (e) A. F. G. Goldberg, N. R. O'Connor, R. A. Craig and B. M. Stoltz, Org. Lett., 2012, 14, 5314–5317 CrossRef CAS PubMed.
- (a) Y. He, J. Li, S. Luo, J. Huang and Q. Zhu, Chem. Commun., 2016, 52, 8444–8447 RSC; (b) X. Tang, Z. Zhu, C. Qi, W. Wu and H. Jiang, Org. Lett., 2016, 18, 180–183 CrossRef CAS PubMed; (c) A. K. Yadav and L. D. S. Yadav, Org. Biomol. Chem., 2015, 13, 2606–2611 RSC; (d) L. Benati, G. Calestani, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo, S. Strazzari and G. Zanardi, J. Org. Chem., 2003, 68, 3454–3464 CrossRef CAS PubMed.
- (a) L.-R. Wen, Q. Dou, Y.-C. Wang, J.-W. Zhang, W.-S. Guo and M. Li, J. Org. Chem., 2017, 82, 1428–1436 CrossRef CAS PubMed; (b) L.-R. Wen, Q.-Y. Shen, W.-S. Guo and M. Li, Org. Chem. Front., 2016, 3, 870–874 RSC; (c) W. Guo, S. Li, L. Tang, M. Li, L. Wen and C. Chen, Org. Lett., 2015, 17, 1232–1235 CrossRef CAS PubMed.
- (a) S. Mahato, S. Santra, R. Chatterjee, G. V. Zyryanov, A. Hajra and A. Majee, Green Chem., 2017, 19, 3282–3295 RSC; (b) S. G. Pharande, A. R. C. Escobosa and R. Gámez-Montaño, Green Chem., 2017, 19, 1259–1262 RSC; (c) V. A. Rassadin, D. P. Zimin, G. Z. Raskil'dina, A. Y. Ivanov, V. P. Boyarskiy, S. S. Zlotskii and V. Y. Kukushkin, Green Chem., 2016, 18, 6630–6636 RSC; (d) P. Chaudhary, S. Gupta, N. Muniyappan, S. Sabiah and J. Kandasamy, Green Chem., 2016, 18, 2323–2330 RSC; (e) T. N. Poudel, Y. R. Lee and S. H. Kim, Green Chem., 2015, 17, 4579–4586 RSC.
- S. Härling, J. Greiser, T. M. A. Al-Shboul, H. Görls, S. Krieck and M. Westerhausen, Aust. J. Chem., 2013, 66, 1264–1273 Search PubMed.
- (a) J. Xie, H. Li, Q. Xue, Y. Cheng and C. Zhu, Adv. Synth. Catal., 2012, 354, 1646–1650 CrossRef CAS; (b) W. Han, P. Mayer and A. R. Ofial, Adv. Synth. Catal., 2010, 352, 1667–1676 CrossRef CAS; (c) O. Baslé and C.-J. Li, Chem. Commun., 2009, 4124–4126 RSC.
- The nucleophilic addition process was further confirmed by the Hammett correlation study. See the ESI, Fig. S3.†.
- The generation of H2S was confirmed by using lead acetate paper. See the ESI, Fig. S4.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1503930. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7gc03101g |
| This journal is © The Royal Society of Chemistry 2018 |