
Synthesis and characterization of isocyanate-free polyureas†
Joseph M.
Dennis
a,
Limor I.
Steinberg
a,
Allison M.
Pekkanen
a,
Jon
Maiz
b,
Maruti
Hegde
a,
Alejandro J.
Müller
 bc and
Timothy E.
Long
*a
bc and
Timothy E.
Long
*a
aVirginia Tech, Department of Chemistry, Macromolecules Innovation Institute (MII), Blacksburg, Virginia 24061, USA. E-mail: telong@vt.edu
bPOLYMAT and Polymer Science and Technology Department, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018 Donostia-San Sebastián, Spain
cIKERBASQUE, Basque Foundation for Science, Bilbao, Spain
First published on 29th November 2017
Abstract
Due to continued health and safety concerns surrounding isocyanates, alternative synthetic routes to obtain urea-containing polymers is gaining much attention. Melt polycondensation of urea with diamines achieved polyureas in the absence of catalyst or solvents. 1H NMR spectroscopy and thermogravimetric analysis confirmed targeted compositions and thermal stability, respectively. Differential scanning calorimetry and dynamic mechanical analysis provided insight into the copolymers’ thermal and morphological behavior. A steady increase in the melting temperature across the range of compositions suggested co-crystallization of the different repeating units, in sharp contrast to non-hydrogen bonded copolymers. Furthermore, tunable melt temperatures and mechanical performance illustrated the versatility of these copolymers in high performance applications. Finally, initial biodegradation studies using a naturally occurring, soil enzyme (urease) demonstrated steady degradation over 4 weeks, releasing ammonia as a potential nitrogen source for agricultural applications.
Introduction
Polyureas represent a unique class of polymers that have found utility in a wide array of applications from protective coatings1,2 to foams and biomedical implants.3,4 The complement of hydrogen bonding and semicrystallinity provides advantageous solvent resistance, and relatively high melting points (Tms) garner attention as thermally resilient thermoplastics.1–7 Furthermore, many synthetic avenues exist to form the urea carbonyl from urea,3,4,8 isocyanates,9,10 phosgene,11 and carbon dioxide.2 The formation of polyurea typically involves the quantitative reaction between a diamine and a diisocyanate.9,10,12 The combination of small and low-Tg, oligomeric diamines with diisocyanates results in thermoplastic elastomers (TPEs) with high extensibility and low hysteresis.9,10,12 However, the use of toxic and highly reactive isocyanates poses many health and environmental concerns.13 Recent advances in the sequestration of carbon dioxide provide an exciting avenue for biorenewable feedstocks. Direct incorporation of CO2 into polyureas with diamines illustrates a promising route to generate polyureas from inexpensive raw materials.2 However, the low polymer yields, high pressures, and exotic solvent requirements hinder wide application, and limit favorable environmental impact. An alternate technique to synthesize polyureas includes microwave irradiation.5,14,15 However, the necessity of high boiling point solvents, e.g., dimethylacetamide or o-dichlorobenzene, raises additional health and environmental concerns.5,14 Removing solvents during the synthesis of polyureas not only highlights a critical step in reducing waste streams, it further aligns these materials with the twelve principles of green chemistry and industrially practiced melt polycondensation methods.In addition to greener feedstocks and synthetic avenues, investigation into the biodegradation of polymeric materials continues to grow rapidly.16,17 As a result of the established biodegradability of urea18,19 and ester20 containing polymers, many poly(urea ester)s provide advantageous mechanical durability while maintaining biodegradability.3,4 Although bulk polycondensation of urea-containing diols with aliphatic diacids generates the product, the synthesis of urea-containing diols requires several steps.3,4 Interestingly, the bulk polycondensation of diamines with urea has received minimal attention.6–8,21–23 Leibler et al. utilized the reaction of highly branched, oligoamines with urea to afford self-healing rubber networks,8 and the remaining literature resides in the patent literature.6,7,21–23
Herein, we report the direct polycondensation of diamines with urea to afford high molecular weight, linear polyurea copolymers. Use of an inexpensive, biological feedstock and solvent free polymerization provides an industrially relevant and environmentally benign synthesis for a novel family of polyureas. Furthermore, the elimination of solvents and isocyanates while enabling biodegradability provides a robust platform for a library of materials with tailored thermomechanical properties and enzyme-catalyzed degradation. The successful design and illustration of enzymatically catalyzed biodegradation in a novel, high performance material illustrates an important intersection of material engineering and life cycle considerations.
Experimental
Materials
Urea (BioReagent), and 2,2-(ethylenedioxy)bis(ethylamine) (98%) were purchased from Sigma Aldrich and used as received. 1,8-Diaminooctane (98%) was purchased from Sigma Aldrich and distilled under vacuum at 140 °C. Urease from Canavalia ensiformis (Jack Bean) Type III (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 U g−1) was purchased from Sigma Aldrich. Phosphate buffered saline (PBS) was purchased from Life Technologies. An ammonia assay kit was purchased from Sigma Aldrich. The urease activity kit was purchased from Sigma Aldrich. All deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc.
000–30000 U g−1) was purchased from Sigma Aldrich. Phosphate buffered saline (PBS) was purchased from Life Technologies. An ammonia assay kit was purchased from Sigma Aldrich. The urease activity kit was purchased from Sigma Aldrich. All deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc.
Analytical methods
1H NMR spectroscopy utilized a Varian Unity 400 spectrometer operating at 399.87 MHz and 23 °C with a 0.1 M LiBr salt solution in DMSO-d6 at approximately 8 mg mL−1. Utilizing a TA Instruments TGA Q500 from room temperature to 600 °C at a heating rate of 10 °C min−1 under constant N2 purge provided thermogravimetric analysis (TGA). Differential scanning calorimetry (DSC) probed thermal transitions with a PerkinElmer 8500 DSC operating under a N2 atmosphere and scanning rates of 5 and 20 °C min−1. The DSC calibration was performed with indium and tin. Dynamic mechanical analysis (DMA) revealed thermomechanical behavior using a TA Instruments DMA Q800 in oscillatory tension mode at 1 Hz and 3 °C min−1. Polyureas were dried at 120 °C under vacuum prior to melt processing. Compression molding between Kapton® sheets at 270 °C with a PHI hydraulic press generated free-standing films, which were cooled in air over 1 h.Synthesis of poly(octamethylene urea)-co-poly(di(ethylene oxide) ethylene urea)s [poly(OMU)-co-poly(DEOEU)s]
The synthesis of isocyanate-free polyureas utilized a facile melt transamidification process, and the synthesis of poly(OMU)50-co-poly(DEOEU)50 follows as an example. Urea (4.805 g, 0.08 mol), 1,8-diaminooctane (DAO, 8.656 g, 0.06 mol), and 2,2-(ethylenedioxy)bis(ethylamine) (EBA, 8.892 g, 0.06 mol) were charged to a 100 mL, single-necked, round-bottomed flask. The flask was equipped with a nitrogen inlet, overhead metal stir rod, and condensing tube with collection flask. Subsequent vacuum and N2 purge cycles (3×) ensured removal of oxygen to provide an inert atmosphere for polymerization. Under a constant N2 flow (circa 10 mL min−1), the contents were heated to 170 °C and held for 1 h. Immediately, ammonia gas was observed during the first 30 min, and condensation of ammonia in a dry ice cooled flask provided insight into reaction progress. After 1 h, the temperature was increased step-wise from 200 °C to 250 °C over 1.5 h. Application of vacuum at 250 °C removed any residual ammonia and enabled the distillation of excess diamines generated through transamidification. The reaction continued for 2 h at 250 °C under vacuum with dramatic increases in melt viscosity observed for all compositions. The polymer was isolated directly and used without further purification. Polymer nomenclature follows poly(OMU)x-co-poly(DEOEU)y where ‘x’ and ‘y’ are the weight percent of the respective urea in the final polymer, as determined by 1H NMR spectroscopy (see ESI†).Ammonia release assay
A 500 U mL−1 solution of urease in phosphate buffered saline was prepared for all ammonia release experiments. Sections of polymer film were placed in the bottom of a 2-dram vial, onto which urease solution or phosphate buffered saline were placed. 20 U of urease per mg of sample were added, and additional phosphate buffered saline was added to maintain similar hydrostatic pressure (2 mL) between all samples. Ammonia release was analyzed each week at 25 °C over a four week period according to manufacturer's protocol. Briefly, 100 μL of sample was placed into 1 mL ammonia assay reagent in a disposable, small volume cuvette. The absorbance at 340 nm was analyzed on a SpectraMax M2 plate reader in absorbance mode. A blank of 100 μL of water in ammonia assay reagent was read for a control sample. L-Glutamate dehydrogenase was added to each cuvette and allowed to react for 5 min. Finally, the solution was mixed and absorbance at 340 nm analyzed. Calculations to determine ammonia concentration were conducted according to manufacturer's protocol. Following the assay each week, fresh solutions containing urease and phosphate buffered saline were added to each film.Statistical analysis
A multiple linear regression fit was performed on ammonia release as a function of composition and time through analysis of variance (ANOVA) in JMP software. Following ANOVA, a Tukey's HSD test determined statistical differences between groups. For percent remaining polymer and total released ammonia, an ANOVA test followed by a Tukey's HSD determined statistical significance between groups.Results and discussion
Most earlier reports for polyurea synthesis utilize diamines with diisocyanates to generate the urea linkage.9,10 Although quantitative and well established, the high reactivity of diisocyanates introduces many hazards.13 Furthermore, the hydrogen bonding of a symmetric urea lends to poor solubility in many common organic solvents, restricting the length of the urea segment in segmented block copolymers.12 To circumvent these concerns, melt polycondensation provided a facile approach to the synthesis of polyureas using a biosourced monomer and solvent-free conditions (Scheme 1).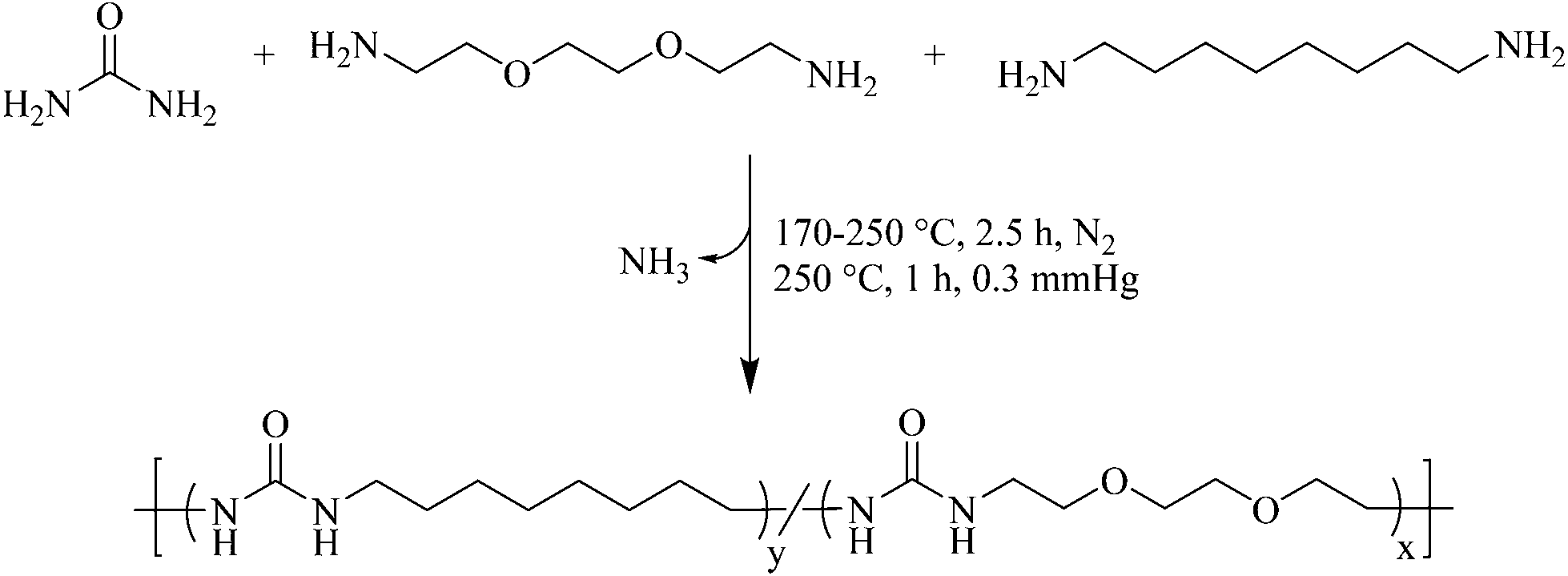 | ||
| Scheme 1 Synthesis of isocyanate-free poly(octamethylene urea)-co-poly(di(ethylene oxide) ethylene urea) [poly(OMU)-co-poly(DEOEU)] utilizing melt polycondensation. | ||
Similar to a poly(ethylene terephthalate) polymerization, heating low molar mass diamines with urea to 170 °C afforded a homogeneous melt. Ammonia gas evolution from the reaction flask provided a visual aid to the reaction progress, and determined reaction temperatures and times. After 1 h, the rate of ammonia generation slowed and the melt viscosity increased significantly. Increasing the reaction temperature to 250 °C reduced the melt viscosity and ensured higher conversions before reducing the pressure. In the final stage of the reaction, vacuum at elevated temperatures enabled the distillation of the excess diamines, and drove the reaction to high conversions and high molecular weights. The reaction proceeded until the melt viscosity remained stable. At this point, the high melt viscosity hindered stirring, and indicated high molecular weight formation. Aggregation, which was determined through dynamic light scattering in most organic solvents, prevented molecular weight determination using size exclusion chromatography.
Detailed evaluations of the reaction mechanism for the formation of dialkyl ureas using amines are well understood.8,24,25 In summary, the reaction proceeds through an initial decomposition of urea into ammonia and isocyanic acid which react with an amine to produce a monosubstituted alkyl urea. Further elimination of ammonia from the alkyl urea and subsequent reactions with another amine provides the disubstituted alkyl urea. Although several side reactions are possible at temperatures below 170 °C, above 170 °C the primary substitution results in the 1,1-dialkyl urea and produces linear polyureas.24
1H NMR spectroscopy in a 0.1 M LiBr DMSO-d6 solution probed polymer compositions for the random copolymers (Fig. S1–4,†Table 1). Unique chemical shifts for the methyl protons adjacent to the urea illustrated a retention of targeted compositions, and the compositional variation permitted the derivation of structure–property relationships for these novel polyureas. Importantly, the elevated temperatures and extended time ensured randomization of the copolymer sequence through transamidification.26 Thermogravimetric analysis (TGA) demonstrated the absence of weight loss up to 320 °C with a minimal weight loss of <1% near 100 °C attributed to absorbed water. As a result of the water absorption, melt processing required a drying procedure to minimize bubbles in the compression molded films.
| 1H NMR | TGA | DSCa | DMAg | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EBA (mol %) | DAO (mol %) | T d,5% (°C) |
T
m1b (°C) |
T
m2b (°C) |
T
cb (°C) |
ΔHcc (J mol−1) |
ΔHccd (J mol−1) |
ΔHme (J mol−1) |
X
cf (%) |
T
gh (°C) |
T
fi (°C) |
|
|
a Heating/cooling/heating thermal cycles of 5 °C min−1, values determined from second heating scans.
b
T
c and Tms are measured as the onset temperature in the exotherm and endotherm, respectively.
c ΔHc determined as area under the exothermic event.
d ΔHcc determined as area under the exothermic event during heating sweep (cold crystallization).
e ΔHm determined as area under the endothermic event.
f
X
c is the degree of crystallinity, [(ΔHm − ΔHcc)/ΔHm,100%]; where (ΔHm − ΔHcc) is the melting enthalpy of the component under consideration and ΔHm,100% is the enthalpy of fusion of a 100% crystalline sample estimated by group contribution theory.
g Heating rate of 3 °C min−1, 1 Hz.
h
T
g reported as peak temperature from tanδ.
i
T
f reported as highest temperature before flow.
|
||||||||||||
| Poly(DEOEU) | 100 | 0 | 325 | 128 | N/A | 96 | 0 | 12.0 | 23.7 | 6.8 | −1.9 | 125 |
| Poly(OMU)22-co-poly(DEOEU)78 | 78 | 22 | 314 | 141 | 150 | 63 | 0 | 11.7 | 22.0 | 5.7 | 16.9 | 133 |
| Poly(OMU)43-co-poly(DEOEU)57 | 57 | 43 | 306 | 178 | 194 | 153 | 35.0 | 0 | 44.8 | 23.7 | 25.2 | 170 |
| Poly(OMU)51-co-poly(DEOEU)49 | 51 | 49 | 319 | 200 | 211 | 178 | 34.3 | 0 | 64.2 | 33.5 | 26.4 | 199 |
| Poly(OMU)57-co-poly(DEOEU)43 | 43 | 57 | 326 | 206 | 216 | 186 | 47.6 | 0 | 68.4 | 35.2 | 37.9 | 210 |
| Poly(OMU)78-co-poly(DEOEU)22 | 22 | 78 | 325 | 228 | 236 | 212 | 72.9 | 0 | 85.0 | 41.9 | 51.3 | 230 |
| Poly(OMU) | 0 | 100 | 326 | 231 | 250 | 224 | 91.9 | 0 | 100.4 | 47.5 | N/A | N/A |
Differential scanning calorimetry (DSC) at 5 °C min−1 provided insight into the thermal transitions of the polyurea copolymers (Fig. 1). Moreover, different sweeps at 20 °C min−1 were also performed in order to compare the crystallization processes of the different copolymers (Fig. S5†). Fig. 1 illustrates the differences in crystallization kinetics under non-isothermal conditions between poly(OMU) and poly(DEOEU). The crystallization exotherm of the poly(OMU) is easily detected during cooling at 5 °C min−1, while poly(DEOEU) did not exhibit an apparent exothermic event. However, during subsequent heating, poly(DEOEU) exhibited cold crystallization and melting events. Importantly, the melting enthalpy is larger than its cold crystallization exotherm, which indicated some crystallinity existed prior to heating (see comparative enthalpies in Table 1).
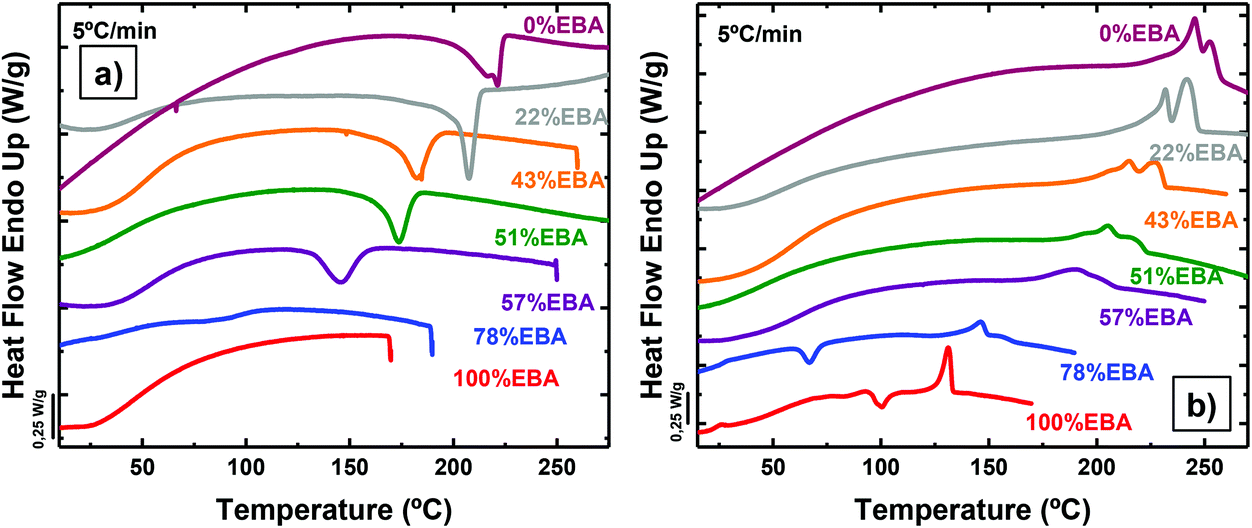 | ||
| Fig. 1 (a) DSC cooling scans from the melt at 5 °C min−1 and (b) subsequent heating scans at 5 °C min−1 for the indicated homopolymers and random copolymer samples. | ||
Interestingly, all copolymers crystallized over the entire compositional range, including symmetric compositions (i.e., 51% EBA). Fig. 2 highlights the compositional dependence of peak melting temperature (Tm) and melt enthalpy (ΔHm) with DAO incorporation. This is in sharp contrast to non-hydrogen bonded, semicrystalline copolymers where the Tm decreases with comonomer incorporation.27,28
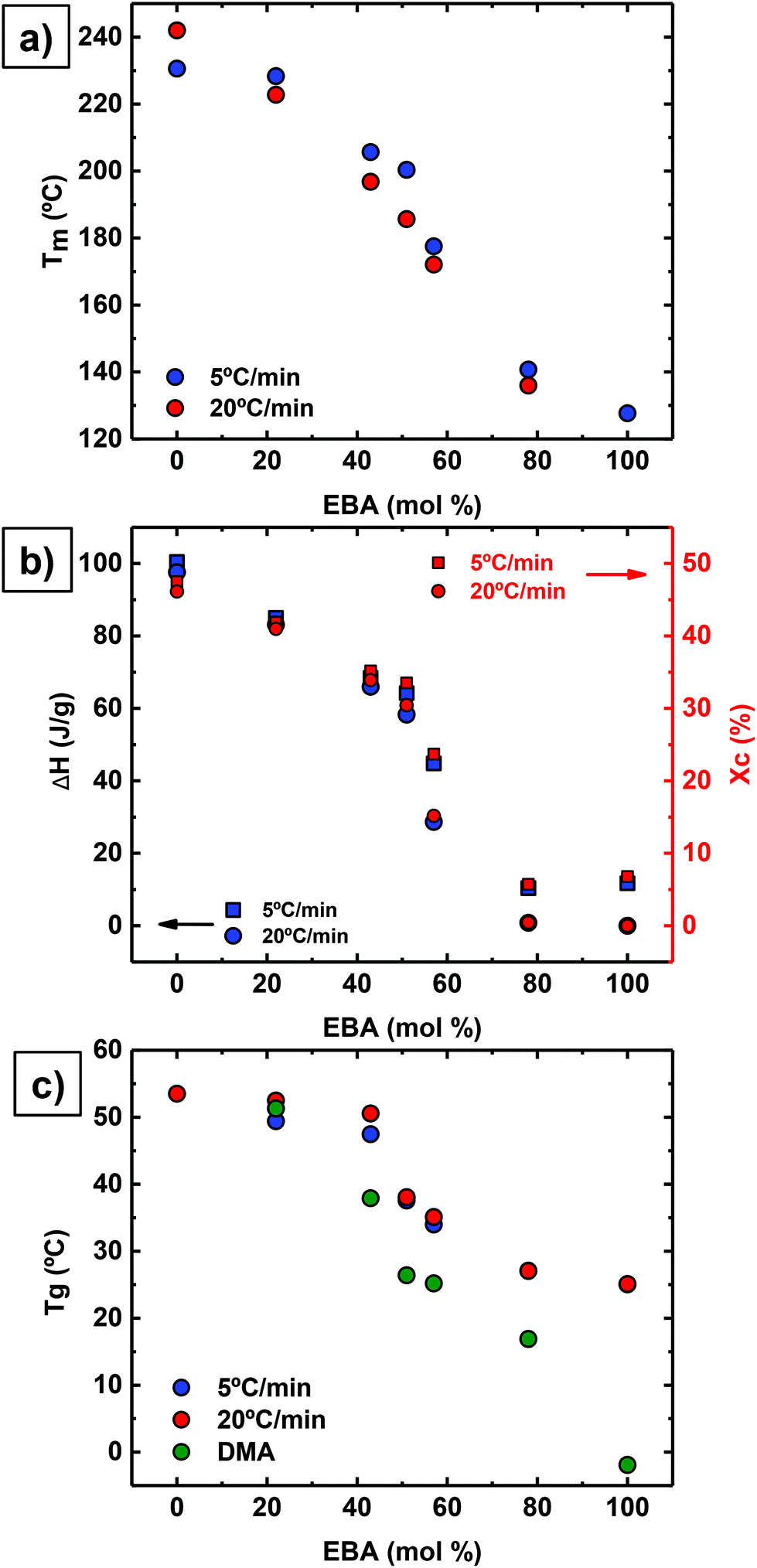 | ||
| Fig. 2 (a) Experimental melting temperatures (Tm); (b) melting enthalpies (ΔHm) and calculated degree of crystallinities (Xc); and (c) glass transition temperatures (Tg) determined by DSC and DMA as a function of EBA content. | ||
Reliable, experimental values for equilibrium melting enthalpies 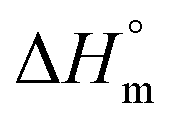 (i.e., the melting enthalpy of 100% crystalline polymers) are only available for a limited number of polymers, and currently there are no values reported for
(i.e., the melting enthalpy of 100% crystalline polymers) are only available for a limited number of polymers, and currently there are no values reported for 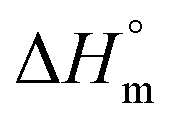 or crystalline structures for the homopolymers reported here. Therefore, the calculation of the degree of crystallinity (Xc), as a first approximation, employed the group contribution, semi-empirical approach reported by Van Krevelen et al.29 Values of 172.2 J g−1 and 211.4 J g−1 were determined for poly(DEOEU) and poly(OMU) homopolymers, respectively, from the group contribution method. Furthermore, calculation of copolymer
or crystalline structures for the homopolymers reported here. Therefore, the calculation of the degree of crystallinity (Xc), as a first approximation, employed the group contribution, semi-empirical approach reported by Van Krevelen et al.29 Values of 172.2 J g−1 and 211.4 J g−1 were determined for poly(DEOEU) and poly(OMU) homopolymers, respectively, from the group contribution method. Furthermore, calculation of copolymer 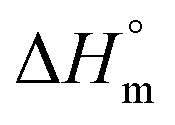 followed by assuming a linear dependence with composition. Fig. 2b shows the correlation between the
followed by assuming a linear dependence with composition. Fig. 2b shows the correlation between the 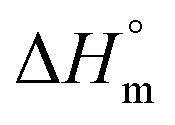 and the Xcversus composition. The crystallinity for the poly(OMU) homopolymer is higher as compared with the poly(DEOEU) homopolymer, while those of the random copolymers exhibited intermediate values that are highly dependent on composition. In fact, tailoring of the crystallinity and melting points with composition provides a unique variable for specific applications. The crystallinity also exhibited a dependence on cooling rates, particularly for copolymers rich in EBA content.
and the Xcversus composition. The crystallinity for the poly(OMU) homopolymer is higher as compared with the poly(DEOEU) homopolymer, while those of the random copolymers exhibited intermediate values that are highly dependent on composition. In fact, tailoring of the crystallinity and melting points with composition provides a unique variable for specific applications. The crystallinity also exhibited a dependence on cooling rates, particularly for copolymers rich in EBA content.
Based on the behavior reported in Fig. 1 and 2, it is plausible that these random copolymers behave as isomorphic materials, where co-crystallization between both types of chains occurs over the entire composition range.30 However, this requires a detailed study of their crystal structure with wide-angle X-ray scattering and crystal structure determination. This is outside the scope of the present paper but is currently underway and will be reported in the future.
Fig. 2c shows a plot of Tg values versus composition. The Tg values decrease with increasing poly(DEOEU) content, as expected, since the ether units in EBA make poly(DEOEU) more flexible in comparison with poly(OMU) homopolymer. The molecular flexibility is reflected in both Tg and Tm values. The Tg values presented in Fig. 3c were determined by DSC (at two different scanning rates, 5 and 20 °C min−1) and by dynamic mechanical analysis (discussed below). Incorporation of up to 43 mol% OMU increased and broadened the Tg. Further increasing DOA content continued to suppress the Tg's endothermic event, and suggested the development of a rigid amorphous phase.31,32 This will be explored in the near future by performing dielectric relaxation experiments.
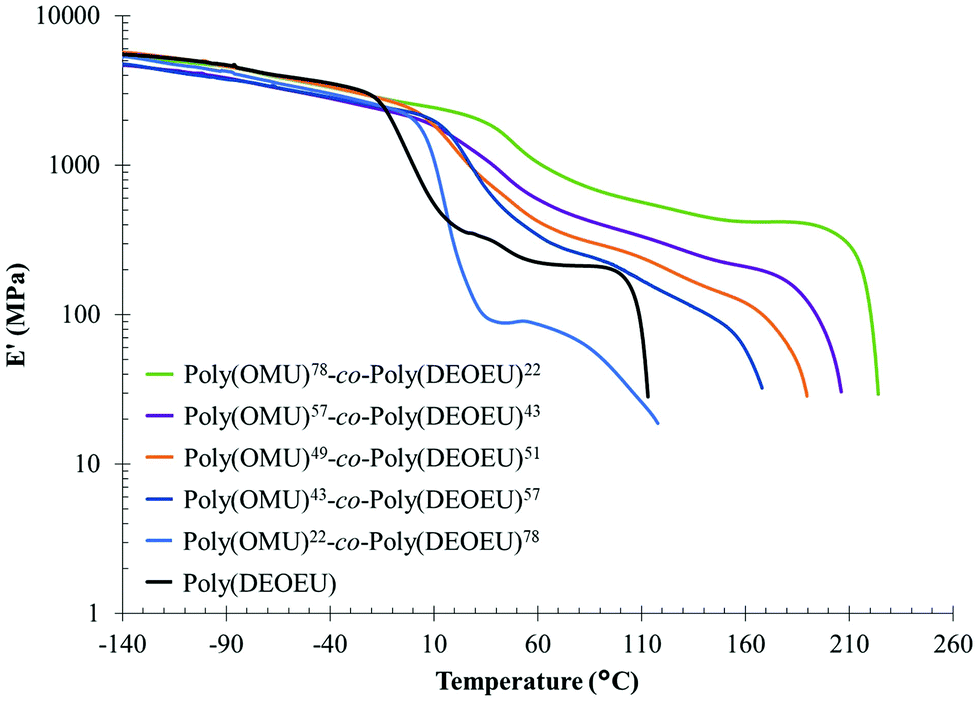 | ||
| Fig. 3 Dynamic mechanical analysis of poly(OMU)-co-poly(DEOEU) copolymers. | ||
Reducing the ether units in the polymer backbone by incorporating the homoatomic 1,8-diaminooctane resulted in an increase in Tg, Tm and enthalpy (crystallinity). Moreover, Tm peak broadening suggested a broad distribution of lamellar sizes. Further studies are underway to understand the crystallization kinetics and the crystallite type formed in these copolymers. In summary, these results demonstrated the significance of ethers in the main chain in modulating polyurea thermal properties and obtaining a melt processable material.
Dynamic mechanical analysis (DMA) using melt pressed films evaluated storage modulus as a function of temperature and confirmed the thermal transitions observed by DSC (Table 1). This complementary analytical technique also provided more distinct transitions for the Tgs as a result of the high sensitivity of dynamic mechanical properties to temperature. The lower heating rate and mechanical deformation at 1 Hz resulted in the slight discrepancy between Tg values obtained by DMA and DSC (see Fig. 2c), but differences of this magnitude are common when comparing these two analytical techniques.
Fig. 3 shows elastic modulus curves as a function of temperature obtained by DMA. Above the Tg, all copolymers exhibited a plateau region associated with the physical crosslinks of crystalline domains, hydrogen bonding interactions, and intermolecular entanglements within chains. In conjunction with the increase in crystallinity, the plateau modulus systematically increased with 1,8-diaminooctane incorporation, and further suggested an increase in rigid amorphous phase and crystalline content. Finally, the absence of a plateau above Tm suggested that the thermally labile, hydrogen-bonding network is highly disassociated at the temperatures required to melt the crystalline network. This correlated well with previous reports on hydrogen bond disassociation temperatures.10,12,33
Mechanical analysis of this novel family of polyureas demonstrated a wide range of properties obtainable (Table 2). Although the Tgs for poly(DEOEU) and poly(OMU)22-co-poly(DEOEU)78 were below ambient temperatures, the combination of hydrogen bonding and crystallinity enabled film formation and tensile analysis. Increasing the DAO content resulted in less ductility in conjunction with an increase in the Tg, Tm, and ΔHm. For poly(DEOEU) and poly(OMU)22-co-poly(DEOEU)78, a strain-induced hardening occurred above 100% strain. Furthermore, the systematic increase in Young's moduli illustrated a predictable dependence with DAO incorporation. However, the compositionally independent yield stress suggested similar plastic deformation mechanisms for chain–chain slippage during tensile deformation.
| Tensile stress at break (MPa) | Tensile strain at break (%) | Tensile stress at yield (MPa) | Young's modulus (MPa) | |
|---|---|---|---|---|
| Poly(OMU)78-co-poly(DEOEU)22 | ||||
| Mean | 38 | 10 | 47 | 1831 |
| SD | 14 | 2 | 15 | 204 |
| Poly(OMU)57-co-poly(DEOEU)43 | ||||
| Mean | 22 | 11 | 35 | 1501 |
| SD | 8 | 4 | 7 | 169 |
| Poly(OMU)49-co-poly(DEOEU)51 | ||||
| Mean | 31 | 6 | 17 | 1550 |
| SD | 8 | 1 | 4 | 134 |
| Poly(OMU)43-co-poly(DEOEU)57 | ||||
| Mean | 42 | 146 | 49 | 1837 |
| SD | 21 | 60 | 4 | 272 |
| Poly(OMU)22-co-poly(DEOEU)78 | ||||
| Mean | 31 | 176 | 30 | 889 |
| SD | 14 | 90 | 1 | 22 |
| Poly(DEOEU) | ||||
| Mean | 31 | 252 | 27 | 494 |
| SD | 15 | 98 | 12 | 90 |
A potential advantage of polyureas over other polymers resides in their biodegradability from natural soil enzymes (e.g., urease). The action of urease requires the cooperative action of a hinged group to sequester urea into the enzyme for subsequent degradation.18,34 Previous studies highlight the wide range of ammonia release profiles obtained in polymeric substrates and microenvironments,35,36 and necessitated additional studies to confirm biodegradability of polyureas. In Fig. 4, released ammonia as a function of time and composition achieved significant levels of ammonia within 4 weeks. Furthermore, acceleration of ammonia release for all compositions in weeks 3 and 4 suggested a significant increase in urea availability (Table S1†). Despite ammonia release at short times, over 99% of the polyureas remained after four weeks (Fig. S6†). The compositionally independent biodegradation suggested similar urea accessibility, permitting material selection based solely on processing condition requirements. Further investigation into the influence of hydrophilicity is underway.
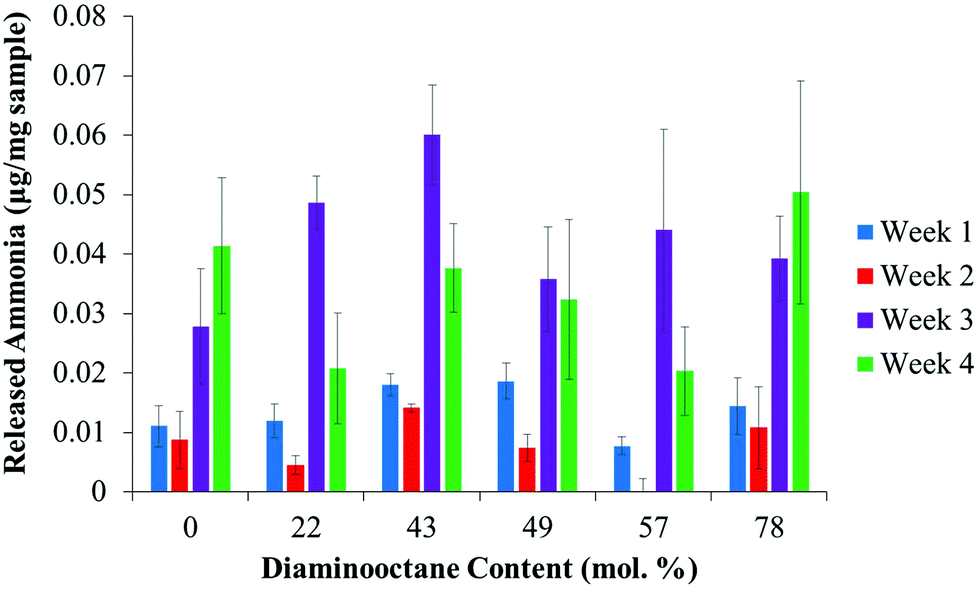 | ||
| Fig. 4 Concentration of ammonia released over each week as a function of composition. Released ammonia increases with increasing time but does not change with composition. Data is presented as average ± standard deviation. | ||
Conclusions
Urea, a biosourced monomer, provided an inexpensive and biorenewable feedstock to generate polyureas under melt polycondensation conditions. Copolymerization with commercially available diamines generated a family of copolymers and illustrated the versatility of this method. A complement of analytical techniques evaluated the thermal and mechanical properties. The steady increase in Tm with 1,8-diaminooctane incorporation suggested co-crystallization of the two repeat units. DMA further confirmed the trends observed in DSC, while tensile testing highlighted a range of mechanical properties for these copolymers. Finally, exploration into environmental stability suggested a potentially slow degradation pathway catalyzed by natural soil enzymes that liberates ammonia for plant uptake.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JM wants to acknowledge “Fomento San Sebastián” in the framework program “Retorno del Talento Local” Donostia up! 2016.References
- Q. Li, P. Wang, S. Liu, Y. Fei and Y. Deng, Green Chem., 2016, 18, 6091–6098 RSC.
- P. Wang, X. Ma, Q. Li, B. Yang, J. Shang and Y. Deng, RSC Adv., 2016, 6, 54013–54019 RSC.
- J. Yu, F. Lin, P. Lin, Y. Gao and M. L. Becker, Macromolecules, 2014, 47, 121–129 CrossRef CAS.
- D. Tang, Z. Chen, F. Correa-Netto, C. W. Macosko, M. A. Hillmyer and G. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(24), 3795–3799 CrossRef CAS.
- A. Banihashemi, H. Hazarkhani and A. Abdolmaleki, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2106–2111 CrossRef CAS.
- C. J. Bouboulis and K. Isidor, US3412072, 1968 Search PubMed.
- C. J. Bouboulis, K. Isidor and C. M. White, United Stats Pat, US3390137, 1968 Search PubMed.
- D. Montarnal, P. Cordier, C. Soulié-Ziakovic, F. Tournilhac and L. Leibler, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7925–7936 CrossRef CAS.
- D. J. Buckwalter, A. G. Hudson, R. B. Moore and T. E. Long, Polym. Int., 2014, 63, 1184–1191 CrossRef CAS.
- D. J. Buckwalter, M. Zhang, D. L. Inglefield Jr., R. B. Moore and T. E. Long, Polymer, 2013, 54, 4849–4857 CrossRef CAS.
- E. L. Wittbecker, United States Pat, US2816879, 1957 Search PubMed.
- D. J. Buckwalter, J. M. Dennis and T. E. Long, Prog. Polym. Sci., 2015, 45, 1–22 CrossRef CAS.
- C. A. Redlich and M. H. Karol, Int. Immunopharmacol., 2002, 2, 213–224 CrossRef CAS PubMed.
- S. Wu, W. Li, M. Lin, Q. Burlingame, Q. Chen, A. Payzant, K. Xiao and Q. M. Zhang, Adv. Mater., 2013, 25, 1734–1738 CrossRef CAS PubMed.
- R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2007, 28, 368–386 CrossRef CAS.
- D. Briassoulis and C. Dejean, J. Polym. Environ., 2010, 18, 384–400 CrossRef CAS.
- S. Kasirajan and M. Ngouajio, Agron. Sustainable Dev, 2012, 32, 501–529 CrossRef CAS.
- C. W. Pratt and K. Cornely, Essential biochemistry, Wiley, Hoboken, N.J., 3rd edn, 2014 Search PubMed.
- B. Krajewska, J. Mol. Catal. B: Enzym., 2009, 59, 9–21 CrossRef CAS.
- Y. Tokiwa and T. Suzuki, Nature, 1977, 270, 76–78 CrossRef CAS PubMed.
- M. Helmut and G. Rudolf, United States Pat, US3388103, 1968 Search PubMed.
- M. Helmut and G. Rudolf, US3223682, 1965 Search PubMed.
- R. D. Mathis, United States Pat, US3857819, 1974 Search PubMed.
- J. G. Erickson, J. Am. Chem. Soc., 1954, 76, 3977–3978 CrossRef CAS.
- W. A. Harold, US2145242, 1939 Search PubMed.
- R. J. Gaymans, in Synthetic Methods in Step-Growth Polymers, John Wiley & Sons, Inc., 2003, pp. 135–195, DOI:10.1002/0471220523.ch3.
- D. A. Schiraldi, M. L. Occelli and S. A. C. Gould, J. Appl. Polym. Sci., 2001, 82, 2616–2623 CrossRef CAS.
- A. Biswas and J. Blackwell, Macromolecules, 1988, 21, 3146–3152 CrossRef CAS.
- D. W. van Krevelen and K. te Nijenhuis, Properties of polymers: their correlation with chemical structure, their numerical estimation and prediction from additive group contributions, Elsevier, Amsterdam, The Netherlands, 4th edn, 2009.
- P. Pan and Y. Inoue, Prog. Polym. Sci., 2009, 34, 605–640 CrossRef CAS.
- J. Menczel and B. Wunderlich, J. Polym. Sci., Polym. Lett. Ed., 1981, 19, 261–264 CrossRef CAS.
- P. Huo and P. Cebe, Colloid Polym. Sci., 1992, 270, 840–852 CAS.
- K. Zhang, G. B. Fahs, M. Aiba, R. B. Moore and T. E. Long, Chem. Commun., 2014, 50, 9145–9148 RSC.
- C. Follmer, Phytochemistry, 2008, 69, 18–28 CrossRef CAS PubMed.
- D. I. Metelitsa, E. I. Tarun, A. V. Puchkaev and Y. P. Losev, Appl. Biochem. Microbiol., 2001, 37, 168–173 CrossRef CAS.
- P. V. Sundaram, Biochim. Biophys. Acta, Enzymol., 1973, 321, 319–328 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02996a |
| This journal is © The Royal Society of Chemistry 2018 |