
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mineralogically-inspired silver–bismuth hybrid material: an efficient heterogeneous catalyst for the direct synthesis of nitriles from terminal alkynes†
Sándor B.
Ötvös
 *ab,
Rebeka
Mészáros
a,
Gábor
Varga
cd,
Marianna
Kocsis
acd,
Zoltán
Kónya
ef,
Ákos
Kukovecz
f,
Péter
Pusztai
f,
Pál
Sipos
dg,
István
Pálinkó
*cd and
Ferenc
Fülöp
*ab
*ab,
Rebeka
Mészáros
a,
Gábor
Varga
cd,
Marianna
Kocsis
acd,
Zoltán
Kónya
ef,
Ákos
Kukovecz
f,
Péter
Pusztai
f,
Pál
Sipos
dg,
István
Pálinkó
*cd and
Ferenc
Fülöp
*ab
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary. E-mail: otvossandor@pharm.u-szeged.hu
bMTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, Eötvös u. 6, H-6720 Szeged, Hungary. E-mail: fulop@pharm.u-szeged.hu
cDepartment of Organic Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary. E-mail: palinko@chem.u-szeged.hu
dMaterial and Solution Structure Research Group, Institute of Chemistry, University of Szeged, Aradi Vértanúk tere 1, H-6720 Szeged, Hungary
eMTA-SZTE Reaction Kinetics and Surface Chemistry Research Group, Hungarian Academy of Sciences, Rerrich B. tér 1, H-6720 Szeged, Hungary
fDepartment of Applied and Environmental Chemistry, University of Szeged, Rerrich B. tér 1, H-6720 Szeged, Hungary
gDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
First published on 8th January 2018
Abstract
The synthesis and characterization of a silver-containing hybrid material is reported as a novel heterogeneous noble metal catalyst. In order to eliminate the need for traditional immobilization techniques, and to create a solid material with structurally-bound silver catalytic centers, the layered structure of a naturally occurring mineral served as the basis of the initial catalyst design. The novel material was prepared by means of the urea-mediated homogeneous precipitation of the corresponding metal nitrates, and was fully characterized by means of diverse instrumental techniques (X-ray diffractometry, Raman, IR, UV-Vis, EPR, X-ray photoelectron spectroscopies, thermal methods as well as atomic force, scanning and transmission electron microscopies). The as-prepared material exhibited outstanding activity in silver-catalyzed C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond activation to yield organic nitriles directly from terminal alkynes with less environmental concerns as compared to the classical synthesis methods. The effects of the reaction time, the temperature, as well as the role of various solvents, nitrogen sources and additives were carefully scrutinized in order to achieve high-yielding and selective nitrile formation. The heterogeneous nature of the reaction was verified and the solid catalyst was recycled and reused numerous times without loss of its activity or degradation of its structure, thereby offering a sustainable synthetic methodology.
C bond activation to yield organic nitriles directly from terminal alkynes with less environmental concerns as compared to the classical synthesis methods. The effects of the reaction time, the temperature, as well as the role of various solvents, nitrogen sources and additives were carefully scrutinized in order to achieve high-yielding and selective nitrile formation. The heterogeneous nature of the reaction was verified and the solid catalyst was recycled and reused numerous times without loss of its activity or degradation of its structure, thereby offering a sustainable synthetic methodology.
Introduction
Silver-mediated transformations have attained much importance in organic syntheses, which is exemplified by the large number of related review articles published in the last couple of years.1 Silver-based heterogeneous catalysts play historical roles in industrial oxidation processes, and they are also important in environmental applications (e.g., in catalytic elimination of pollutants).2 Fine-chemical syntheses (either in industry or in academic research) typically rely on Ag(I) salts or (in situ generated) complexes as homogeneous sources for the catalytically active metal.1,3Because of environmental and economic reasons, there is increasing need for heterogeneous noble metal catalysts. A literature survey indicates that amongst heterogeneous silver-mediated processes, the application of supported nanoparticles predominates.4 This can be explained by the fact that silver nanoparticles can readily be immobilized on various surfaces, such as activated charcoal,5 silica,6 alumina,7 carbon nanotubes,8 hydrotalcite materials,9 graphene10 or graphene oxide.11 The main limitation of supported nanoparticles is that the catalytic metal is typically attached to the surface via weak forces,12 which often results in limited catalyst stability and robustness, especially under demanding reaction conditions, such as high-temperature, high-pressure and continuous-flow conditions or in the presence of coordinating ligands and bases.13 Alternatively, there are only a few examples for heterogenized silver complexes and silver-exchanged materials (e.g., heteropoly acids) as reusable silver catalysts.14
Due to the excellent transformability of CC bonds and because of their excellent availability, alkynyl compounds have emerged as versatile intermediates for the atom-economic synthesis of a wide array of valuable products.15 Alkynes are important components in numerous catalytic transformations, such as diverse coupling and cyclization reactions16 including the well-established click chemistry.17 Because of these beneficial features, alkyne-based catalytic reactions are generating continuously increasing interest.15 Due to its d10 electronic configuration, silver plays a marked role in the activation of CC bonds of alkynyl compounds.1g As a consequence of this pronounced alkynophilicity, a large number of practical synthesis methods have been devised via silver-mediated σ- or π-activation of terminal or internal alkynes.1a,1b,1g,18 Most of these important reactions rely on homogeneous silver catalysts and employ relatively large amounts of the precious metal. Very recently, Duan and co-workers reported a pioneering methodology for the heterogeneous catalytic activation of internal alkynes by introducing a porous silver coordination polymer for the cyclization of propargylic alcohols with CO2.19
Because of their incredible versatility, aromatic and aliphatic nitriles are of outstanding importance in fine chemical, pharmaceutical and natural product syntheses. Organic nitriles are essential precursors for amines, amides, aldehydes and various carboxylic acid derivatives,20 and they are particularly important as intermediates for the production of valuable heterocycles.21 In addition, the cyano group represents a vital motif in various biologically and medicinally relevant structures, including numerous FDA-approved drugs like letrozole and rilpivirine.22 Traditional strategies in nitrile synthesis involve Sandmeyer23 and metal cyanide (or metalloid cyanide)-mediated cyanation reactions,24 dehydration of amides and oximes,25 ammoxidation of aromatic hydrocarbons26 and the Schmidt reaction of aldehydes with azides.27 However, these methods suffer from multiple limitations, including serious environmental concerns (e.g., cyanide salts and toxic metal waste), the use of expensive catalysts or harsh reaction conditions, selectivity issues and limited scope. To circumvent these drawbacks, Jiao and co-workers recently demonstrated a silver-catalyzed CC bond cleavage reaction to yield organic nitriles directly from terminal alkynes in the presence of a suitable nitrogen source and under reasonably mild conditions.28
In chemical industry, the concept of catalytic materials from naturally occurring minerals has notable historical roots,29 with the family of synthetic zeolites as the best-known examples.30 Due to economic and environmental reasons, the application of synthetic minerals and mineral-derived materials as designer catalysts has drawn significant attention in organic syntheses. For example, layered double hydroxides (LDHs) have emerged in diverse catalytic applications as catalyst supports or catalyst precursors or as the actual catalysts.31 Unlike silicate-based cationic clays, only relatively few anionic clays occur naturally;32 however, most of them can easily be synthesized.33 These layered materials can be comfortably modified, and they frequently offer peculiar catalytic properties due to the intrinsic characteristics of the layers (surface acid–base character), the interlayer modifier (e.g., redox-active intercalated complexes) and the sterically constrained environment, which may give rise to various selectivity effects.34 Thus, LDHs may be tailored to specific needs with relative ease.35 Traditional immobilization of metal catalysts on various prefabricated supports is often accompanied by negative effects such as poor accessibility, random anchoring or disturbed geometry of the active sites, which may result in reduced activity and/or selectivity; and in the case of unsatisfactory catalyst-support interactions, the possible leaching of the metal component may lead to serious environmental concerns.36 In contrast, catalytic materials with structurally bound active centers (e.g. LDHs with layers of active metals) exhibit increased thermodynamic stability and robustness as compared with the classically immobilized ones.37
In recent years, we have been active in this field concentrating on the synthesis, modification, structural characterization and catalytic applications of LDHs.38 During this work, we were able to prepare a thus far unknown Ba(II)Fe(III)-LDH,39 which refuted the generally accepted wisdom that for the formation of an LDH material, the ionic radii of the cationic components must be similar in size. As concerns catalytic applications, we not only exploited synthetic LDHs as catalyst carriers (intercalated catalysts),40 but we also showed that a copper-containing primary LDH can act as a highly active and robust solid catalyst even under demanding reaction conditions (high-temperature/high-pressure continuous-flow conditions).41
Although silver-containing minerals are scarce in nature, we speculated that a mineralogically-inspired catalyst design may aid the development of a robust heterogeneous silver catalyst. Our aim was to eliminate the need for classical immobilization techniques, and to prepare a solid catalyst containing Ag(I) as a constituent. As a proof of the concept, we were intrigued to explore the direct nitrogenation of alkynes to organic nitriles. To the best of our knowledge, this transformation has not yet been achieved by using a heterogeneous noble metal catalyst. Thus, we wish to present our results on the synthesis and characterization of a novel silver-containing hybrid material as a recyclable and highly selective heterogeneous catalyst for the sustainable synthesis of organic nitriles from terminal alkynes.
Experimental
Synthesis of the silver–bismuth hybrid material
All chemicals used were analytical grade Sigma-Aldrich products, and were applied without further purification.In a typical synthesis using the urea hydrolysis method,42 appropriate amounts of Bi(NO3)3·5H2O (5.36 g) and AgNO3 (3.73 g) were dissolved in 50–50 mL 5 wt% nitric acid. After mixing, urea (7.05 g) dissolved in 100 mL of deionized water was added to the solution and stirred for 72 h at 130 °C. The obtained colored product was filtered, washed with aqueous thiosulfate solution, water and ethanol four times, and dried at 60 °C to obtain the final product (material C). The syntheses of all byproducts (materials A and B) were carried out the same way except leaving out either the silver or the bismuth salt.
For control purposes, the catalyst synthesis was also carried out using high purity (99.999%) metal salts (Sigma-Aldrich).
Characterization of the silver–bismuth hybrid material
The X-ray diffraction (XRD) patterns were recorded on a Rigaku XRD-MiniFlex II instrument applying CuKα radiation (λ = 0.15418 nm), 40 kV accelerating voltage at 30 mA.The structure-building inorganic components were identified by Raman microscopy and IR spectroscopy. Raman spectra were recorded with a Thermo Scientific™ DXR™ Raman microscope at an excitation wavelength of 635 nm applying 10 mW laser power and averaging 20 spectra with an exposure time of 6 seconds. The instruments for recording the IR spectra were a BIO-RAD Digilab Division FTS-65A/896 (mid-range spectra) and a BIO-RAD Digilab Division FTS-40 vacuum FT-IR spectrophotometer (far-range spectra) with 4 cm−1 resolution. The 4000–600 and 600–100 cm−1 wavenumber ranges were recorded, but only the most relevant parts are displayed and discussed. 256 scans were collected for each spectrum. The mid IR spectra were recorded in diffuse reflectance mode, while for the far IR spectra the Nujol mull technique was used between two polyethylene windows (the suspension of 10 mg sample and a drop of Nujol mull).
UV–vis spectra were registered on an Ocean Optics USB4000 spectrometer with a DH-2000-BAL UV–vis-NIR light source measuring diffuse reflectance and using BaSO4 as reference. The spectra were analyzed with the SpectraSuite package.
Electron paramagnetic resonance (EPR) spectra were recorded with a BRUKER EleXsys E500 spectrometer (microwave frequency 9.51 GHz, microwave power 12 mW, modulation amplitude 5 G, modulation frequency 100 kHz) in quartz EPR tubes at room temperature. Approximately 10 mg of samples were used for each measurement, and the spectra were recorded without any additional sample preparation. All recorded EPR spectra were simulated with a home-made computer program.43
X-ray photoelectron spectra (XPS) were recorded using a SPECS instrument equipped with a PHOIBOS 150 MCD 9 hemispherical electron energy analyzer using AlKα radiation (hν = 1486.6 eV). The X-ray gun was operated at 210 W (14 kV, 15 mA). The analyzer was operated in the FAT mode, with the pass energy set to 20 eV. The step size was 25 meV, and the collection time in one channel was 250 ms. Typically, 5–10 scans were added up to acquire a single spectrum. Energy referencing was not applied. In all cases the powder-like samples were evenly laid out on one side of a double-sided adhesive tape, the other side being attached to the sample holder of the XPS instrument. The samples were evacuated at room temperature, and then inserted into the analysis chamber of the XPS instrument.
The topology of the as-prepared sample was studied by atomic force microscopy (AFM) on an NT-MDT SOLVER scanning probe microscope. Measurement was performed in the tapping mode at room temperature and atmospheric pressure in air. The non-contact silicon cantilevers had typical force constant of 42 N m−1 and resonance frequency of 278.8 kHz. For imaging, an SSS-NCH-10 tip (nominal radius of curvature: 2 nm, length: 15 μm) manufactured by NANOSENSORS was used with 0.43 kHz scanning rate and 700 × 700 pixel resolution.
The morphology of the as-prepared sample was studied by scanning electron microscopy (SEM). The SEM image was registered on an S-4700 scanning electron microscope (SEM, Hitachi, Japan) with accelerating voltage of 10–18 kV.
More detailed images, both of the as-prepared and the used samples, were captured by transmission electron microscopy (TEM), and information on the crystal structures was gathered by selected area diffraction (SAED). For these measurements, an FEI Tecnai™ G2 20 X-Twin type instrument was used, operating at an acceleration voltage of 200 kV.
The thermal behavior of the hybrid sample was investigated by thermogravimetry (TG) and differential thermogravimetry (DTG). The silver-containing sample was studied with a Setaram Labsys derivatograph operating in air at 5 °C min−1 heating rate. For the measurements, 20–30 mg of the samples were applied.
The specific surface areas were measured by the Brunauer–Emmett–Teller (BET) method (adsorption of N2 at 77 K).44 For these measurements, a NOVA3000 instrument was applied (Quantachrome, USA). The samples were flushed with N2 at 100 °C for 5 h to clean the surface of any adsorbents.
The amount of metal ions was measured by ICP–AES on a Thermo Jarell Ash ICAP 61E instrument. Before measurements, a few milligrams of the samples measured with analytical accuracy were digested in 1 cm3 cc. nitric acid; then, they were diluted with distilled water to 50 cm3 and filtered.
General procedure for the catalytic reactions
All the materials and reagents were of analytical grade and were used as received without purification.A typical procedure for the catalytic alkyne-nitrile transformation is as follows. Dimethyl sulfoxide (DMSO, 2 mL), the corresponding alkyne (0.5 mmol, 1 equiv.), trimethylsilyl azide (TMSN3, 1 mmol, 2 equiv.) and the silver-containing hybrid material as catalyst (44.2 mg, corresponding to 5 mol% Ag loading) were combined in an oven-dried Schlenk tube equipped with a magnetic stir bar. The reaction mixture was stirred for 24 h at 80 °C. Then, the mixture was cooled to room temperature, and the catalyst was filtered off. Brine (10 mL) was next added, and the resultant liquid was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The crude samples were checked by 1H NMR and GC-MS (in order to determine conversion and product ratio) and were purified by chromatographic techniques to isolate the desired products. Flash column chromatographic purification (performed on Merck silica gel 60, particle size 63–200 mm) and analytical thin-layer chromatography (TLC, performed on Merck silica gel 60 F254 plates) were carried out using mixtures of n-hexane/EtOAc as eluent. When using TLC, the compounds were visualized by means of UV or KMnO4.
Investigation of the catalyst reusability
For investigation of the catalyst reusability, the reaction of p-methoxy phenylacetylene with TMSN3 was carried out multiple times utilizing a single portion of catalyst. DMSO (5 mL), p-methoxy phenylacetylene (1.25 mmol, 1 equiv.), TMSN3 (2.5 mmol, 2 equiv.) and the heterogeneous silver catalyst (110.5 mg, corresponding to 5 mol% Ag loading) were combined in an oven-dried Schlenk tube equipped with a magnetic stir bar. The reaction mixture was stirred for 24 h at 80 °C. The mixture was next cooled to room temperature, and the solid material was removed by centrifugation. The liquid phase was extracted, dried and evaporated as detailed above. The removed catalyst was washed with DMSO (four times), and was dried in nitrogen flow before the next cycle. The conversion and selectivity were determined after each cycle by using 1H NMR.Characterization of the reaction products
The nitrile products were characterized by NMR spectroscopy and mass spectrometry. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance DRX 400 spectrometer, in CDCl3 as solvent, with trimethylsilane as internal standard at 400.1 and 100.6 MHz, respectively. GC-MS analyses were performed on a Thermo Scientific Trace 1310 Gas Chromatograph coupled with a Thermo Scientific ISQ QD Single Quadrupole Mass Spectrometer using a Thermo Scientific TG-SQC column (15 m × 0.25 mm ID × 0.25 μm film). Measurement parameters were as follows. Column oven temperature: from 50 to 300 °C at 15 °C min−1; injection temperature: 240 °C; ion source temperature: 200 °C; electrospray ionization: 70 eV; carrier gas: He at 1.5 mL min−1; injection volume: 2 μL; split ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33.3; and mass range: 25–500 m/z. The analytical data can be found in the ESI.†
:33.3; and mass range: 25–500 m/z. The analytical data can be found in the ESI.†
Results and discussion
The proof of the concept – catalytic application of the silver–bismuth hybrid material in direct alkyne-nitrile transformations and comparison to other silver-based catalysts
In order to show that it is worthwhile to spend time and energy on the characterization of the newly developed silver–bismuth hybrid material (AgBi HM), let us demonstrate its performance in a catalytic model reaction (Scheme 1) in comparison with other silver-based catalysts (Fig. 1). | ||
| Scheme 1 Model reaction: Nitrogenation of p-methoxyphenylacetylene with TMSN3 as nitrogen source. | ||
 | ||
| Fig. 1 Investigation of various catalysts in the direct nitrogenation of p-methoxyphenylacetylene with TMSN3 as nitrogen source (see Scheme 1). Reaction conditions: 1 equiv. (0.25 M) alkyne, 2 equiv. TMSN3, 10 mol% catalyst, solvent: DMSO, 80 °C, 24 h reaction time. | ||
The direct nitrogenation of p-methoxyphenylacetylene with TMSN3 as nitrogen source was chosen as a model reaction (Scheme 1).
DMSO was the initial choice of solvent, and the reaction mixture, containing 1 equiv. of alkyne (0.25 M) and 2 equiv. of TMSN3, was stirred at 80 °C for 24 h.
We were delighted to find complete conversion in the presence of 10 mol% AgBi HM. 1H NMR analysis of the crude material indicated an excellent selectivity of 91% towards the formation of 4-methoxybenzonitrile (2). The side product obtained at an extent of 9% was found to be 5-(4-methoxyphenyl)-1H-1,2,3-triazole (3), which was presumably formed via a thermal azide–alkyne cycloaddition as a side reaction.45 This was corroborated by the finding that without the silver catalyst under otherwise identical conditions, a conversion of merely 6% occurred and triazole 3 was formed exclusively.
The catalytic performance of the AgBi HM was directly compared with commercially available Ag(I) salts (Fig. 1). Ag2CO3 and AgBF4 gave comparable results to the hybrid material: the conversion was quantitative in both cases and the nitrile/triazole ratio was 88:12 and 87:13, respectively. Further Ag(I) salts furnished incomplete conversions and/or lower product selectivities. Among the commercial catalysts tested, AgOAc and AgOTf proved less effective. In these instances, traces of 1-(1-azidovinyl)-4-methoxybenzene (1) could also be detected in the crude product mixture. On the basis of the reaction mechanism proposed by Jiao and co-workers (Scheme 2),28a following a silver-mediated alkyne activation and the attack of the azide anion, nitrile formation occurs via a vinyl azide intermediate.46 The presence of the corresponding vinyl azide byproduct in the reaction mixture therefore indicates the incompleteness of the reaction.
 | ||
| Scheme 2 Possible mechanism of the silver-mediated formation of nitriles from terminal alkynes.28a | ||
As corroborated by a test reaction carried out with 10 mol% of Bi(NO3)3·5H2O as catalyst, the Bi(III) component of the hybrid material is inactive as concerns nitrile formation. When the AgBi HM-catalyzed nitrogenation of p-methoxyphenylacetylene was repeated under argon atmosphere, a significant conversion decrease occurred verifying the oxidative nature of the reaction.
Synthesis and characterization of the silver–bismuth hybrid material
Our previous experiences with LDH-based materials served as the basis of the catalyst synthesis. An LDH-like structure was desired, because organized layers allow easier control over the catalytic properties, and Ag(I) prefers octahedral coordination in solid materials.47 Although LDH layers typically consist of di- and trivalent metal cations,31 our initial concept was supported by the fact that an LDH with a monovalent cationic component (instead of the divalent one) is already known.48 Bi(III) was chosen as a possible trivalent cation, since it also prefers octahedral coordination in solids,49 and the ionic radii of Ag(I) and Bi(III) are comparable: 129 and 117 pm, respectively.50 (As was referred to above, we were able to prepare LDHs with much larger differences in ionic radii.) Moreover, we identified a mineral called beyerite (CaBi2O2(CO3)2), which has a layered structure, and contains Ca(II) ions fixed among the layers of BiO(CO)3.51 It was reckoned that if we could incorporate Ag(I) ions instead of Ca(II), we may well form a basis for success, since we would have a structure having Ag(I) ions immobilized in a lattice which is less ordered than that of beyerite, which thus may have considerable catalytic activity. All these factors gave enough inspiration to start with the synthesis.The urea method was chosen for the co-hydrolysis of the metal salts, since in this way the pH can precisely be controlled through the temperature of hydrolysis.42 Appropriate amounts of Bi(NO3)3·5H2O and AgNO3 were thus dissolved in nitric acid and urea was added to the solution. After stirring for 72 h, the precipitate was filtered, washed, dried and then carefully characterized.
The X-ray diffractograms indicated that the most suitable temperature for the co-hydrolysis was 130 °C. At this temperature, a novel material (assigned as AgBi HM) was formed (Fig. 2, trace C). The diffractogram obtained was very different from those of the components (Fig. 2, traces A and B), and it is not their sum either. It was also found that using a temperature of 100 °C for the co-hydrolysis yielded a material having similar features to Ag2O–Ag2CO3–AgNO3 after hydrolysis (Fig. 2, trace D), i.e., it was probably an Ag2O–Ag2CO3 mixture supported over material A. For catalytic purposes, especially in the liquid phase, this substance would be inappropriate, as the silver component would surely be leached out, since it is not a constituent of the crystal lattice of the support.
 | ||
| Fig. 2 The X-ray patterns of the hydrolyzed Bi(NO3)3·5H2O (A),50 the hydrolyzed mixture of Ag2O–Ag2CO3–AgNO3 (B, where + denotes Ag2O, indices denote Ag2CO3),53 the Bi(NO3)3·5H2O–AgNO3 mixture co-hydrolyzed at 130 °C (C)51 and the Bi(NO3)3·5H2O–AgNO3 mixture co-hydrolyzed at 100 °C (D, where + denotes Ag2O, indices denote Ag2CO3). | ||
Material A was indexed as Bi2O2CO3,52 material B is a mixture of Ag2O and Ag2CO3,53 and material D is Ag2O–Ag2CO3 supported over BiO2CO3. Carbonation is due mainly to urea as well as the airborne CO2. In LDH chemistry, carbonation is disadvantageous in most cases, since the carbonate ion is firmly attached to the positively charged layers, preventing its modification via anion exchange. In this instance, however, it is not a problem, since our goal is fixing Ag(I), while keeping its catalytic activity on one hand, and keeping it in a position which is non-leachable on the other. As has been mentioned already, the X-ray pattern of material C differs from those of the constituents, and is not their sum either. It resembles the X-ray pattern of beyerite (a mineral with various polymorphs but an approximate formula of CaBi2O2(CO3)2), but it is definitely not the same.
UV–vis–near IR DRS spectra as well as the EPR spectra also revealed that material C is a novel substance, and is not a physical mixture of materials A and B (Fig. S1 and S2†). Regarding this work, this is important information, and these spectra are only used as further support for the decisive X-ray diffractograms.
Raman spectroscopy is the method of choice to identify the anionic component(s) (Fig. 3). The observed shifts were assigned as given in Table 1. It is seen that the anion is carbonate in the hybrid material, and there is no nitrate present. One can arrive at the same conclusion from studying the FT-IR spectra (Fig. S3 and Table S1†). The IR spectrum of material C reveals that there is no surface OH.
 | ||
| Fig. 3 The Raman spectra of the hydrolyzed Bi(NO3)3·5H2O (A), the hydrolyzed mixture of Ag2O–Ag2CO3–AgNO3 (B), the Bi(NO3)3·5H2O–AgNO3 mixture co-hydrolyzed at 130 °C (C) and the Bi(NO3)3·5H2O–AgNO3 mixture co-hydrolyzed at 100 °C (D). | ||
| Samples | Raman shift (cm−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | d | e | f | g | h | i | j | k | |
a: Ag–O(carbonate) bending,54 b: Bi–O–Bi stretching,55 c: bridging Bi–O–Bi bonds in [BiO6],56 d: Bi![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching,57 e: Bi–O stretching,58 f: ν4 in-plane deformation of the nitrate anion,59 g: ν4 in-plane deformation of the carbonate anion,57 h: ν2 out-of-plane bending mode of the nitrate anion,59 i: ν1 symmetric mode of the nitrate anion,59 j: Ag–O stretching/bending modes,60 k: ν3 antisymmetric mode of the nitrate anion.60 O stretching,57 e: Bi–O stretching,58 f: ν4 in-plane deformation of the nitrate anion,59 g: ν4 in-plane deformation of the carbonate anion,57 h: ν2 out-of-plane bending mode of the nitrate anion,59 i: ν1 symmetric mode of the nitrate anion,59 j: Ag–O stretching/bending modes,60 k: ν3 antisymmetric mode of the nitrate anion.60 |
|||||||||||
| A | — | 312 | 446 | 542 | — | — | — | — | — | — | — |
| B | 284 | — | — | — | — | 668 | — | 802 | 1038 | 1072 | 1337 |
| C | 260 | — | 460 | — | 620 | — | 714 | — | — | — | — |
| D | 260 | — | 470 | — | — | 674 | 717 | 810 | 1034 | 1064 | 1339 |
In order to learn more about the novel AgBi HM, elemental analysis was performed by the ICP–AES method and a formula of Ag0.17Bi0.88O2CO3 was obtained; for the data, see Table S2.†
Since the intended use of the hybrid substance is a heterogeneous catalyst, its thermal stability is an important feature. Thermal analysis revealed that the original structure was kept up to 380 °C, then, weight losses occurred in three endothermic steps, and no more weight losses could be observed from 510 °C until 700 °C, the maximum temperature applied (Fig. 4). The three steps altogether resulted in 7.5 wt% weight loss (4 wt% + 2 wt% + 1.5 wt%). Since there is no surface OH, all the weight loss should be due to the evolution of CO2. On a molar basis, it can be calculated that 22 g CO2, i.e., half of the carbon content, remained in the heat-treated sample. Consequently, the residue should contain a mixture of oxides, as well as carbonates. Nevertheless, the most important message is that the temperature in the hybrid material-catalyzed reactions must be kept under 380 °C. The three-step weight loss may give some structural information as well.
 | ||
| Fig. 4 Thermal analytical curve obtained during the heat treatment of the AgBi HM. | ||
As far as the cationic components of material C are concerned, the XPS spectra attest that the silver and the bismuth components of the hybrid material are in the +1 and +3 oxidation states, respectively (Fig. S4†).60 The shift of ∼0.2 eV in the hybrid catalyst material indicates some electron transfer towards Ag+; this seemed to be advantageous in our probe reaction of oxidative nature, which might be the reason for outstanding catalytic activity.
The AFM measurement indicates a smooth outer surface (Fig. S5†), and the SEM image displays a lamellar morphology (Fig. 5a), which is verified by the high-resolution TEM measurement (Fig. 5b). On the enlarged version of the TEM image (Fig. S6a†), it was possible to measure the basal spacing (0.645 nm, the thickness of one layer plus the interlayer space), which is the d002 value attested by the SAED image (Fig. S6b†). This value was determined because it can be directly compared to the one calculated from X-ray diffraction using the reflection at the lowest angle (Table 2). The table also contains the BET surface areas as well as the particle size. It is seen that the calculated basal spacing compares well to the one measured on the TEM image.
 | ||
| Fig. 5 The SEM (a) and the high-resolution TEM (b) images of the AgBi HM. (SEM – scanning electron microscopy, TEM – transmission electron microscopy.) | ||
| Sample | d (nm) | Particle thickness (nm) | BET surface area (m2 g−1) |
|---|---|---|---|
| AgBi HM | 0.680 | 29.61 | 3.0 |
The basal spacing is small (note that it also includes the thickness of one layer) and the low BET surface indicates that the interlayer space (i.e. the inner surface) is not accessible even for the nitrogen molecules. In our model reaction, the sizeable organic molecules (Scheme 1 and the compounds in Table 5) can only use the outer surface of the AgBi HM, and as the activities observed attest, they can use it efficiently.
Let us sum up what we have learnt from these measurements. In our hands, we have a hybrid material containing Ag(I) and Bi(III) cationic and carbonate anionic components, the latter in three possible positions. The silver ion is the minor cationic component, probably a guest in the bismuth-carbonate lattice. (Indeed, the X-ray diffractogram resembles that of beyerite [CaBi2O2(CO3)2].) This is a material with a layered structure, keeping this structure up to 380 °C. The Bi(III) ions are constituents of the layers and they are connected via one of the oxygen atoms of the carbonate ions. The silver ions are attached to the carboxylate end of the carbonate ions, thus forming a separate layer residing among the Bi(III) containing layers.
Optimization and extension of the probe reaction
Encouraged by the promising preliminary results, we set out to investigate the effects of all the important reaction parameters in order to determine the optimum conditions for a sustainable methodology. As the next step, the role of the reaction medium was explored. Strong dependence was registered on the nature of the solvent applied. Besides DMSO, only dipolar aprotic solvents exhibiting an amide moiety proved acceptable: N,N-dimethylacetamide (DMA) worked similarly well as DMSO (the conversion was 90% and the nitrile/triazole ratio was 87:13, Table 3, entries 1 and 3), whereas the application of N,N-dimethylformamide (DMF) and N-methylpyrrolidone (NMP) resulted in lower conversions (72% and 41%, respectively) but still good selectivities towards the formation of nitrile 2 (84% and 76%, respectively, Table 3, entries 2 and 4). In other solvents investigated, including polar and non-polar ones, only very low conversions were detected even upon prolonging the reaction time to 72 h (Table 3, entries 5–11). This remarkable dependence is possibly related to the nucleophilicity of the azide anion and the stability of the trans alkenyl silver complex key intermediate, which are strongly dependent on the solubilizing properties of the solvent applied.61 The choice of DMSO as preferable reaction medium was further corroborated by the fact that, according to Pfizer's solvent selection guide for medicinal chemistry, it is a usable solvent, whereas most amide solvents are designated as undesirable.62
|
|
||||||
|---|---|---|---|---|---|---|
| # | Solvent | Time (h) | Conv.a (%) | Product selectivitya (%) | ||
| 1 | 2 | 3 | ||||
| a Determined by 1H NMR analysis of the crude product. b Not determined. c THF = tetrahydrofuran. | ||||||
| 1 | DMSO | 24 | 100 | 0 | 91 | 9 |
| 2 | DMF | 24 | 72 | 0 | 84 | 16 |
| 3 | DMA | 24 | 90 | 0 | 87 | 13 |
| 4 | NMP | 24 | 41 | 2 | 76 | 22 |
| 5 | EtOAc | 72 | 7 | n.d.b | ||
| 6 | THFc | 72 | 5 | n.d.b | ||
| 7 | MeCN | 72 | Traces | n.d.b | ||
| 8 | Toluene | 72 | Traces | n.d.b | ||
| 9 | CH2Cl2 | 72 | Traces | n.d.b | ||
| 10 | Acetone | 72 | Traces | n.d.b | ||
| 11 | n-Hexane | 72 | 0 | — | ||
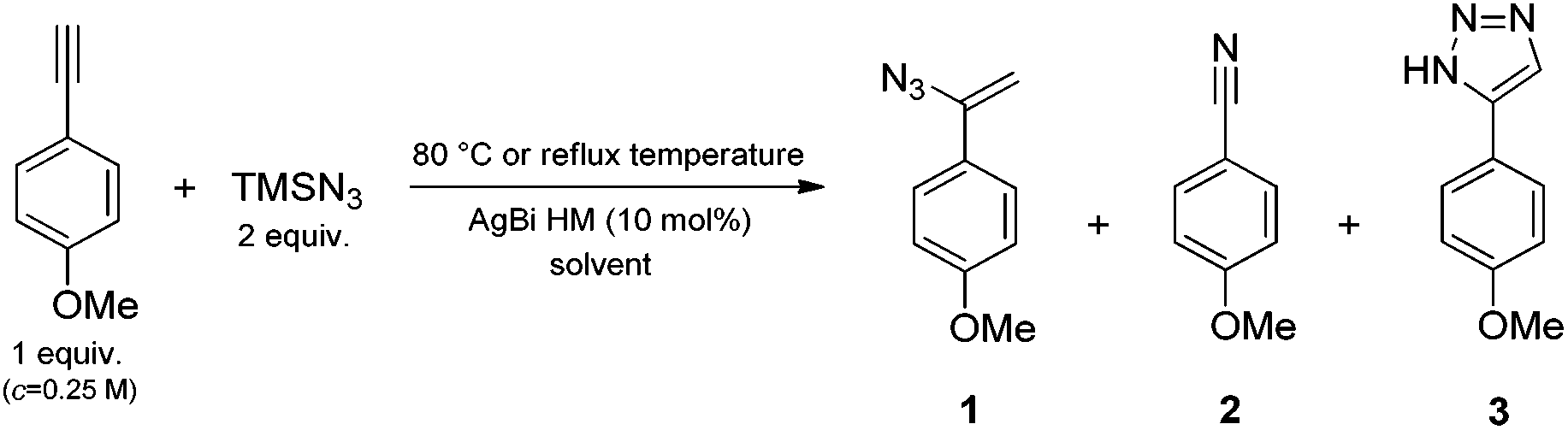
Fig. 6 displays the effects of the reaction time on the progress of the model reaction at 80 °C. As was expected, the total conversion increases gradually with the reaction time reaching completion after slightly longer than 16 h. In the initial stages of the reaction, a substantial amount of vinyl azide 1 can be detected. It is clearly visible that as intermediate 1 is consumed, the amount of the nitrile product grows continuously. After 6 h, only traces of vinyl azide remain, and the ratio of nitrile 2 becomes constant at around 90%. It was also found that triazole 3 appeared as a side product after 1 h of stirring at 80 °C, and its ratio in the reaction mixture became steady around 10% after 6 h. These results led us to conclude that the reaction time should be in the range of 16–24 h to achieve optimum results.
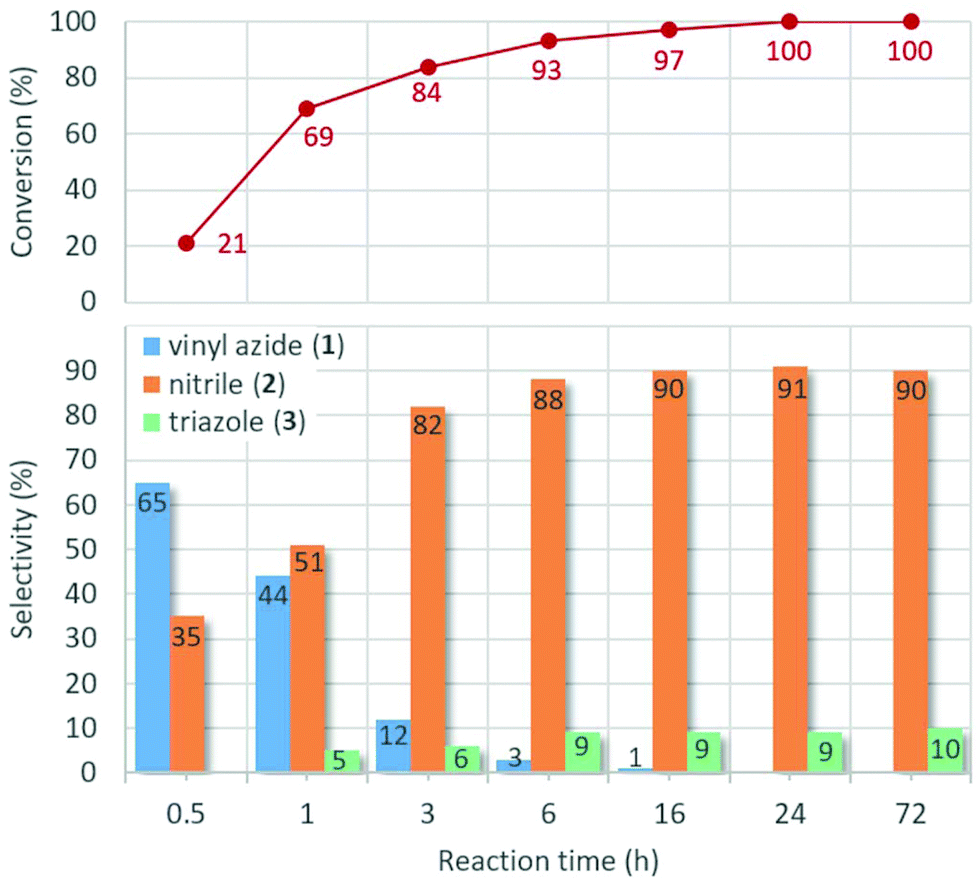 | ||
| Fig. 6 Investigation of the effects of the reaction time on the AgBi HM-catalyzed nitrogenation of p-methoxyphenylacetylene with TMSN3 as nitrogen source (see Scheme 1). Reaction conditions: 1 equiv. (0.25 M) alkyne, 2 equiv. TMSN3, 10 mol% catalyst, solvent: DMSO, 80 °C. | ||
In attempts to improve the rate and the selectivity of the reaction, the role of the temperature was examined carefully (see Fig. S7† for the detailed representation of the experimental data). Elevation of the temperature resulted in a substantial increase in the reaction rate (the conversion of p-methoxyphenylacetylene was 91% after 1 h at 130 °C); however, at the expense of the product selectivity: higher temperatures promoted the formation of the triazole side product via thermal azide–alkyne cycloaddition. For example, at 130 °C, a nitrile/triazole ratio of 80:20 occurred after 24 h of stirring, whereas at 50 °C, triazole 3 was barely formed (to an extent of merely 3%, although conversion was only 79%). 80 °C was selected as the reaction temperature of choice with an excellent nitrile/triazole ratio of 91:9 and quantitative conversion in 24 h.
According to the suggested reaction mechanism of the direct alkyne nitrogenation (Scheme 2), trace amounts of H2O are required for the successful nitrile formation.28a First of all, after the attack of the azide anion, the resulting trans alkenyl silver intermediate should be hydrolyzed to give the corresponding vinyl azide, and a small portion of H2O may also be crucial to generate some HN3 necessary for the final vinyl azide-nitrile transformation. We thus speculated that H2O added to the reaction mixture might have a beneficial effect on the reaction rate. In contrast to our assumption, the presence of excess H2O (2 equiv.) had no significant effects on the reaction progress. In a 0.5–72 h reaction time window, similar conversions were measured as earlier without H2O (see Fig. S8†vs.Fig. 6); however, a small amount of p-methoxyacetophenone appeared among the reaction products as a consequence of competitive silver-catalyzed alkyne hydration.63 These results suggest that none of the reaction steps involving the presence of H2O is rate determining.
Besides TMSN3, NaN3 and diphenylphosphoryl azide (DPPA) were also investigated as nitrogen sources. However, no conversion was detected in the case of DPPA, and NaN3 did not work well either resulting in a conversion of merely 8% with triazole 3 as the main product (Table 4, entries 2 and 3). We tried to reduce the excess of TMSN3 applied, but it was found that 2 equiv. were necessary for the reaction to complete: lower amounts resulted in a sharp decrease in conversion (Table 4, entries 4 and 5). Gratifyingly, under the previously set conditions (80 °C, 24 h, DMSO as solvent, 2 equiv. of TMSN3 as nitrogen source), we were able to lower the catalyst loading to 5 mol% without detectable change in conversion or selectivity (Table 4, entry 6).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| # | N. source | N. source amount (ekv) | Catalyst loading (mol%) | Conv.a (%) | Prod. selectivitya (%) | ||
| 1 | 2 | 3 | |||||
| a Determined by 1H NMR analysis of the crude product. b Not determined. c Ultra-high purity AgBi HM was used as catalyst (synthesized from 99.999% pure metal salts). | |||||||
| 1 | TMSN3 | 2 | 10 | 100 | 0 | 91 | 9 |
| 2 | NaN3 | 2 | 10 | 8 | 0 | 18 | 82 |
| 3 | DPPA | 2 | 10 | 0 | n.d.b | ||
| 4 | TMSN3 | 1.5 | 10 | 91 | 0 | 87 | 13 |
| 5 | TMSN3 | 1.1 | 10 | 80 | 0 | 89 | 11 |
| 6 | TMSN3 | 2 | 5 | 100 | 0 | 90 | 10 |
| 7 | TMSN3 | 2 | 1 | 76 | 2 | 89 | 9 |
| 8c | TMSN3 | 2 | 5 | 100 | 0 | 90 | 10 |
It is well-known that Cu(I) compounds are excellent catalysts for the 1,3-dipolar cycloaddition of organic azides and alkynes to yield 1,2,3-triazoles.17 In order to exclude the possibility of copper contaminations in the catalytic material contributing consistently to triazole side product formation, the synthesis of the AgBi HM was repeated using ultra-high purity (99.999%) silver and bismuth salts. The model reaction was then carried out with the high purity catalyst under optimized reaction conditions. The amount of triazole present as a side product was exactly the same as with the ordinary AgBi HM as catalyst (Table 4, entries 6 vs. 8). This finding, in combination with the fact that Ag(I) itself is not active as catalyst in azide–alkyne cycloadditons,64 suggests that the observed triazole formation (the ratio of triazole 3 was 10% under optimized conditions) can indeed be explained by thermal cycloaddition as a competing reaction pathway.
A range of alkynes exhibiting diverse substitution patterns were next submitted to the optimized reaction conditions. We were delighted to find quantitative conversions and selectivities around 90% in reactions of phenylacetylene and its substituted derivatives containing methyl, tert-butyl or bromo moieties (Table 5, entries 1–5). Even multisubstituted phenylacetylene derivatives and 3-ethynylthiophene gave outstanding results (Table 5, entries 6–8), although they are known to be less reactive in the silver-mediated nitrogenation.28a Phenyl propargyl ether, benzoic acid propargyl ester and p-nitrobenzoic acid propargyl ester displayed excellent reactivities (conversions were in the range 95–100%); however, enhanced triazole formation led to slightly lower selectivities (in the range of 73–81%, Table 5, entries 9–11). The nitrogenation of aliphatic alkynes went smoothly to open chain alkynes with varying chain length and a cyclic alkyne were transformed into the corresponding nitriles with excellent conversions (in the range of 96–100%) and selectivities around 90% (Table 5, entries 12–15). Reactions of internal alkynes (such as ethyl phenylpropiolate or oct-4-yne) were also attempted; however, in these cases no nitrile formation occurred.
|
|
|||||
|---|---|---|---|---|---|
| # | Alkyne | Conv.a (%) | Prod. selectivitya (%) | ||
| I | II | III | |||
| a Determined by 1H NMR or GC-MS analysis of the crude product. | |||||
| 1 |
|
100 | 0 | 90 | 10 |
| 2 |
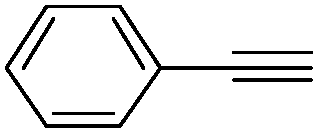
|
100 | 0 | 91 | 9 |
| 3 |

|
100 | 0 | 92 | 8 |
| 4 |

|
100 | 0 | 90 | 10 |
| 5 |
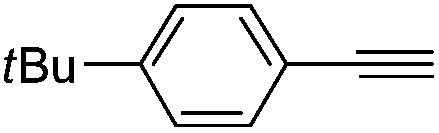
|
100 | 0 | 93 | 7 |
| 6 |
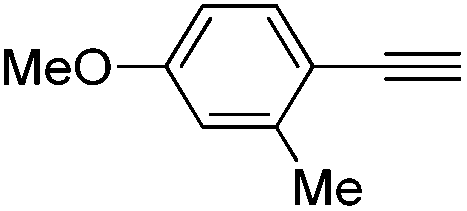
|
100 | 0 | 94 | 6 |
| 7 |
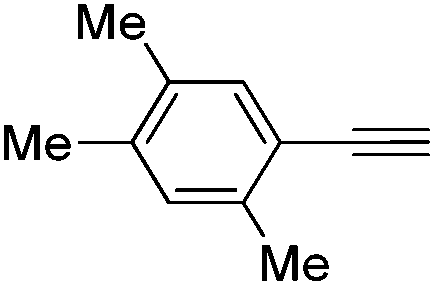
|
100 | 0 | 94 | 6 |
| 8 |

|
100 | 0 | 90 | 10 |
| 9 |
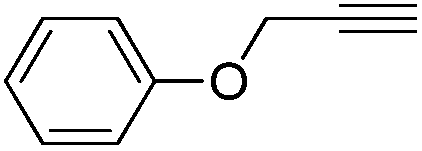
|
100 | 0 | 81 | 19 |
| 10 |

|
100 | 0 | 74 | 26 |
| 11 |

|
95 | 0 | 73 | 27 |
| 12 |
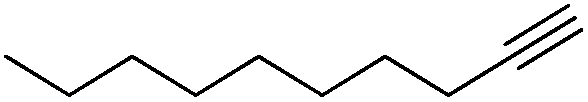
|
100 | 0 | 89 | 11 |
| 13 |
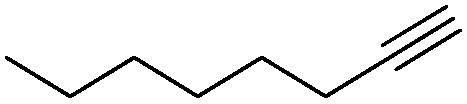
|
100 | 0 | 90 | 10 |
| 14 |

|
96 | 0 | 93 | 7 |
| 15 |

|
98 | 0 | 87 | 13 |
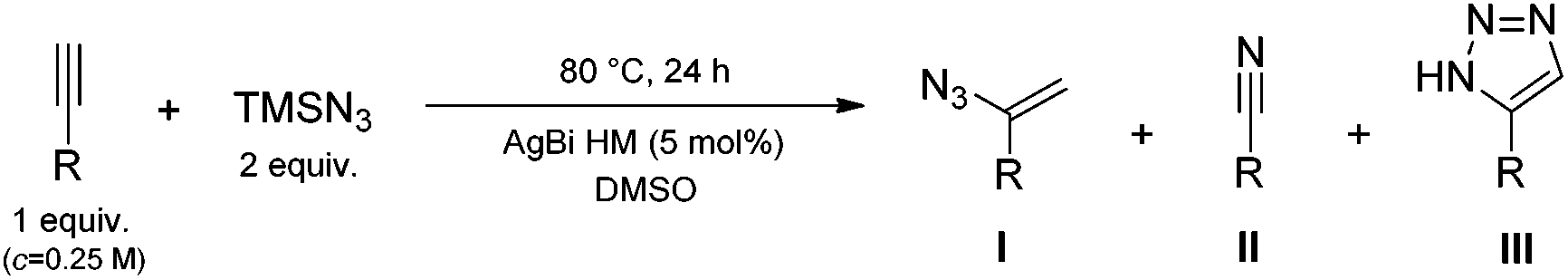
In order to verify the heterogeneous nature of the reaction, the hot filtration test was attempted. The nitrogenation of p-methoxy phenylacetylene with TMSN3 as nitrogen source was carried out under the optimized reaction conditions (80 °C, DMSO as solvent, 2 equiv. of TMSN3, 5 mol% catalyst loading). After 1 h, the AgBi HM catalyst was filtered off, and the filtrate was stirred at 80 °C for another 23 h. As can be seen in Fig. 7, the conversion of p-methoxy phenylacetylene was 65% after 1 h, and remained the same after 1 + 23 h. It is worth mentioning that after 1 h, the product distribution of the reaction was as follows: 47% vinyl azide 1, 49% nitrile 2 and 4% triazole 3. According to our NMR measurements, after 1 + 23 h, all vinyl azide intermediate was consumed (as nicely demonstrated by the disappearance of the alkenyl CH2 signals at 4.9 and 5.3 ppm in Fig. 7) and thermal triazole formation proceeded somewhat further to yield a final nitrile/triazole ratio of 89:11. These results are in good accordance with the mechanistic observations of Jiao and co-workers that it is only the initial hydroazidation step which is silver-mediated, while the final vinyl azide-nitrile transformation requires no silver catalyst.28a
 | ||
| Fig. 7 1H NMR spectroscopic analysis (400.1 MHz, CDCl3) of the AgBi HM-catalyzed reaction of p-methoxyphenylacetylene with TMSN3 as nitrogen source (see Scheme 1). Reaction conditions: 1 equiv. (0.25 M) alkyne, 2 equiv. TMSN3, 5 mol% catalyst, solvent: DMSO, 80 °C. After 1 h, the catalyst was removed, and the filtrate was stirred for another 23 h (hot filtration). Ar–H signals are marked with blue rectangles. | ||
Theoretically, one of the primary benefits of heterogeneous catalysis is the possibility of recycling and reusing the catalytic material, but, in practice, negative effects such as leaching of the active components, erosion of the catalyst structure and irreversible substrate deposition may significantly limit the sustainable application of these materials.29 Although the hot filtration test verified the predominantly heterogeneous nature of the AgBi HM-catalyzed nitrogenation reaction, we were intrigued to explore the robustness and reusability of our catalyst. The reaction of p-methoxyphenylacetylene with TMSN3 was carried out repeatedly (conditions were as follows: 80 °C, DMSO as solvent, 2 equiv. of TMSN3, 5 mol% catalyst loading) utilizing the same portion of AgBi HM as catalyst. Between each cycle, the catalyst was removed by centrifugation and reused after washing and drying. We were delighted to find that no decrease in activity or selectivity occurred even after 10 consecutive catalytic cycles (Fig. 8). The conversion of p-methoxyphenylacetylene was steady in the range of 97–100%, and the selectivity towards the anticipated nitrile product was found to be around 90% in all reactions. Besides small amounts of triazole 3, no vinyl azide byproduct was detected in any of the crude product mixtures analyzed.
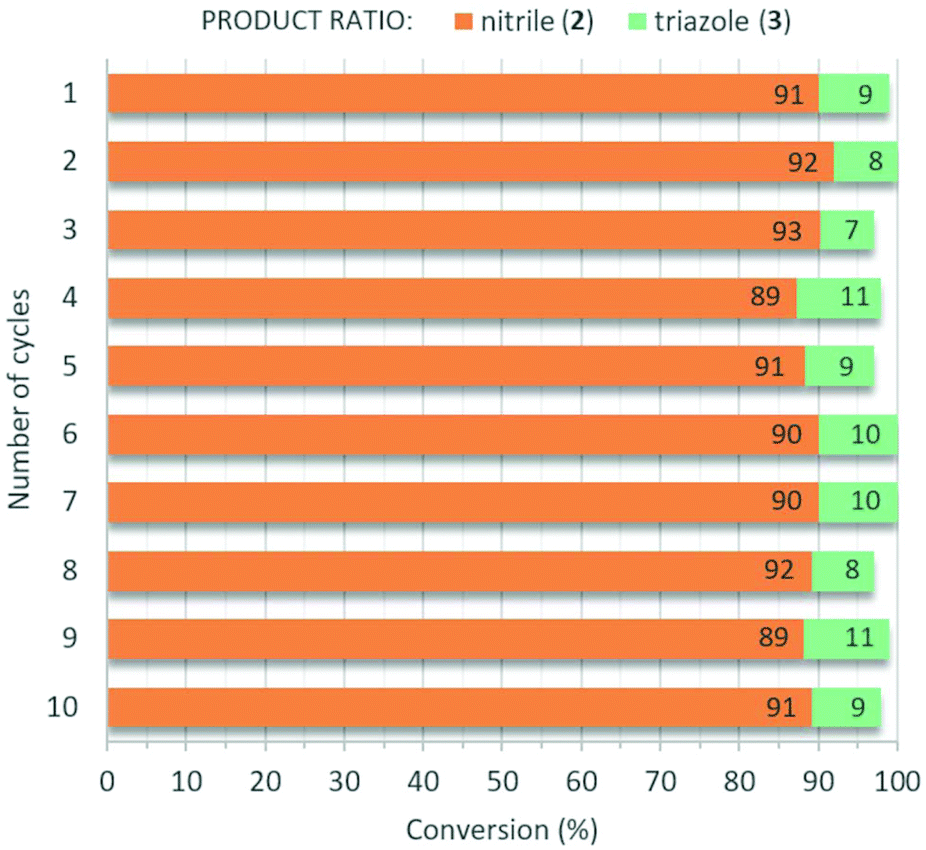 | ||
| Fig. 8 Testing the reusability of the AgBi HM catalyst in the direct nitrogenation of p-methoxyphenylacetylene with TMSN3 as nitrogen source (see Scheme 1). Reaction conditions: 1 equiv. (0.25 M) alkyne, 2 equiv. TMSN3, 5 mol% catalyst, solvent: DMSO, 80 °C, 24 h reaction time. | ||
The catalyst sample used in repeated runs was structurally characterized by many of the methods applied for the freshly-made material. The results of the most important methods, which are decisive in determining whether our material is a truly efficiently recyclable catalyst with high stability, are as follows. The post-reaction X-ray pattern (Fig. 9a) looks the same as trace C in Fig. 2. Indexing may even be attempted using that of beyerite as a known close analog. The Raman spectra of the freshly-made and the used samples are also very similar (compare trace C in Fig. 3 to the spectrum in Fig. S9†) indicating that the structure did not change, the anionic component (the carbonate ion), and the silver ion attachment to the carbonate oxygen all remained the same. The ICP–AES measurement performed on the used catalyst gave a formula of Ag0.17Bi0.91O2CO3 (Table S3,† last row), which is practically the same as the freshly-made one, i.e., the sample survived the recycling test without losing any of its components. The XPS spectra revealed that there was no change in the oxidation states of any of the cationic components (Fig. S10†). The high-resolution TEM image indicates (Fig. 9b) that the layered structure is preserved in the used catalyst sample as well. Taking into account all the characterization data of the used sample, we can conclude that the hybrid material proved to be a highly robust heterogeneous catalyst preserving its structure even after extended use.
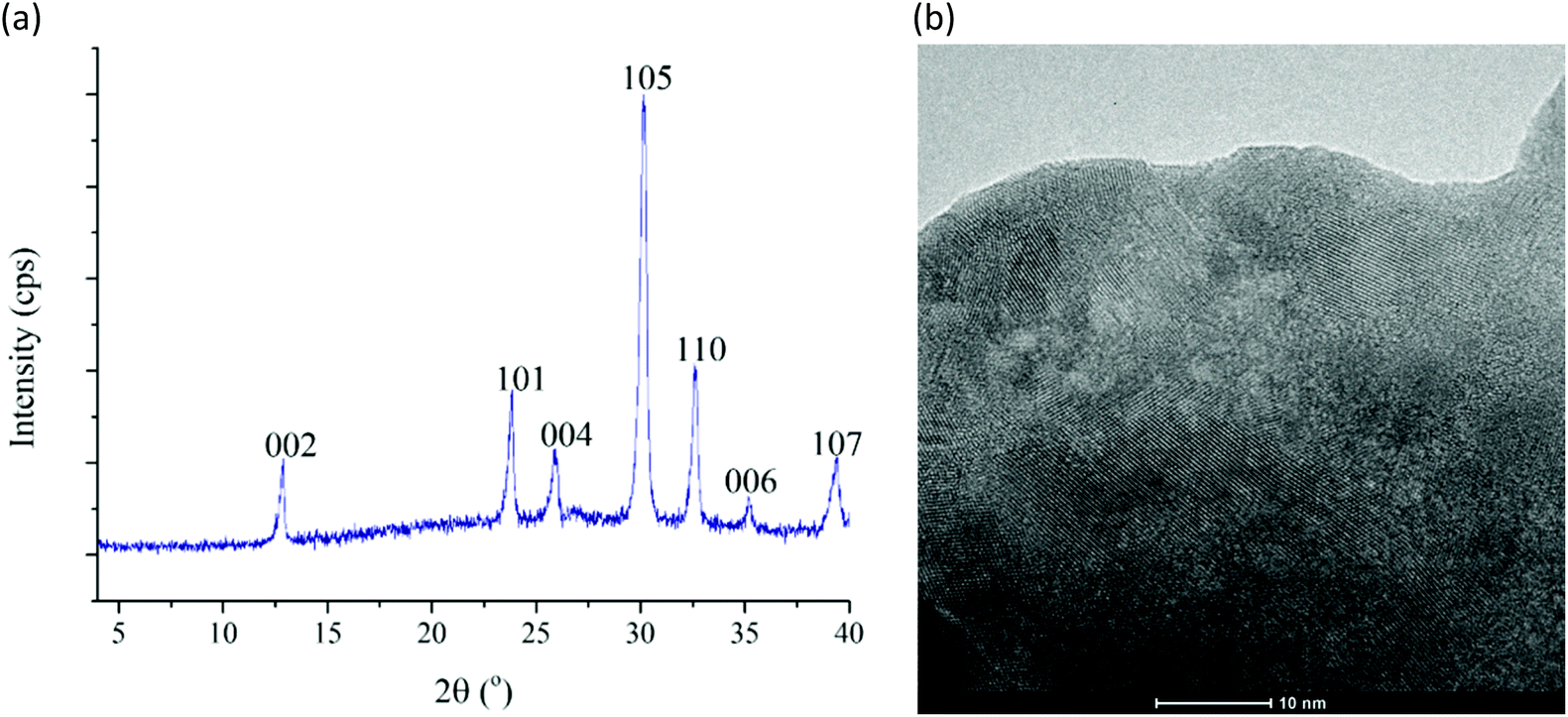 | ||
| Fig. 9 The X-ray pattern (a) and high-resolution TEM image (b) of the used catalyst sample. (TEM – transmission electron microscopy.) | ||
Conclusions
An Ag(I)Bi(III)-containing hybrid material (Ag0.17Bi0.88O2CO3) was synthesized as inspired by a naturally occurring mineral and on the analogy of layered double hydroxides. It proved to be a robust, efficiently recyclable, highly active catalyst in the direct synthesis of organic nitriles from terminal alkynes. The most important reaction conditions determining the reaction rate and the product selectivity were carefully optimized in order to achieve high-yielding nitrile formation and minimal waste generation. A diverse set of aromatic as well as aliphatic alkynes were successfully transformed into the corresponding nitriles with small amounts of triazole as the only unwanted product. The heterogeneous nature of the reaction was demonstrated by the completion of the hot filtration test. Importantly, the solid catalyst could be recycled and reused ten times without loss of activity and without detectable structural degradation. To the best of our knowledge, the first example of a heterogeneous noble metal catalyzed nitrile synthesis via alkyne activation has been presented herein. Due to its outstanding activity, selectivity, reusability and robustness, the catalyst reported may contribute towards the development of sustainable silver-mediated processes, and may find applications in the synthesis of valuable organic compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Fund of Hungary through grants OTKA NKFI K 115731 and GINOP-2.3.2-15-2016-00013. SBÖ acknowledges the Premium Post Doctorate Research Program of the Hungarian Academy of Sciences. RM and GV were supported by the UNKP-17-3 New National Excellence Program of the Ministry of Human Capacities. The financial help is highly appreciated. Bertalan Oszkó (University of Szeged) and László Korecz (Research Center for Natural Sciences of the Hungarian Academy of Sciences, Budapest) are thanked for registering the X-ray photoelectron and the electron paramagnetic spectra, respectively.Notes and references
- (a) Q.-Z. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590–4627 RSC; (b) K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524–4532 RSC; (c) R. Karmakar and D. Lee, Chem. Soc. Rev., 2016, 45, 4459–4470 RSC; (d) H. Pellissier, Chem. Rev., 2016, 116, 14868–14917 CrossRef CAS PubMed; (e) Y. Wang, R. K. Kumar and X. Bi, Tetrahedron Lett., 2016, 57, 5730–5741 CrossRef CAS; (f) V. K.-Y. Lo, A. O.-Y. Chan and C.-M. Che, Org. Biomol. Chem., 2015, 13, 6667–6680 RSC; (g) G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC; (h) G. Abbiati and E. Rossi, Beilstein J. Org. Chem., 2014, 10, 481–513 CrossRef PubMed; (i) T. Xu and G. Liu, Synlett, 2012, 955–958 CAS; (j) Y. Yamamoto, Chem. Rev., 2008, 108, 3199–3222 CrossRef CAS PubMed; (k) M. Naodovic and H. Yamamoto, Chem. Rev., 2008, 108, 3132–3148 CrossRef CAS PubMed; (l) M. Álvarez-Corral, M. Muñoz-Dorado and I. Rodríguez-García, Chem. Rev., 2008, 108, 3174–3198 CrossRef PubMed; (m) J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149–3173 CrossRef CAS PubMed.
- (a) X. E. Verykios, F. P. Stein and R. W. Coughlin, Catal. Rev., 1980, 22, 197–234 CrossRef CAS; (b) C. Wen, A. Yin and W.-L. Dai, Appl. Catal., B, 2014, 160–161, 730–741 CrossRef CAS.
- Silver in organic chemistry, ed. M. Harmata, John Wiley & Sons Inc., 2010 Search PubMed.
- X.-Y. Dong, Z.-W. Gao, K.-F. Yang, W.-Q. Zhang and L.-W. Xu, Catal. Sci. Technol., 2015, 5, 2554–2574 CAS.
- S. F. Cai, H. P. Rong, X. F. Yu, X. W. Liu, D. S. Wang, W. He and Y. D. Li, ACS Catal., 2013, 3, 478–486 CrossRef CAS.
- A. K. Clarke, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 13798–13802 CrossRef CAS PubMed.
- X. Y. Toy, I. I. B. Roslan, G. K. Chuah and S. Jaenicke, Catal. Sci. Technol., 2014, 4, 516–523 CAS.
- J. Safari and S. Gandomi-Ravandi, RSC Adv., 2014, 4, 11654–11660 RSC.
- T. Mitsudome, Y. Mikami, H. Funai, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 138–141 CrossRef CAS PubMed.
- N. Salam, A. Sinha, A. S. Roy, P. Mondal, N. R. Jana and S. M. Islam, RSC Adv., 2014, 4, 10001–10012 RSC.
- J. D. Kim, T. Palani, M. R. Kumar, S. Lee and H. C. Choi, J. Mater. Chem., 2012, 22, 20665–20670 RSC.
- (a) C. T. Campbell, Acc. Chem. Res., 2013, 46, 1712–1719 CrossRef CAS PubMed; (b) G. Prieto, J. Zečević, H. Friedrich, K. P. de Jong and P. E. de Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef CAS PubMed; (c) R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- (a) B. Ballarin, D. Barreca, E. Boanini, M. C. Cassani, P. Dambruoso, A. Massi, A. Mignani, D. Nanni, C. Parise and A. Zaghi, ACS Sustainable Chem. Eng., 2017, 5, 4746–4756 CrossRef CAS; (b) R. Ricciardi, J. Huskens and W. Verboom, ChemSusChem, 2015, 8, 2586–2605 CrossRef CAS PubMed; (c) L. D. Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- (a) J. Cao, G. Xu, P. Li, M. Tao and W. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 3438–3447 CrossRef CAS; (b) X. Tang, C. Qi, H. He, H. Jiang, Y. Ren and G. Yuan, Adv. Synth. Catal., 2013, 355, 2019–2028 CrossRef CAS; (c) M. Jeganathan, A. Dhakshinamoorthy and K. Pitchumani, ACS Sustainable Chem. Eng., 2014, 2, 781–787 CrossRef CAS; (d) A. H. Jadhav, A. Chinnappan, R. H. Patil, W.-J. Chung and H. Kim, Chem. Eng. J., 2014, 236, 300–305 CrossRef CAS; (e) K. T. Venkateswara Rao, P. S. Sai Prasad and N. Lingaiah, Green Chem., 2012, 14, 1507–1514 RSC; (f) K. Mohan Reddy, N. Seshu Babu, I. Suryanarayana, P. S. Sai Prasad and N. Lingaiah, Tetrahedron Lett., 2006, 47, 7563–7566 CrossRef.
- Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, ed. B. M. Trost and C.-J. Li, Wiley-VCH Verlag GmbH & Co. KGaA, 2014 Search PubMed.
- (a) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed; (b) X. Zeng, Chem. Rev., 2013, 113, 6864–6900 CrossRef CAS PubMed; (c) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790–3807 RSC; (d) R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- R. K. Kumar and X. Bi, Chem. Commun., 2016, 52, 853–868 RSC.
- Z. Zhou, C. He, L. Yang, Y. Wang, T. Liu and C. Duan, ACS Catal., 2017, 7, 2248–2256 CrossRef CAS.
- The Chemistry of the Cyano Group, ed. Z. Rappoport, John Wiley & Sons Ltd, 1970 Search PubMed.
- (a) L.-G. Xie, S. Niyomchon, A. J. Mota, L. González and N. Maulide, Nat. Commun., 2016, 7, 10914 CrossRef PubMed; (b) Y. Wang, L.-J. Song, X. Zhang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 9704–9708 CrossRef CAS PubMed; (c) B. Gutmann, J.-P. Roduit, D. Roberge and C. O. Kappe, Angew. Chem., Int. Ed., 2010, 49, 7101–7105 CrossRef CAS PubMed.
- (a) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed; (b) F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597–606 RSC.
- H. H. Hodgson, Chem. Rev., 1947, 40, 251–277 CrossRef CAS PubMed.
- (a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779–794 CrossRef CAS; (b) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948–11959 CrossRef CAS PubMed; (c) N. Kurono and T. Ohkuma, ACS Catal., 2016, 6, 989–1023 CrossRef CAS.
- (a) W. E. Dennis, J. Org. Chem., 1970, 35, 3253–3255 CrossRef CAS; (b) M. K. Singh and M. K. Lakshman, J. Org. Chem., 2009, 74, 3079–3084 CrossRef CAS PubMed.
- A. Martin and B. Lücke, Catal. Today, 2000, 57, 61–70 CrossRef CAS.
- B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2012, 77, 5364–5370 CrossRef CAS PubMed.
- (a) T. Shen, T. Wang, C. Qin and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 6677–6680 CrossRef CAS PubMed; (b) P. Bisseret, G. Duret and N. Blanchard, Org. Chem. Front., 2014, 1, 825–833 RSC.
- Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH Verlag GmbH, 2nd edn, 2008 Search PubMed.
- (a) A. Primo and H. Garcia, Chem. Soc. Rev., 2014, 43, 7548–7561 RSC; (b) J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- (a) G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC; (b) J. Feng, Y. He, Y. Liu, Y. Du and D. Li, Chem. Soc. Rev., 2015, 44, 5291–5319 RSC.
- Z. P. Xu, J. Zhang, M. O. Adebajo, H. Zhang and C. Zhou, Appl. Clay Sci., 2011, 53, 139–150 CrossRef CAS.
- (a) J. Qu, Q. Zhang, X. Li, X. He and S. Song, Appl. Clay Sci., 2016, 119, 185–192 CrossRef CAS; (b) F. L. Theiss, G. A. Ayoko and R. L. Frost, Appl. Surf. Sci., 2016, 383, 200–213 CrossRef CAS.
- (a) K. Yan, Y. Liu, Y. Lu, J. Chai and L. Sun, Catal. Sci. Technol., 2017, 7, 1622–1645 RSC; (b) V. Prevot and Y. Tokudome, J. Mater. Sci., 2017, 52, 11229–11250 CrossRef CAS.
- C. H. Zhou, Appl. Clay Sci., 2011, 53, 87–96 CrossRef CAS.
- Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322–359 CrossRef CAS PubMed.
- S. He, Z. An, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2013, 49, 5912–5920 RSC.
- P. Sipos and I. Pálinkó, Catal. Today DOI:10.1016/j.cattod.2016.12.004 , in press.
- D. Srankó, A. Pallagi, E. Kuzmann, S. E. Canton, M. Walczak, A. Sápi, Á. Kukovecz, Z. Kónya, P. Sipos and I. Pálinkó, Appl. Clay Sci., 2010, 48, 214–217 CrossRef.
- (a) G. Varga, Á. Kukovecz, Z. Kónya, L. Korecz, S. Muráth, Z. Csendes, G. Peintler, S. Carlson, P. Sipos and I. Pálinkó, J. Catal., 2016, 335, 125–134 CrossRef CAS; (b) G. Varga, Z. Timár, S. Muráth, Z. Kónya, Á. Kukovecz, S. Carlson, P. Sipos and I. Pálinkó, Catal. Today DOI:10.1016/j.cattod.2016.12.005, in press; (c) G. Varga, S. Ziegenheim, S. Muráth, Z. Csendes, Á. Kukovecz, Z. Kónya, S. Carlson, L. Korecz, E. Varga, P. Pusztai, P. Sipos and I. Pálinkó, J. Mol. Catal. A: Chem., 2016, 423, 49–60 CrossRef CAS; (d) M. Szabados, R. Mészáros, S. Erdei, Z. Kónya, Á. Kukovecz, P. Sipos and I. Pálinkó, Ultrason. Sonochem., 2016, 31, 409–416 CrossRef CAS PubMed.
- (a) S. B. Ötvös, Á. Georgiádes, R. Mészáros, K. Kis, I. Pálinkó and F. Fülöp, J. Catal., 2017, 348, 90–99 CrossRef; (b) S. B. Ötvös, Á. Georgiádes, M. Ádok-Sipiczki, R. Mészáros, I. Pálinkó, P. Sipos and F. Fülöp, Appl. Catal., A, 2015, 501, 63–73 CrossRef.
- A. Inayat, M. Klumpp and W. Schwieger, Appl. Clay Sci., 2011, 51, 452–459 CrossRef CAS.
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef; (b) Y. Tanaka, S. R. Velen and S. I. Miller, Tetrahedron, 1973, 29, 3271–3283 CrossRef CAS.
- Z. Liu, P. Liao and X. Bi, Org. Lett., 2014, 16, 3668–3671 CrossRef CAS PubMed.
- A. F. Cotton and G. Wilkinson, Advanced inorganic chemistry, John Wiley & Sons, 5th edn, 1988, p. 940 Search PubMed.
- A. V. Besserguenev, A. M. Fogg, R. J. Francis, S. J. Price, D. O'Hare, V. P. Isupov and B. P. Tolochko, Chem. Mater., 1997, 9, 241–247 CrossRef CAS.
- A. F. Cotton and G. Wilkinson, Advanced inorganic chemistry, John Wiley & Sons, 5th edn, 1988, p. 383 Search PubMed.
- A. F. Cotton and G. Wilkinson, Advanced inorganic chemistry, John Wiley & Sons, 5th edn, 1988, p. 1387 Search PubMed.
- V. Malik, M. Pokhriyal and S. Uma, RSC Adv., 2016, 6, 38252–38262 RSC.
- T. Selvamani, A. M. Asiri, A. O. Al-Youbi and S. Anandan, Mater. Sci. Forum, 2013, 764, 169–193 CrossRef.
- (a) H. Xia and G. Yang, J. Mater. Chem., 2012, 22, 18664–18670 RSC; (b) H. Xu, J. Zhu, Y. Song, W. Zhao, Y. Xu, Y. Song, H. Ji and H. Li, RSC Adv., 2014, 4, 9139–9147 RSC.
- C.-B. Wang, G. Deo and I. E. Wachs, J. Phys. Chem. B, 1999, 103, 5645–5656 CrossRef CAS.
- G. Zhao, Y. Tian, H. Fan, J. Zhang and L. Hu, J. Mater. Sci. Technol., 2013, 29, 209–214 CAS.
- F. Chen, B. Song, C. Lin, S. Dai, J. Cheng and J. Heo, Mater. Chem. Phys., 2012, 135, 73–79 CrossRef CAS.
- F. Dong, S. C. Lee, Z. Wu, Y. Huang, M. Fu, W.-K. Ho, S. Zou and B. Wang, J. Hazard. Mater., 2011, 195, 346–354 CrossRef CAS PubMed.
- P. Dararutana, S. Pongkrapan, N. Sirikulrat, M. Thawornmongkolkij and P. Wathanakul, Spectrochim. Acta, Part A, 2009, 73, 440–442 CrossRef CAS PubMed.
- B. M. Gatehouse, S. E. Livingstone and R. S. Nyholm, J. Chem. Soc., 1958, 3137–3142 RSC.
- W. Wang, Y. Liu, H. Zhang, Y. Qian and Z. Guo, Appl. Surf. Sci., 2017, 396, 102–109 CrossRef CAS.
- T. B. Phan and H. Mayr, J. Phys. Org. Chem., 2006, 19, 706–713 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- M. B. T. Thuong, A. Mann and A. Wagner, Chem. Commun., 2012, 48, 434–436 RSC.
- T. U. Connell, C. Schieber, I. P. Silvestri, J. M. White, S. J. Williams and P. S. Donnelly, Inorg. Chem., 2014, 53, 6503–6511 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis, IR, EPR, XPS, ICP-AES spectra and spectral data, AFM, TEM and SAED images on the fresh AgBi HM sample; Raman, XPS and ICP-AES data on the used catalyst sample; additional reaction optimization data; NMR and MS data of the reaction products. See DOI: 10.1039/c7gc02487h |
| This journal is © The Royal Society of Chemistry 2018 |