 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is zeroth order crystal structure prediction (CSP_0) coming to maturity? What should we aim for in an ideal crystal structure prediction code?
Sarah L.
Price

Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: s.l.price@ucl.ac.uk
First published on 27th July 2018
Abstract
Crystal structure prediction based on searching for the global minimum in the lattice energy (CSP_0) is growing in use for guiding the discovery of new materials, for example, new functional materials, new phases of interest to planetary scientists and new polymorphs relevant to pharmaceutical development. This Faraday Discussion can assess the progress of CSP_0 over the range of types of materials to which CSP is currently and could be applied, which depends on our ability to model the variety of interatomic forces in crystals. The basic hypothesis, that the outcome of crystallisation is determined by thermodynamics, needs examining by considering methods of modelling relative thermodynamic stability not only as a function of pressure and temperature, but also of size, solvent and the presence of heterogeneous templates or impurities (CSP_thd). Given that many important materials persist, and indeed may be formed, when they are not the most thermodynamically stable structure, we need to define what would be required of an ideal CSP code (CSP_aim).
Introduction
Intellectual curiosity as to whether we can predict crystal structures predates the computer era, with Kitaigorodsky’s mechanical structure seeker fitting the “projections” (bumps) formed by the atoms of one molecule into the hollows of another so that the molecules dovetailed into a close packed structure, or various radius ratio rules for simple inorganic crystals, that also are based on the principle of close packing. Maddox’s famous quote1 in 1981, “One of the continuing scandals in the physical sciences is that it remains in general impossible to predict the structure of even the simplest crystalline solids from a knowledge of their chemical composition”, can be seen as reflecting the common assumption at the time, that a given molecule or ionic composition always crystallised in the same crystal structure. The term Crystal Structure Prediction (CSP) originates from the days when they were seeking to predict the crystal structure, which was that of the first crystal that could be grown to a size and quality suitable for a crystallographic determination. The crystallography was usually done to prove that the correct molecule had been synthesised. As McCrone said in 1965 “In spite of the fact that different polymorphs of the same compound are, in general, as different in structure and properties as the crystals of two different compounds, most chemists are almost completely unaware of the nature of polymorphism and the potential usefulness of knowledge of this phenomenon in research”.2Computers make it possible to test whether we really understand what determines how a molecule will crystallise, by programming a theory and applying it to test the “predictions” against experiment. There is a big distinction between a theory or program that seeks to predict the crystal structure and one which seeks to predict all polymorphs. This distinction needs to be clear in this discussion. Most CSP programs are based on the theory that the crystal structure is the most thermodynamically stable structure, and, at least initially, assume that the relative thermodynamic stability can be approximated by the lattice energy, the energy of the static lattice relative to infinitely separated molecules in their lowest energy conformation (or relative to all electrons and nuclei at infinite separation).
I would like to define CSP_0, the zeroth order model, as the attempt to predict the most likely (thermodynamically stable) crystal structure as the most stable in lattice energy. That some of the competitive local minima in the lattice energy correspond to polymorphs is fortunate for the practical interest in CSP.
We can then define CSP_thd as the full implementation of the assumption that realistically estimates the relative thermodynamic stability of the crystal structures, and so will predict the most thermodynamically stable crystal structure under given conditions of temperature and pressure and any other thermodynamic variables that are relevant. The pharmaceutical industry would really welcome such a code, but currently the calculation of the phase diagram of crystalline methanol represents a major step forward in this regard.3 The situation is more advanced for other materials, with a revision of the phase diagram of CaCO3 over half the pressure range within the earth’s mantle,4 leading to the discovery of a new phase with implications for carbon storage in the deep mantle.5 Differences in the approximations required for estimating the appropriate relative thermodynamic stability for different material types are currently important.
If we calculate the free energy minima by calculating the true thermodynamic stabilities of the CSP_0 structures, we expect that some lattice energy minima will have merged into the same free energy minimum. This structure could have a higher symmetry on average than any lattice energy minimum, if it is a dynamically disordered structure. The structures that are within the likely energy range of possible polymorphism constitute the crystal energy landscape, the set of thermodynamically plausible structures. Thus CSP_thd methods will undoubtedly improve the prediction of polymorphism at practically relevant temperatures, but how do we define which local minima on the crystal energy landscape are possible, practically relevant polymorphs?
One practical justification for developing CSP methods is in the design of new materials, to guide synthetic work. The main benefit of such studies is to avoid the synthesis of molecules or materials that will not readily crystallise in a structure which has the desired properties. This requires the calculation of the property of interest from the computer-generated structures, with sufficient realism to convince the experimental group that the synthetic effort is worth making. If more than the thermodynamically stable structure is of interest, which will generally be the case if metastable polymorphs (or variants in chiral composition) are likely to complicate product manufacture, then the property needs to be calculated for all the structures on the crystal energy landscape, giving crystal-structure–property maps. CSP_0 may be adequate for excluding an organic molecule from the synthetic program, but CSP_thd is desirable to test whether the desired novel organic material is the most thermodynamically stable at ambient. Other materials can survive over a much greater range of temperatures and pressures, and for these CSP_thd can be essential, with pressure being particularly widely used for planetary science.
The second practical justification for developing CSP is polymorphism. The importance of polymorphism for the quality control of industrial materials, which is particularly acute for the pharmaceutical industry, has inspired the development and commercialisation of CSP. However, intellectually, it messes up the ability to test the theory for predicting crystal structures, as it is unclear what crystal structures have to be predicted. Conclusions from the Cambridge Crystallographic Data Centre’s blind tests of organic crystal structure prediction6 have always been qualified by the possibility of unreported polymorphs. Over the lifetime of CSP, the question has changed from which systems are polymorphic to which are not. Systems, such as aspirin, that used to be quoted as examples of monomorphic systems now have polymorphs in the Cambridge Structural Database (CSD).7
It is now fifteen years since a group of UK scientists convinced Research Councils UK that a computational method of crystal structure prediction would be a “Basic Technology” for many areas of science. It is gratifying that one of our industrial contributions raises the question as to whether CSP is changing from basic science to applied technology (DOI: 10.1039/c8fd00033f). I would like to start this discussion meeting by asking a few questions about the state of CSP_0 and CSP_thd and considering what we would expect of a genuine crystal structure prediction code (CSP_aim) in terms of polymorph prediction. Here we must recognise that over-predicting polymorphs is not helpful, though it is more useful than having no computational guidance about the completeness of the experimental polymorph screen. Should we be aiming for a code that only produces the polymorphs that can be experimentally found, along with sufficiently reliable predicted properties to ensure that they could be found or safely dismissed as irrelevant?
The other dimension to this discussion is the extension of CSP methods to the widest range of materials. The ability to predict the crystal structures of benzene will not guarantee that all pharmaceuticals can be predicted by the same methods, any more than a CSP method that works for NaCl will work for zeolites. We now are developing CSP for many new forms of materials such as MOFs (metal organic frameworks), organometallics, and anything that the synthetic materials chemist can develop, including interface structures. This discussion should exchange ideas between the CSP communities working on different types of materials. The differences arise from the nature of the building blocks being used, and different interatomic interactions being balanced. A lot of the challenges in organic CSP for flexible molecules come from trying to balance the different interactions within the molecules, such as flexible torsions, with the variety of possible intermolecular interactions. When the molecules are linked by a great diversity of atoms, such as metals, the distinctions between inter- and intramolecular, ionic and covalent, oxidation and spin states may become less clear-cut and so cause inaccuracy for a given type of lattice energy modelling. As the coverage of the periodic table increases, the possibility of chemical substitutions in the lattice increases, as different ions can be exchanged with less effect on structures and relative energies than changing organic functional groups. When CSP is applied to geological materials there is a problem of defining the composition. The different classes of materials have different plausible starting assumptions in CSP, such as (approximately) rigid building blocks, but the complexity and difficulty comes as we combine different types of interactions of different strength and directionality. Hence “variety of interatomic interactions” is a major aspect of the material complexity axis on a two dimensional diagrammatic scheme on which to assess our progress as shown in Fig. 1.
 | ||
| Fig. 1 Schematic of the two directions of progress required for a universal crystal structure prediction code, which predicts only all the experimentally observable polymorphs of a system, and sufficient properties to enable them to be found. | ||
Dirac’s famous quote, “The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known”, is essentially saying that we have the physical laws for a universal CSP code. Developments in computer power are allowing us to make huge strides in applying CSP_0, however we are all limited by resources in testing the theoretically more correct CSP_thd. How long will it be necessary to make material and application-dependent short cuts? Against the huge ambition of a universal CSP_aim code, we can plot our progress on Fig. 1. At each point, we have to tension reliability for all materials in that class against McCrone’s famous quote “every compound has different polymorphic forms, and that, in general, the number of forms known for a given compound is proportional to the time and money spent in research on that compound”.2
CSP_0, finding the structure that has the lowest lattice energy
Accurate determination of the relative lattice energies
Most theoretical chemists will see an analogy between Fig. 1 and the diagram for systematically improving approximate wavefunctions as a function of basis set size (one axis) and electron correlation (the other axis) to reach true solution to the Schrödinger equation for a molecule or solid. Density functional methods have lost the advantages of the variation theorem, but there is still a hierarchy of methods8–10 and the methods widely used in CSP are on the lower rungs. The hierarchy of what method of calculating the lattice energy is good enough for CSP is very system dependent. To what extent is the quantum mechanical method good enough for the type of material that the final CSP_0 energies can be relied upon as being accurate enough? For inorganic systems, are the possible variants in oxidation states, spin states and even relativistic effects being adequately represented? For some materials, theory shows that there is no hope of a worthwhile CSP study without evaluating the lattice energy by using an electronic structure theory accuracy calculation on all competitive structures (Ψcrys).Electronic structure theory methods suffer from the intimate link between the dispersion energy and electron correlation. Hence a high level of theory is required to incorporate dispersion realistically ab initio. Modelling the dispersion (DOI: 10.1039/c8fd00066b) is very important for organics, particularly if you are contrasting crystal structures which have only dispersion between layers with those that have stronger forces, such as hydrogen bonding in all three directions. Structures such as molecular ionic cocrystals (DOI: 10.1039/c8fd00036k) have many competing forces. This can lead to complex systems e.g. a pharmaceutical salt which exhibits many solid forms, including a series of alcohol solvates, where the alcohol interdigitates between the ionic and molecular layers.11 At one level this is a simple series of salt solvates, but there are subtle differences that can give rise to disorder in the alcohol hydrocarbon tail layer (causing problems in crystal growth and characterisation) and disorder in Cl− ions only detectable with the most accurate crystallography. In addition, this study that started from a CSP on the pharmaceutical salt, was further complicated by the late appearance of a highly metastable polymorph.11 This illustrates the issue of the level of atomic detail and energetic accuracy at which we want to predict crystal structures, in systems where there can be considerable stabilisation by solvents in a disordered form (DOI: 10.1039/c8fd00031j), or multiple similar ions can occupy the same position. The level of atomic detail required is intimately linked with the quality of the lattice energy model needed to accurately balance all the different types of intermolecular interactions present.
We now know that calculating the lattice energy by electronic modelling (Ψcrys) is very expensive, if it will be applied to a sufficient number of structures. We have different approaches to this challenge. One innovative approach is to use data-driven learning of the potential energy surface. There are applications of this approach to the CSP of medium and large-sized boron clusters (DOI: 10.1039/c8fd00055g) and to phosphorus (DOI: 10.1039/c8fd00034d) starting our conference.
The more traditional approach is to have a hierarchical scheme, increasing in accuracy as it is possible to focus on the more plausible structures. This needs care but CSP_0 is a valuable test for the developers of different lattice energy codes, from the variety of lattice energy models that can be used, to the vital details that improve efficiency and accuracy, such as use of symmetry, optimisation methods, lattice summations and convergence criteria. We should be careful. Tests made on the X23 set of molecular crystal lattice energies, such as the recent one using 4 codes and 18 functionals12 are valuable, but this is a data set of very small, virtually rigid molecules. For pharmaceuticals or other molecules with significant flexibility and considerable variation in the intramolecular dispersion and packing density, then the reliability of different methods could be different (DOI: 10.1039/c8fd00010g). Balancing the effects of flexibility/conformational change with the other forces is a problem for a wide range of materials, from pharmaceuticals, to novel inorganic materials such as ultra-flexible boron oxide frameworks (DOI: 10.1039/c8fd00052b) but the forces differ according to the types of atoms and structures involved.
The demands of the search mean that many methods will use atomistic modelling somewhere in the hierarchy, with an appropriate force-field. The accuracies of force-fields for different applications vary according to the crystals, but they are widely used for ionic and molecular systems. The use of molecules or fixed ionic groups makes a lot of sense for restricting the search to the molecule or structural type of interest. For molecular systems, it seems that a molecule-specific electrostatic model is usually essential. For flexible molecules, this requires wavefunction calculations on the molecule covering the possible range of conformations in the crystal (Ψmol), which also provides the conformational energy penalty. Atomistic modelling of molecular crystals can then be performed with the addition of a set of empirically fitted repulsion–dispersion intermolecular atom–atom potentials. We are now revisiting the early work of Williams in empirically parameterising such atom–atom potentials for organics, with Day’s group recently showing that such parameterizations combined with distributed multipoles can rival popular DFT-D methods in accuracy.13 A new scheme is being proposed for empirical repulsion–dispersion potentials for use with a specific level of Ψmol (DOI: 10.1039/c8fd00064f). Empirically fitted potentials have the advantage of absorbing some of the approximations into the parametrisation, but that can also be a disadvantage in preventing extrapolation to other regions, e.g. predicting a high pressure phase of pyridine required a non-empirical anisotropic atom–atom intermolecular potential.14 We also risk double counting thermal effects when these potentials are used in Molecular Dynamics (MD) approximations to CSP_thd. On the other hand, empirically fitted potentials, or other CSP schemes that are based on experimental crystal structures, could be more effective than CSP_0 from implicitly absorbing non-thermodynamic effects, i.e. partially moving towards CSP_thd.
Any system of CSP_0 is reliant on relative lattice energies, and relies on cancelation of errors between different crystal structures of the same system. This accounts for the huge successes that have been achieved by CSP_0, when the absolute lattice energies may be very inaccurate and the Ψcrys or Ψmol calculations are so far from even state-of-the-art approximate solutions to the real wavefunction. Hence the need to be aware of the theoretical basis of the different approximations used as we increase the variety of types of interatomic interactions in our crystals.
Search coverage in CSP_0 structure generation
Choices are made in defining the search space to be covered. We can use sophisticated methods to try to ensure that the search is complete within a defined range, but a lack of computer and human time often prevents this limit of confidence being reached. Refining our methods to make them more efficient can potentially extend the scope of CSP studies, but to what extent are these refinements universal or material dependent, for example using genetic algorithms for molecular crystals (ref. 15, DOI: 10.1039/c8fd00067k) or zeolites (DOI: 10.1039/c8fd00035b)? Recognised patterns in the appropriate class of materials may be incorporated into the structure generation methodology very explicitly for the class of materials, from oxidation states (DOI: 10.1039/c8fd00032h), to properties at given site-symmetries in zeolites (DOI: 10.1039/c8fd00040a), optimised blueprints for MOFs (DOI: 10.1039/c8fd00051d), or probe structure for the inorganic compositional space (DOI: 10.1039/c8fd00045j). Ensuring that this provides an efficient encapsulation of the possible chemistry without restricting the ability to propose exciting new materials, particularly those that bridge traditional classes, is the real challenge in improving the complexity of structures being created first in the computer by CSP (Fig. 1). We need to ensure that assumptions made for computational convenience do not become outdated. For example, when CSP_0 was first instigated for organic molecules, the assumption of Z′ = 1 (one molecule in the asymmetric unit cell) was more reasonable than it is now, as more Z′ > 1 structures are determined from better crystallographic methods and Z′ > 1 is common for metastable polymorphs.16 Disorder plays a different role in different types of materials, but it is notable that one prediction made at the CCDC’s 50th party17 was that in another 50 years, all organic crystal structures would be disordered. Does using data on known crystal structures (such as the Cambridge Structural Database)18,19 risk bias from sociological influences on research and reporting or historic limitations in analytical capabilities?We need to be careful in defining our search and the expectations of users of the CSP codes. There is a history of claims being made that CSP has shown that all the polymorphs of a molecule are already known, which is what industry would love the codes to be able to do, but the earliest commercial CSP code had problems when claiming this for paracetamol, when many experimental polymorphism experts were aware of the literature evidence of another form. Subsequent CSP work has identified models for form III of paracetamol,20 and there are now claims of more polymorphs formed under pressure21 requiring a structure determination. More recently, CSP led to the experimental finding of the room temperature stable form of creatine22 whereas an earlier CSP had helped determine the first structure and claimed that there would be no polymorphs.23
Currently, the most effective search strategy and the type of lattice energy evaluation that is good enough is very dependent on the type of material being studied. The shortcuts allowed by choices of composition and stoichiometry also differ – different elements can substitute into the same inorganic crystal structure, but even the best functional group swap favoured by crystal engineers, methyl for chloro in organic molecules, leads to a change of crystal structure unless there are no close or strong (e.g. hydrogen bonding) interactions involved at the substitution site.24 The importance of polymorphism appears to differ between materials, though this may reflect the range of crystallisation/synthesis methods that can be applied, or the motivation for experimental screening for polymorphs. However, the ease of computation means that CSP_0 is applied to the widest range of materials. Whilst generally focussing on the global minimum in the lattice energy, we need to look with care at the energy gap to alternative structure(s) to determine whether it is within the likely accuracy of the method, i.e. a reasonable approximation to CSP_thd. For organic crystals, this is sometimes the case, but this monomorphism does require a uniquely good packing defining the structure in all three dimensions, which will rarely be true of all members of a class of materials that adopts low symmetry crystal structures.
CSP_thd; defining the crystal energy landscape
CSP_thd is calculating the thermodynamics accurately, so that you are genuinely predicting the most thermodynamically stable crystal structure at a given set of thermodynamic conditions. If thermodynamics was the only determinant of crystal structures, then the global minimum in CSP_thd at ambient would be the only structure observed at ambient conditions. Since this is often not the case, with strictly metastable polymorphs being sufficiently long-lived to appear to be stable, we interpret the crystal energy landscape, the set of all thermodynamically plausible structures, and expect that this would show all the polymorphs that would be stable under those thermodynamic conditions.How this relates to the assumption that CSP_0 should be adequate, as a starting point, can be seen by considering the thermodynamics of chiral separation by crystallisation, i.e. the ability to predict when you would get a mixture of enantiopure crystals from a racemic solution. Using the sublimation thermodynamic cycle, where you separate your molecules by subliming the crystal and then solvate them to form the solution, we can predict the solubility ratio of the racemic crystal (RS) and the enantiopure crystal (S):
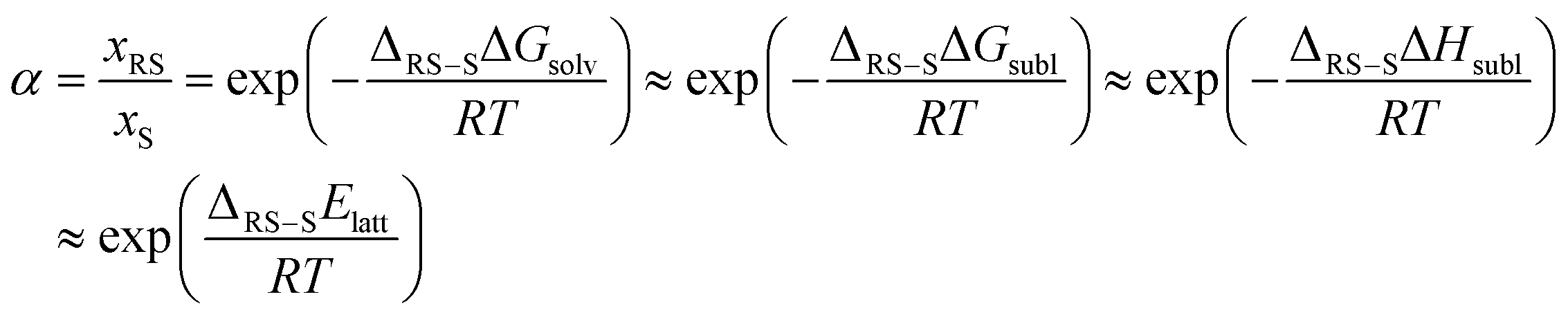
Since the solutions are composed of the same molecule (apart from 50% of the RS solution being the mirror image molecule) the energy of solvation should be the same, assuming the solutions are ideal or non-ideal to the same extent, so the solubility difference is determined by the crystal thermodynamics. Assuming the thermal entropy terms are the same, the free energy of sublimation differences reduce to the enthalpy of sublimation differences, and that, ignoring heat capacity and zero-point energy differences, leads to the lattice energy determining the solubility ratio. Thus CSP_0 covering both chiral and enantiopure space groups provides the most stable structure for each to determine whether chiral separation is possible. If this ratio is turned into the eutectic excess, then the thermodynamic prediction of the crystal that is formed is shown to be very sensitive to the approximated energy difference, as shown in Fig. 2.
 | ||
| Fig. 2 The proportion of enantiopure and racemic crystal structures as a function of their energy difference ΔG = ΔG(RS) − ΔG(S), plotted for three temperatures. The asymmetry comes from the definition of one mole of the racemic crystal corresponding to 1/2 a mole of each enantiomer, as used often in experimental work25 but not in CSP where the reference state is a mole independent of chirality.26 The spread of energies that changes the crystallisation outcome is not dependent on the reference state. | ||
The steepness of the curve in Fig. 2 shows that if the lattice energy difference is large, you can confidently predict which crystal will form by CSP_0, but if it is small, the outcome is horribly dependent on the energy difference and hence the cancellation of errors in all the other energy terms. Our recent comparison of measured heat capacities at low and ambient temperatures for three pairs of enantiopure and racemic crystals of diverse organic molecules, and other measured thermodynamic quantities,25 shows that the approximations listed above are not good enough for any of these molecules. The contributions differ in their sensitivity to the molecular and structural differences, particularly the extent to which the molecular vibrational frequencies are unchanged on crystallisation. Differences in the hydrogen bonding motif and consequent frequency shifts in the IR spectra between the two crystals, can lead to a temperature dependence of the heat capacity difference around ambient. The performance of our attempts to predict thermal corrections using the harmonic approximation,25 using rigid molecule Ψmol or Ψcrys methods similar to those used for larger systems (DOI: 10.1039/c8fd00010g), shows the challenge of predicting free energies or relative solubility at a useful accuracy.
These arguments also apply to polymorphs and can be extended. What are the free energy difference implications of thermal expansion properties (DOI: 10.1039/c8fd00048d), which can differ so much in their anisotropy between polymorphs or systems? Correcting experimental quantities back to give an “experimental” lattice energy, as done in benchmarking set X23, will be limited by the accuracy of the calculated27 or experimental28 thermal corrections (which are nevertheless a big recent improvement on the 2RT correction that initially justified the use of CSP_0 for chiral resolution). The cancellation of the small thermodynamic terms between different structures of the same compound will be very dependent on the structural differences.
Perhaps a more significant implication of Fig. 2 is a limit on the energy differences between polymorphs obtained by solution crystallisation, which is comparable to the estimated range of polymorphic lattice energy differences of observed polymorphs,29 and the cutoffs often used for progressing CSP_0 to more demanding calculations. This can mean that polymorphs obtained by other methods, for example by desolvating solvates11 or the solid state synthesis of the molecule, could be much more metastable than polymorphs obtained by solution crystallisation. The concept of a crystal energy landscape requires a cut-off of thermodynamic plausibility. If this is dependent on the methods of crystallisation that can be applied, then it could be a context-determined parameter.
There are other thermodynamic contributions that should be included, given that real crystals are not perfect and infinite, such as disorder. Configurational disorder is an energy term which has been estimated to account for why the low temperature phase of caffeine is statically disordered.30 Going from CSP generating potential disorder components to realistic modelling of the thermodynamics of disorder, let alone estimating disorder that is not thermodynamic but frozen in during crystallisation, is important for pharmaceuticals (DOI: 10.1039/c8fd00072g), and yet order–disorder phase transitions are challenging to experimentally characterise or model (DOI: 10.1039/c8fd00042e). Some forms of disorder, such as polytypism, imply that coverage of the search space will never be complete.
The size of a crystallite and the presence of solvent have recently been shown to be important in thermodynamic stability by the demonstration of a reversible cross over in relative stability of the polymorphs of a cocrystal and an aromatic disulphide compound in liquid-assisted grinding experiments.31 These were rationalised by considering solvent-dependent surface energies relative to the bulk lattice energy. The effect that size can have on the relative stability of nuclei of different forms comes out clearly from consideration of the nano-cluster modelling of inorganic oxides (DOI: 10.1039/c8fd00060c).
The development of accurate calculations of the relative thermodynamic stability for CSP_thd will be a great step forward for the theoretical modelling of important properties, such as the solubility, morphology and stability range for pharmaceuticals. Doing this accurately for all properties related to thermodynamics for all known phases, let alone the entire crystal energy landscape, provides a significant aim for theory and computer modelling. However, a major benefit of the move from CSP_0 to CSP_thd is to eliminate structures that are artefacts of the use of a static lattice in CSP_0, i.e. are not minima in the free energy crystal landscape. The number of lattice energy minima that are eliminated by some form of molecular dynamics, from just a short shake-up32 to a metadynamics treatment,33–35 is very dependent on the molecule and structures concerned. A realistic CSP_thd should produce the more symmetrical structures that arise from dynamic disorder averaging over a variety of lattice energy minima (such polymorphs do not correspond to a lattice energy minimum and so are not produced by CSP_0). Experimental identification of such plastic crystals may be aided by simulations, allowing insight into the energetic and mechanistic forces that drive order–disorder phase transitions in plastic crystals (DOI: 10.1039/c8fd00042e). There may be experimentally observed intermediate phases that we may feel are beyond the scope of CSP, for example, CSP gave the low temperature ordered phase III of cyclopentane, and MD the hexagonal plastic phase I, but the size of the MD cell probably contributed to the inability of the dynamical simulations to get good agreement with the experimental powder pattern of the intermediate plastic phase II.36 The development of sophisticated MD based methods to explore the free energy surface and study phase transitions, such as meta-shooting (DOI: 10.1039/c8fd00053k) will improve our understanding of the effects of the potential energy surface of CSP_thd.
CSP_aim
What do we consider that a genuine crystal structure prediction code (CSP_aim) should do?
Most people would want a CSP code to predict all practically important crystal structures that could be found for a defined system, and those involved in material synthesis or solid form screening would also want a recipe for how to find them. Full CSP_thd would be adequate for systems where you expect to form the most stable structure at a given set of thermodynamic conditions, so experimentally you should just move to the relevant thermodynamic conditions.CSP is undoubtedly leading to an increase in the number of polymorphs whose structures are now known. The use of CSP has helped to solve the structures of extremely fibrous polymorphs of coumarin grown from the melt,80 new needle forms of resorcinol37 (another example of a new form being found despite published predictions that there were no more forms), and another form of aspirin38 from the melt. The system ROY, with its various colours, is one where it has long been known that there are further uncharacterised forms39 that exist at ambient, but CSP produces too many candidates.40 The use of CSP to increase the number of polymorphs that have their structures determined, and the challenges involved will come up in discussion of the 8th solved structure (DOI: 10.1039/c8fd00039e). Can CSP also, by proposing new experiments or encouraging more detailed analysis, increase the number of polymorphs that are detected (DOI: 10.1039/c8fd00069g), as well as aiding their characterisation?
Disappearing polymorphs, where there is an inability to maintain control of the crystallisation of an apparently stable form after a more thermodynamically stable form appears, have been a major justification for developing CSP because of their practical importance to the pharmaceutical industry.41 CSP_0 must generate any structure that is significantly more stable than the known forms, as well as the known polymorphs, with CSP_thd helping confirm the relative thermodynamic stability at ambient. However, the scientific rationalisation of disappearing polymorphs would conclude that you ought to be able to reproduce any polymorph that has ever been observed, using identical crystallisation conditions, even if this requires a fresh laboratory and student41 to avoid seeding. Having identical crystallisation conditions may be practically unachievable, as a 50 year old sample of the disappearing form II of progesterone contained a cocktail of impurities not found in the modern material.42 How does the issue of elusive or disappearing polymorphs, and the effects of impurities on the polymorph formed, impact the computational prediction of metastable polymorphs?
This is illustrated by the new polymorph (γ) of succinic acid that was found in an attempt to purify a synthesiser-made peptide from the resultant soup of impurities, by cocrystallisation with succinic acid (Fig. 3).43 We were challenged to predict the structure. It was readily generated as a metastable polymorph by CSP. Periodic DFT-D lattice energy calculations had γ slightly more stable than the other known polymorphs for most dispersion corrections, though using the PBE + TS model, harmonic phonon estimates of the free energy differences made the β form more stable at room temperature. This closeness in energy is consistent with the γ form being found concomitantly with β in the failed cocrystallisation experiment, and illustrates the improvement of CSP_thd on CSP_0. This case is relevant to defining CSP_aim because the γ form has not been crystallised again, despite a large series of experiments testing the different impurities.43
 | ||
| Fig. 3 The three polymorphs of succinic acid, showing the prediction of the novel γ form by CSP_0 using a Ψmol approach, and the relative lattice energies by a variety of Ψcrys methods. Despite its relative thermodynamic stability, the γ polymorph has only been crystallised once in a failed cocrystallisation experiment.43 | ||
The serendipitously-found γ crystal is a conformational polymorph of succinic acid, with the conformation that NMR and MD simulations in water show as being dominant in solution.43 MD on the γ form does not give a solid state transformation, but reproduces the structure with a small change in the monoclinic angle, which, if forced by metadynamics, leads to extensive defect formation. That is the best explanation we can find to the problem of reproducing the new polymorph: the γ form can be rather susceptible to defect formation that may lead to a ready transformation to the β form. Perhaps γ succinic acid will be found again, and it is in the CSD as a challenge to anyone who can design a method of finding it. The high-temperature α phase of succinic acid was once considered elusive at normal temperatures, but has been found as a contaminant after grinding44,45 and in liquid- (but not air-) segmented flow crystallisation46 or by spray-drying from water.47 This suggests that the α polymorph is observed when it forms first and there has not been sufficient time for the solvent mediated transformation to β. Gradually we are beginning to see the effects of kinetics and the role of impurities in catalysing or inhibiting polymorphic transformations. The serendipitous observation of γ succinic acid raises the question of what is wanted from CSP_aim: a recipe for producing the γ form and predicting its stability as a function of crystal size? If it had not been observed, would its apparent feasibility in the computational modelling have appeared to be an over-prediction of polymorphs? An early CSP study had noted that the planar conformation of succinic acid was less stable than that in the γ form, but had used the dominance of the planar conformation in the many crystal structures of succinic acid, and a very limited search in the alternative conformation, to dismiss the possibility of any conformational polymorphs.48
Thus, if we are to achieve CSP_aim, we need to work with a knowledge of the variables in crystallisation/synthesis conditions that can lead to the formation of different structures. This will be very much a function of the types of materials and the range of interatomic interactions involved, i.e. going up the vertical axis of Fig. 1 and will essentially encapsulate the whole field of crystal engineering into our CSP theories and eventually codes. The possible synthetic methods in every field are evolving rapidly, and CSP plays the crucial role of predicting structures that should be achievable, and there may be structure dependent methods that can help find them. For example, CSP has inspired an isomorphous heterogeneous solution seeding experiment to produce a CSP predicted cocrystal which could not be made in four expert labs without the seeds initiating the first cocrystallisation.49 Metastable pharmaceutical polymorphs have been successfully targeted by sublimation onto an isomorphous template crystal.50 Will we ever have the control to be able to make a crystal by placing each atom in the position corresponding to the CSP-generated desired crystal structure? This is more possible for inorganic materials than pharmaceuticals. The pharmaceutical industrial scientists recognise that there is no such thing as a standard solid form screen; not only can the basic workflow vary between companies,11,29 but also between molecules from the same drug discovery program, e.g. if one is able to obtain an amorphous starting material for one, and not for the other51 then there may be a difference in the extent to which the starting point of the crystallisation has lost the memory of the input material structure.
This sensitivity of crystal structure outcome to exact material synthesis conditions probably extends to all types of materials, which is why CSP is most effective when there is very close collaboration between the people doing the calculations and those in the laboratory. In the aim for a code that can predict all the crystal structures that can be found, but not those that could never be made, we will have to consider relative nucleation and growth rates. Nucleation has been the focus of another recent Faraday Discussion.52 There is an increasing realisation of the variety of mechanisms for crystallisation by particle attachment, and how this can vary in synthetic, biogenic and geological environments.53 The aspect of whether crystallisation occurs by monomer attachment, let alone according to the models of classical nucleation theory, is up for debate and observation. Atomic force microscopy (AFM) showed dense hillocks forming at ledges of the dominant surface of olanzapine in water.54 This two-step nucleation process, with a dense disordered solution forming, can be rationalised as the suspected growth unit (a dimer) has a variety of attachment sites on the ledge that are more stable than docking in the crystallographic site on the surface. The growth unit will not often dock directly into the most stable crystallographic ledge site when it can get caught in so many ledge, or solute–solute, and solute–water complexes. The AFM also showed that the dense blobs on the surface could grow into orientated crystals of the thermodynamically stable olanzapine dihydrate D, whereas dissolution of olanzapine in water usually resulted in the metastable dihydrate B. We can computationally rationalise the better registry (in the correct orientation) of a small nanocluster of the stable dihydrate D on the specific olanzapine anhydrate surface than the metastable dihydrate B, representing the support the surface gives to nucleating one form over another.54
As we consider interfaces and growth rates, the problem of distinguishing thermodynamics from kinetics gets worse. Morphology predictions based on minimising the surface energy give unrealistically spherical structures (consistent with classical nucleation theory), which led to the attachment energy model, which is strictly only appropriate for vapour grown crystals if all surfaces are below their roughening temperature. The attachment energy model is the cheap model for morphology–structure–energy landscapes, with relative growth volume rates being a possible means of eliminating unlikely polymorphs.55 Predicting the morphology in different solvents uses models based on kinetic rates of attachment, dissolution, loss of solvent shell etc. derived from MD.56 If we need to be reliant on MD to predict nucleation, growth and transformation rates, we are starting to enter the realm of multi-scale approximations to solving the appropriate nuclear and electronic time-dependent Schrödinger equation for the real system. We have lost the simplification of distinguishing between thermodynamics and kinetics.
The question of the extent to which CSP_aim will need to consider kinetic effects arising from differences in the experimental growth conditions is very problem dependent. The organic electronic community are obviously interested in substrate-induced polymorphs.57 Are these cases where full thermodynamic treatment including the interfacial effects would predict these structures? Concomitant polymorphs raise further questions37,58,59 as to whether we could ever devise a computational method that would allow the confident prediction of the conditions needed to crystallise phase pure samples. Extensive crystallisation work could not obtain phase pure samples of the metastable forms II and III of olanzapine. After the structure of form II was solved from a serendipitous single crystal and CSP suggested a structure for form III, the degree of similarity, (differing only in the stacking of layers), made it rather improbable that the crystallisation could ever be controlled to crystallise the forms separately.60
The question as to how changing crystallisation or synthesis conditions can affect which structures are formed, and how readily they transform to the most thermodynamically stable structure at the specified temperature and pressure, brings us back to the original assumption behind CSP. Does the thermodynamically most stable form have to be obtainable? It is the central tenet of CSP methods, and yet when so many molecules are difficult to crystallise at all, the first structure formed is quite unlikely to be the most stable by Ostwald’s rule.61 (Indeed, if CSP could guarantee that a pharmaceutical would never crystallise, so that the amorphous form could be confidently developed, that would be a valuable application.) It is quite conceivable that the most stable form could be so kinetically hindered that it would not form. The monohydrate of 4-aminoquinaldine62 is an important example, as CSP predicted that a more thermodynamically stable structure should exist. It was later found by careful experimentation using either hydrothermal conditions or an impurity, but the metastable polymorph is kinetically favoured in both nucleation and growth. There are examples where analysis of the CSP_0 most stable structure, in contrast with the observed forms, can show that its formation seems rather unlikely on either crystal engineering or statistical likelihood.63 It is noteworthy that despite extensive polymorph screening, a significant number of drugs are being marketed on the false assumption (as shown by CSP_0) that they are in the most stable thermodynamic form (DOI: 10.1039/c8fd00069g). Predicting polymorphs that could never be found also has major industrial implications (DOI: 10.1039/c8fd00033f). If CSP_0 is now an applied technology for the pharmaceutical industry (DOI: 10.1039/c8fd00033f), we must have a clear concept of the limitations of the technology, which involves determining how it differs from CSP_aim for this class of material.
Practicality: CSP_aim and energy–structure–function maps
The field of CSP as an aid to the discovery of new materials is becoming a reality across many types of materials. For example, in the field of the 18-valence-electron ABX family, consideration of the unreported family members led to the first synthesis of 15 compounds, including HfIrAs, a topological semi-metal of interest in quantum electronics, ZrNiPb, a small-gap semiconductor with a large Seebeck coefficient suitable for thermoelectric applications, and ZrIrSb, a rare example of a transparent p-type conductor with a high conductivity of holes.64 This approach has also led to the design and discovery of the transparent conductor TaIrGe.65 The use of computation for the accelerated discovery of new crystal structures (rather than just changing elements within known structures) in the complex inorganic Y–Sr–Ca–Ga–O phase field has led to new structures with ordered variants Sr2Ca3Ga6O14 and SrCa2Ga2O6.66 CSP on varying stoichiometries has also impacted energy materials research, with Li7Ge3 being predicted67 and then found in research on germanium anodes in lithium batteries.68The progress in the CSP-led design and realisation of the lowest density porous organic cage crystal, with its gas storage and selectivity properties69 is a particular landmark, as a case when an organic molecule was synthesised following CSP prediction. However, it is noteworthy that the synthesis was targeting a highly metastable structure on the energy–structure–function map. The most thermodynamically stable form (according to the CSP_0) was not reported. This aspect of solvents stabilising porous structures (DOI: 10.1039/c8fd00031j) further illustrates how crystallisation conditions affect the observed structure. Zeolites, another important porous material (DOI: 10.1039/c8fd00035b, DOI: 10.1039/c8fd00040a), can also be more specifically templated,70 though the organic molecules that direct the structure are burnt off afterwards.
Organic functional materials were already expanding in scope at the start of CSP, when the emphasis was on the most stable possible structure, with density being the key property for energetic materials, or non-centrosymmetric packing for non-linear optically active materials. Nowadays, energy–structure–function maps can be made for advanced properties, both in terms of technological applications and the challenge of calculating the property such as charge carrier mobility in organic semiconductors.71 The differences between an organic crystal or film and the traditional silicon semi-conductors illustrates how spanning different types of materials can combine other properties. If a material is being produced industrially, there are a myriad of properties that are relevant to its crystallisation, storage, use, and the quality control procedures that need to be in place. Our CSP studies are just the start of the multi-scale modelling that is involved in the digital design agenda, requiring information on the behaviour of the material on a range of length and time scales, as the pharmaceutical materials science tetrahedron72 relates the structure, properties, performance and processing of a drug. Hence the role of CSP in polymorph screening, including the design of experiments to find new forms, but the number of published examples contrasting the number of polymorphs found by an industrial screening process with those found by CSP is limited.73 This iterative work where CSP may inspire experiments that find more forms, versus the problem of over-prediction, really illustrates the need to define what is required from CSP. Pressure is becoming increasingly important in the discovery of CSP “predicted” forms for pharmaceuticals,14,74,75 whereas CSP at pressure has long been established as a route to finding new phases of interest to planetary scientists, including changes in bonding, such as ionic ammonia.76,77
Hence, CSP is playing a huge role in our ability to make new crystalline solids for many purposes. The combination of CSP and calculating (non-thermodynamic) properties, whether for targeting new functional materials or for structural characterisation, such as various diffraction78 or solid state NMR experiments,79 is really contributing to science. The question though is how much this relies on human scientific understanding interpreting the results of the CSP_0 (or CSP_thd) for experimental collaborators, or whether we can have a black-box computer code.
Conclusion
CSP_0 searches for the lowest lattice energy structures are becoming an established technique for a wide range of materials research, with an increasing diversity of types of interatomic forces within the materials. Hence the area of coverage of different materials in this discussion of the CSP_0 column in Fig. 1 is very wide. However, the most stable structure in CSP_0 is not always observed, and certainly is often not the only structure that can be found. This sometimes reflects the limitations of the thermodynamic modelling accuracy (i.e. really needing CSP_thd) but also the basic assumption that thermodynamics determines crystallisation. If the energy gaps are sufficiently large, then CSP_0 is good for determining whether a material is sufficiently likely to crystallise with a desired property that it is worth some experimental work.However, when CSP_0 produces many structures that are close in energy, then eliminating those that are artefacts of approximating the free energy is more challenging. This is not only a challenge in obtaining the free energy of the perfect infinite crystal at the practically relevant temperatures and pressures. There is also correctly modelling the thermodynamics related to size, solvent, and the presence of specific surfaces or other molecules in solution. How realistic are thoughts of a CSP_thd code, spanning a wide diversity of interatomic interactions?
A CSP_aim code would reliably output the crystal structures that could be formed, and no more. It seems likely that calculating the true relative thermodynamic stability will sometimes incorporate information relevant to the experiments to find them, but much more understanding is needed of the mechanisms for nucleation catalysts and growth inhibitors.
CSP is maturing in the sense that an increasing number of experimental groups, including in the pharmaceutical industry, are taking it seriously enough to invest in CSP. It has been a long haul to get to this state. Over-prediction could lose this impetus. We need to be realistic about how much the codes can deliver, and the extent to which it is the experience of the scientist in interpreting the results in conjunction with the experimentalists that makes CSP practically useful. Hopefully this discussion will establish the current state of the art.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
There are too many people that have contributed to forming the views in this lecture, through discussions or through collaborative work, to be acknowledged but I am very grateful to them. Some are authors of the papers listed on http://www.cposs.org.uk where we have been developing CSP in collaboration with experimental or other computational groups, and this list extends far beyond those funded through GR/S24114, EPSRC EP/F03573X/1, EP/K039229/1 E.U. Horizon 2020 736899 and Eli Lilly.Notes and references
- J. Maddox, Crystals from First Principles, Nature, 1988, 335(6187), 201 CrossRef
.
-
W. C. McCrone, Polymorphism, in Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M. Labes and A. Weissberger, Wiley Interscience, New York, 1965, Vol. II, pp. 725–767 Search PubMed
- C. Cervinka and G. J. O. Beran, Ab initio prediction of the polymorph phase diagram for crystalline methanol, Chem. Sci., 2018, 4622–4629 RSC
- C. J. Pickard and R. J. Needs, Structures and stability of calcium and magnesium carbonates at mantle pressures, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91(10), 104101 CrossRef
- D. Smith, K. V. Lawler, M. Martinez-Canales, A. W. Daykin, Z. Fussell, G. A. Smith, C. Childs, J. S. Smith, C. J. Pickard and A. Salamat, Postaragonite phases of CaCO3 at lower mantle pressures, Phys. Rev. Mater., 2018, 2(1), 013605 CrossRef
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell, R. Car, D. H. Case, R. Chadha, J. C. Cole, K. Cosburn, H. M. Cuppen, F. Curtis, G. M. Day, R. A. DiStasio Jr, A. Dzyabchenko, B. P. van Eijck, D. M. Elking, J. A. van den Ende, J. C. Facelli, M. B. Ferraro, L. Fusti-Molnar, C.-A. Gatsiou, T. S. Gee, R. de Gelder, L. M. Ghiringhelli, H. Goto, S. Grimme, R. Guo, D. W. M. Hofmann, J. Hoja, R. K. Hylton, L. Iuzzolino, W. Jankiewicz, D. T. de Jong, J. Kendrick, N. J. J. de Klerk, H.-Y. Ko, L. N. Kuleshova, X. Li, S. Lohani, F. J. J. Leusen, A. M. Lund, J. Lv, Y. Ma, N. Marom, A. E. Masunov, P. McCabe, D. P. McMahon, H. Meekes, M. P. Metz, A. J. Misquitta, S. Mohamed, B. Monserrat, R. J. Needs, M. A. Neumann, J. Nyman, S. Obata, H. Oberhofer, A. R. Oganov, A. M. Orendt, G. I. Pagola, C. C. Pantelides, C. J. Pickard, R. Podeszwa, L. S. Price, S. L. Price, A. Pulido, M. G. Read, K. Reuter, E. Schneider, C. Schober, G. P. Shields, P. Singh, I. J. Sugden, K. Szalewicz, C. R. Taylor, A. Tkatchenko, M. E. Tuckerman, F. Vacarro, M. Vasileiadis, A. Vazquez-Mayagoitia, L. Vogt, Y. Wang, R. E. Watson, G. A. de Wijs, J. Yang, Q. Zhu and C. R. Groom, Report on the sixth blind test of organic crystal structure prediction methods, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(4), 439–459 CrossRef PubMed
- P. Vishweshwar, J. A. McMahon, M. Oliveira, M. L. Peterson and M. J. Zaworotko, The Predictable Elusive Form II of Aspirin, J. Am. Chem. Soc., 2005, 127(48), 16802–16803 CrossRef PubMed
- J. Klimes, D. R. Bowler and A. Michaelides, van der Waals density functionals applied to solids, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(19), 195131 CrossRef
- L. Goerigk and S. Grimme, A thorough benchmark of density functional methods for general main
group thermochemistry, kinetics, and noncovalent interactions, Phys. Chem. Chem. Phys., 2011, 13(14), 6670–6688 RSC
- Y. Zhao and D. G. Truhlar, Benchmark databases for nonbonded interactions and their use to test density functional theory, J. Chem. Theory Comput., 2005, 1(3), 415–432 CrossRef PubMed
- D. E. Braun, S. R. Lingireddy, M. D. Beidelschies, R. Guo, P. Muller, S. L. Price and S. M. Reutzel-Edens, Unraveling Complexity in the Solid Form Screening of a Pharmaceutical Salt: Why so Many Forms? Why so Few?, Cryst. Growth Des., 2017, 17(10), 5349–5365 CrossRef PubMed
- L. M. LeBlanc, A. Otero-de-la-Roza and E. R. Johnson, Composite and Low-Cost Approaches for Molecular Crystal Structure Prediction, J. Chem. Theory Comput., 2018, 14(4), 2265–2276 CrossRef PubMed
- J. Nyman, O. S. Pundyke and G. M. Day, Accurate force fields and methods for modelling organic molecular crystals at finite temperatures, Phys. Chem. Chem. Phys., 2016, 18(23), 15828–15837 RSC
- A. A. Aina, A. J. Misquitta and S. L. Price, From dimers to the solid-state: Distributed intermolecular force-fields for pyridine, J. Chem. Phys., 2017, 147(16), 161722 CrossRef PubMed
- F. Curtis, X. Y. Li, T. Rose, A. Vazquez-Mayagoitia, S. Bhattacharya, L. M. Ghiringhelli and N. Marom, GAtor: A First-Principles Genetic Algorithm for Molecular Crystal Structure Prediction, J. Chem. Theory Comput., 2018, 14(4), 2246–2264 CrossRef PubMed
- K. M. Steed and J. W. Steed, Packing Problems: High Z′ Crystal Structures and Their Relationship to Cocrystals, Inclusion Compounds, and Polymorphism, Chem. Rev., 2015, 115(8), 2895–2933 CrossRef PubMed
- C. R. Groom and F. H. Allen, The Cambridge Structural Database in Retrospect and Prospect, Angew. Chem., Int. Ed., 2014, 53(3), 662–671 CrossRef PubMed
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef PubMed
- J. C. Cole, C. R. Groom, M. G. Read, I. Giangreco, P. McCabe, A. M. Reilly and G. P. Shields, Generation of crystal structures using known crystal structures as analogues, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 530–541 CrossRef PubMed
- M. A. Perrin, M. A. Neumann, H. Elmaleh and L. Zaske, Crystal structure determination of the elusive paracetamol form III, Chem. Commun., 2009, 22, 3181–3183 RSC
- S. J. Smith, M. M. Bishop, J. M. Montgomery, T. P. Hamilton and Y. K. Vohra, Polymorphism in Paracetamol: Evidence of Additional Forms IV and V at High Pressure, J. Phys. Chem. A, 2014, 118(31), 6068–6077 CrossRef PubMed
- D. E. Braun, M. Orlova and U. J. Griesser, Creatine: Polymorphs Predicted and Found, Cryst. Growth Des., 2014, 14(10), 4895–4900 CrossRef PubMed
- M. D. King, T. N. Blanton, S. T. Misture and T. M. Korter, Prediction of the Unknown Crystal Structure of Creatine Using Fully Quantum Mechanical Methods, Cryst. Growth Des., 2011, 11(12), 5733–5740 CrossRef
- M. K. Corpinot, R. Guo, D. A. Tocher, A. B. M. Buanz, S. Gaisford, S. L. Price and D. K. Bucar, Are Oxygen and Sulfur Atoms Structurally Equivalent in Organic Crystals?, Cryst. Growth Des., 2017, 17(2), 827–833 CrossRef
- H. K. Buchholz, R. K. Hylton, J. G. Brandenburg, A. Seidel-Morgenstern, H. Lorenz, M. Stein and S. L. Price, Thermochemistry of Racemic and Enantiopure Organic Crystals for Predicting Enantiomer Separation, Cryst. Growth Des., 2017, 17(9), 4676–4686 CrossRef
- A. Otero-de-la-Roza, B. H. Cao, I. K. Price, J. E. Hein and E. R. Johnson, Predicting the Relative
Solubilities of Racemic and Enantiopure Crystals by Density-Functional Theory, Angew. Chem., Int. Ed., 2014, 53(30), 7879–7882 CrossRef PubMed
- A. M. Reilly and A. Tkatchenko, Understanding the role of vibrations, exact exchange, and many-body van der Waals interactions in the cohesive properties of molecular crystals, J. Chem. Phys., 2013, 139(2), 024705 CrossRef PubMed
- J. Yang, W. Hu, D. Usvyat, D. Matthews, M. Schutz and H. Chan, Ab initio determination of the lattice energy in crystalline benzene to sub-kilojoule per mole accuracy, Science, 2014, 345(6197), 640–643 CrossRef PubMed
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Facts and fictions about polymorphism, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC
- M. Habgood, Form II Caffeine: A Case Study for Confirming and Predicting Disorder in Organic Crystals, Cryst. Growth Des., 2011, 11(8), 3600–3608 CrossRef
- A. M. Belenguer, G. I. Lampronti, A. J. Cruz-Cabeza, C. A. Hunter and J. K. M. Sanders, Solvation and surface effects on polymorph stabilities at the nanoscale, Chem. Sci., 2016, 7(11), 6617–6627 RSC
- W. T. M. Mooij, B. P. van Eijck, S. L. Price, P. Verwer and J. Kroon, Crystal structure predictions for acetic acid, J. Comput. Chem., 1998, 19(4), 459–474 CrossRef
- P. Raiteri, R. Martonak and M. Parrinello, Exploring Polymorphism: The Case of Benzene, Angew. Chem., Int. Ed., 2005, 44, 3769–3773 CrossRef PubMed
- T. Zykova-Timan, P. Raiteri and M. Parrinello, Investigating the Polymorphism in PR179: A Combined Crystal Structure Prediction and Metadynamics Study, J. Phys. Chem. B, 2008, 112(42), 13231–13237 CrossRef PubMed
- P. G. Karamertzanis, P. Raiteri, M. Parrinello, M. Leslie and S. L. Price, The Thermal Stability of Lattice Energy Minima of 5-Fluorouracil: Metadynamics as an Aid to Polymorph Prediction, J. Phys. Chem. B, 2008, 112(14), 4298–4308 CrossRef PubMed
- A. Torrisi, C. K. Leech, K. Shankland, W. I. F. David, R. M. Ibberson, J. Benet-Buchholz, R. Boese, M. Leslie, C. R. A. Catlow and S. L. Price, The solid phases of cyclopentane: a combined experimental and simulation study, J. Phys. Chem. B, 2008, 112(12), 3746–3758 CrossRef PubMed
- Q. Zhu, A. G. Shtukenberg, D. J. Carter, T. Q. Yu, J. X. Yang, M. Chen, P. Raiteri, A. R. Oganov, B. Pokroy, I. Polishchuk, P. J. Bygrave, G. M. Day, A. L. Rohl, M. E. Tuckerman and B. Kahr, Resorcinol Crystallization from the Melt: A New Ambient Phase and New “Riddles”, J. Am. Chem. Soc., 2016, 138(14), 4881–4889 CrossRef PubMed
- A. G. Shtukenberg, C. H. T. Hu, Q. Zhu, M. U. Schmidt, W. Q. Xu, M. Tan and B. Kahr, The Third Ambient Aspirin Polymorph, Cryst. Growth Des., 2017, 17(6), 3562–3566 CrossRef
- L. Yu, Polymorphism in Molecular Solids: An Extraordinary System of Red, Orange, and Yellow Crystals, Acc. Chem. Res., 2010, 43(9), 1257–1266 CrossRef PubMed
- M. Vasileiadis, A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman and C. C. Pantelides, The polymorphs of ROY: application of a systematic crystal structure prediction technique, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 677–685 CrossRef PubMed
- D. K. Bucar, R. W. Lancaster and J. Bernstein, Disappearing Polymorphs Revisited, Angew. Chem., Int. Ed., 2015, 54(24), 6972–6993 CrossRef PubMed
- R. W. Lancaster, L. D. Harris and D. Pearson, Fifty-year old samples of progesterone demonstrate the complex role of synthetic impurities in stabilizing a metastable polymorph, CrystEngComm, 2011, 13(6), 1775–1777 RSC
- P. Lucaioli, E. Nauha, I. Gimondi, L. S. Price, R. Guo, L. Iuzzolino, I. Singh, M. Salvalaglio, S. L. Price and N. Blagden, Serendipitous isolation of a disappearing conformational polymorph of succinic acid challenges computational polymorph prediction, CrystEngComm, 2018, 20, 3971–3977 RSC
- V. Chikhalia, R. T. Forbes, R. A. Storey and M. Ticehurst, The effect of crystal morphology and mill type on milling induced crystal disorder, Eur. J. Pharm. Sci., 2006, 27(1), 19–26 CrossRef PubMed
- A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. H. Feng, R. B. H. Tan and K. J. Carpenter, Selective polymorph transformation via solvent-drop grinding, Chem. Commun., 2005, 7, 880–882 RSC
- K. Robertson, P. B. Flandrin, A. R. Klapwijk and C. C. Wilson, Design and Evaluation of a Mesoscale Segmented Flow Reactor (KRAIC), Cryst. Growth Des., 2016, 16(8), 4759–4764 CrossRef
- K. M. Carver and R. C. Snyder, Unexpected Polymorphism and Unique Particle Morphologies from Monodisperse Droplet Evaporation, Ind. Eng. Chem. Res., 2012, 51(48), 15720–15728 CrossRef
- P. G. Karamertzanis, A. V. Kazantsev, N. Issa, G. W. A. Welch, C. S. Adjiman, C. C. Pantelides and S. L. Price, Can the Formation of Pharmaceutical Cocrystals Be Computationally Predicted? 2. Crystal Structure Prediction, J. Chem. Theory Comput., 2009, 5(5), 1432–1448 CrossRef PubMed
- D. K. Bucar, G. M. Day, I. Halasz, G. G. Z. Zhang, J. R. G. Sander, D. G. Reid, L. R. MacGillivray, M. J. Duer and W. Jones, The curious case of (caffeine) (benzoic acid): how heteronuclear seeding allowed the formation of an elusive cocrystal, Chem. Sci., 2013, 4(12), 4417–4425 RSC
- V. K. Srirambhatla, R. Guo, S. L. Price and A. J. Florence, Isomorphous template induced crystallisation: a robust method for the targeted crystallisation of computationally predicted metastable polymorphs, Chem. Commun., 2016, 52(46), 7384–7386 RSC
- D. E. Braun, J. A. McMahon, L. H. Koztecki, S. L. Price and S. M. Reutzel-Edens, Contrasting Polymorphism of Related Small Molecule Drugs Correlated and Guided by the Computed Crystal Energy Landscape, Cryst. Growth Des., 2014, 14(4), 2056–2072 CrossRef
- In Nucleation – A transition state to the directed assembly of materials: Faraday Discussion, 2015 Search PubMed.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Z. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Colfen and P. M. Dove, Crystallization by particle attachment in synthetic, biogenic, and geologic environments, Science, 2015, 349(6247), aaa6760 CrossRef PubMed
- M. Warzecha, R. Guo, R. M. Bhardwaj, S. M. Reutzel-Edens, S. L. Price, D. A. Lamprou and A. J. Florence, Direct Observation of Templated Two-Step Nucleation Mechanism during Olanzapine Hydrate Formation, Cryst. Growth Des., 2017, 17(12), 6382–6393 CrossRef
- T. Beyer, G. M. Day and S. L. Price, The prediction, morphology, and mechanical properties of the polymorphs of paracetamol, J. Am. Chem. Soc., 2001, 123(21), 5086–5094 CrossRef PubMed
- Y. Y. Sun, C. J. Tilbury, S. M. Reutzel-Edens, R. M. Bhardwaj, J. J. Li and M. F. Doherty, Modeling Olanzapine Solution Growth Morphologies, Cryst. Growth Des., 2018, 18(2), 905–911 CrossRef
- A. O. F. Jones, B. Chattopadhyay, Y. H. Geerts and R. Resel, Substrate-Induced and Thin-Film Phases: Polymorphism of Organic Materials on Surfaces, Adv. Funct. Mater., 2016, 26(14), 2233–2255 CrossRef
- J. Bernstein, R. J. Davey and J. O. Henck, Concomitant Polymorphs, Angew. Chem., Int. Ed., 1999, 38(23), 3440–3461 CrossRef PubMed
- R. I. Goldstein, R. Guo, C. Hughes, D. P. Maurer, T. R. Newhouse, T. J. Sisto, R. R. Conry, S. L. Price and D. M. Thamattoor, Concomitant conformational dimorphism in 1,2-bis(9-anthryl)acetylene, CrystEngComm, 2015, 17(26), 4877–4882 RSC
- R. M. Bhardwaj, L. S. Price, S. L. Price, S. M. Reutzel-Edens, G. J. Miller, I. D. H. Oswald, B. Johnston and A. J. Florence, Exploring the Experimental and Computed Crystal Energy Landscape of Olanzapine, Cryst. Growth Des., 2013, 13(4), 1602–1617 CrossRef
- T. Threlfall, Structural and thermodynamic explanations of Ostwald’s rule, Org. Process Res. Dev., 2003, 7(6), 1017–1027 CrossRef
- D. E. Braun, H. Oberacher, K. Arnhard, M. Orlova and U. J. Griesser, 4-Aminoquinaldine monohydrate polymorphism: prediction and impurity aided discovery of a difficult to access stable form, CrystEngComm, 2016, 18(22), 4053–4067 RSC
- R. K. Hylton, G. J. Tizzard, T. L. Threlfall, A. L. Ellis, S. J. Coles, C. C. Seaton, E. Schulze, H. Lorenz, A. Seidel-Morgenstern, M. Stein and S. L. Price, Are the Crystal Structures of Enantiopure and Racemic Mandelic Acids Determined by Kinetics or Thermodynamics?, J. Am. Chem. Soc., 2015, 137(34), 11095–11104 CrossRef PubMed
- R. Gautier, X. Zhang, L. Hu, L. Yu, Y. Lin, T. O. L. Sunde, D. Chon, K. R. Poeppelmeier and A. Zunger, Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds, Nat. Chem., 2015, 7, 308 CrossRef PubMed
- F. Yan, X. Zhang, Y. G. Yu, L. Yu, A. Nagaraja, T. O. Mason and A. Zunger, Design and discovery of a novel half-Heusler transparent hole conductor made of all-metallic heavy elements, Nat. Commun., 2015, 6, 7308 CrossRef PubMed
- C. Collins, M. S. Dyer, M. J. Pitcher, G. F. S. Whitehead, M. Zanella, P. Mandal, J. B. Claridge, G. R. Darling and M. J. Rosseinsky, Accelerated discovery of two crystal structure types in a complex inorganic phase field, Nature, 2017, 546, 280 CrossRef PubMed
- A. J. Morris, C. P. Grey and C. J. Pickard, Thermodynamically stable lithium silicides and germanides from density functional theory calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90(5), 054111 CrossRef
- H. Jung, P. K. Allan, Y. Y. Hu, O. J. Borkiewicz, X. L. Wang, W. Q. Han, L. S. Du, C. J. Pickard, P. J. Chupas, K. W. Chapman, A. J. Morris and C. P. Grey, Elucidation of the Local and Long-Range Structural Changes that Occur in Germanium Anodes in Lithium-Ion Batteries, Chem. Mater., 2015, 27(3), 1031–1041 CrossRef
- A. Pulido, L. J. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Functional materials discovery using energy-structure-function maps, Nature, 2017, 543(7647), 657–664 CrossRef PubMed
- D. W. Lewis, D. J. Willock, C. R. A. Catlow, J. M. Thomas and G. J. Hutchings, De novo design of structure-directing agents
for the synthesis of microporous solids, Nature, 1996, 382(6592), 604–606 CrossRef
- B. Rice, L. M. LeBlanc, A. Otero-de-la-Roza, M. J. Fuchter, E. R. Johnson, J. Nelson and K. E. Jelfs, A computational exploration of the crystal energy and charge-carrier mobility landscapes of the chiral [6] helicene molecule, Nanoscale, 2018, 10(4), 1865–1876 RSC
- C. C. Sun, Material Science Tetrahedron-A Useful Tool for Pharmaceutical Research and Development, J. Pharm. Sci., 2009, 98, 1671–1687 CrossRef PubMed
- S. L. Price and S. M. Reutzel-Edens, The potential of computed crystal energy landscapes to aid solid-form development, Drug Discovery Today, 2016, 21(6), 912–923 CrossRef PubMed
- M. A. Neumann, J. V. de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Combined crystal structure prediction and high-pressure crystallization in rational pharmaceutical polymorph screening, Nat. Commun., 2015, 6, 7793 CrossRef PubMed
- I. D. H. Oswald, I. Chataigner, S. Elphick, F. P. A. Fabbiani, A. R. Lennie, J. Maddaluno, W. G. Marshall, T. J. Prior, C. R. Pulham and R. I. Smith, Putting pressure on elusive polymorphs and solvates, CrystEngComm, 2009, 11(2), 359–366 RSC
- C. J. Pickard and R. J. Needs, Highly compressed ammonia forms an ionic crystal, Nat. Mater., 2008, 7(10), 775–779 CrossRef PubMed
- S. Ninet, F. Datchi, P. Dumas, M. Mezouar, G. Garbarino, A. Mafety, C. J. Pickard, R. J. Needs and A. M. Saitta, Experimental and theoretical evidence for an ionic crystal of ammonia at high pressure, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89(17), 174103 CrossRef
- S. Habermehl, P. Morschel, P. Eisenbrandt, S. M. Hammer and M. U. Schmidt, Structure determination from powder data without prior indexing, using a similarity measure based on cross-correlation functions, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 347–359 CrossRef PubMed
- M. Zilka, D. V. Dudenko, C. E. Hughes, P. A. Williams, S. Sturniolo, W. T. Franks, C. J. Pickard, J. R. Yates, K. D. M. Harris and S. P. Brown, Ab initio random structure searching of organic molecular solids: assessment and validation against experimental data, Phys. Chem. Chem. Phys., 2017, 19(38), 25949–25960 RSC
- A. G. Shtukenberg, Q. Zhu, D. J. Carter, L. Vogt, J. Hoja, E. Schneider, H. Song, B. Pokroy, I. Polishchuk, A. Tkatchenko, A. R. Oganov, A. L. Rohl, M. E. Tuckerman and B. Kahr, Chem. Sci., 2017, 8(7), 4926–4940 RSC
| This journal is © The Royal Society of Chemistry 2018 |