 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal structure prediction of flexible pharmaceutical-like molecules: density functional tight-binding as an intermediate optimisation method and for free energy estimation†
Luca
Iuzzolino
a,
Patrick
McCabe
b,
Sarah L.
Price
 a and
Jan Gerit
Brandenburg‡
*a
a and
Jan Gerit
Brandenburg‡
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: gerit.brandenburg@chemie.uni-goettingen.de
bThe Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK
First published on 27th February 2018
Abstract
Successful methodologies for theoretical crystal structure prediction (CSP) on flexible pharmaceutical-like organic molecules explore the lattice energy surface to find a set of plausible crystal structures. The initial search stages of CSP studies use relatively simple lattice energy approximations as hundreds of thousands of minima have to be considered. These generated crystal structures often have poor molecular geometries, as well as inaccurate lattice energy rankings, and performing reasonably accurate but computationally affordable optimisations of the crystal structures generated in a search would be highly desirable. Here, we seek to explore whether semi-empirical quantum-mechanical methods can perform this task. We employed the dispersion-corrected tight-binding Hamiltonian (DFTB3-D3) to relax all the inter- and intra-molecular degrees of freedom of several thousands of generated crystal structures of five pharmaceutical-like molecules, saving a large amount of computational effort compared to earlier studies. The computational cost scales better with molecular size and flexibility than other CSP methods, suggesting that it could be extended to even larger and more flexible molecules. On average, this optimisation improved the average reproduction of the eight experimental crystal structures (RMSD15) and experimental conformers (RMSD1) by 4% and 23%, respectively. The intermolecular interactions were then further optimised using distributed multipoles, derived from the molecular wave-functions, to accurately describe the electrostatic components of the intermolecular energies. In all cases, the experimental crystal structures are close to the top of the lattice energy ranking. Phonon calculations on some of the lowest energy structures were also performed with DFTB3-D3 methods to calculate the vibrational component of the Helmholtz free energy, providing further insights into the solid-state behaviour of the target molecules. We conclude that DFTB3-D3 is a cost-effective method for optimising flexible molecules, bridging the gap between the approximate methods used in CSP searches for generating crystal structures and more accurate methods required in the final energy ranking.
Introduction
In computational materials science, approaches that are based on the fundamental laws of quantum mechanics (QM) are now integral to almost any materials design initiative in academia or industry.1–3 The prevalence of polymorphism in organic molecules4 and its importance in determining the physical properties of organic solids1,5–8 has led to an interest in using simulation methods to predict the range of possible crystal structures and their properties, possibly to design molecular materials with desired characteristics, like high porosity.9 CSP studies aim to predict all the possible putative polymorphs (PPMs) of a molecule, starting from the chemical diagram only.10 This is particularly important for pharmaceutical development when performed as a complement to solid form screening, which attempts to establish the range of solid forms that could either be developed into the pharmaceutical product or must not appear during manufacturing and storage.8,11 Several successful CSP studies on large but relatively rigid systems, such as organic porous cages,12,13 have been reported. For inorganic solids, ab initio random structure searches have been used successfully.14–16 Pharmaceuticals are more challenging since they tend to have a considerable conformational flexibility, being designed for their biological activity, and so most molecules in drug development can crystallise in a variety of conformations.8 For pharmaceuticals with few intramolecular degrees of freedom, several successful CSP studies have been conducted.17–24 However, the CSP search space grows exponentially with conformational flexibility and hence more efficient methods are needed for application to modern pharmaceuticals. The challenge of dealing with molecular flexibility is compounded by small changes in some bond or torsion angles having a significantly greater effect on the overall molecule shape in large pharmaceuticals than in smaller molecules.25 Hence, the number of conformational degrees of freedom that may change from isolated molecule (ab initio) values because of the crystal packing forces can be larger than expected26 from combining the flexibility of fragment model molecules. Whilst optimising all atomic positions simultaneously with the crystal lattice positions avoids the problem of selecting the crystal packing-dependent conformational degrees of freedom, the need to balance the inter- and intramolecular forces sufficiently accurately makes this exceptionally demanding. This has been demonstrated by the challenges posed by the largest molecules in the recent Blind Tests of CSP.1,27,28 These molecules, though large enough to generate interest in CSP methods from industry, are still small and of limited flexibility compared with pharmaceuticals in development. Hence, if CSP is to fulfil its promise for pharmaceutical development, innovations that allow CSP studies on larger molecules within a restricted time frame11 are needed.All CSP methods begin with a search step in which the lattice energy (Elatt) surface is explored to locate all the possible local minima, each corresponding to a separate possible crystal structure.1,29 Since the surface that must be explored is extremely wide, especially for flexible molecules, a large number of candidate crystal structures are produced, at least of the order of hundreds of thousands.22,29 Hence, cheaper but less accurate energy models must be utilised in the search. They can take the form of tailor-made force fields,30 exp-6 atomistic intermolecular potentials combined with quantum-mechanical calculations of the conformational energy,31,32 or transferrable empirical force fields.33
The lattice energy evaluations used in the crystal structure generation (“search”) stage are rarely accurate enough to give a reliable energy ranking, since the lattice energy differences between observed polymorphs are usually less than 5 kJ mol−1. Hence, thousands of generated crystal structures must be optimised and re-ranked to obtain a meaningful list of PPMs. Since this final step requires the use of accurate and expensive models, such as periodic dispersion-corrected density functional theory (DFT-D)34–40 on all crystal structures (the Ψcrys method), or the calculation of a high-quality wave-function for each molecular conformer (the Ψmol method),8,41,42 the computational cost can easily become unfeasible. A possible solution is the use of an intermediate optimisation method, which can bridge the gap between the simple models used to generate the crystal structures and the more accurate ones used to generate an accurate final energy ranking. For flexible molecules, several intermediate methods are used in different CSP workflows, such as optimising some torsion angles with a transferable force-field to improve the molecular conformation geometries,43,44 single-point energy calculations with a more accurate description of the intra- and intermolecular interactions18,20 or partial lattice energy optimisations.1,22
Recently, semi-empirical QM methods have been applied to large chemical and biochemical systems;45,46 these methods do not have the transferability problems of standard force fields but are significantly less demanding than other QM methods.45,47,48 In this work, the interest is focused on dispersion-corrected density functional tight binding methods (DFTB3-D3), which are up to three orders of magnitude cheaper than DFT-D, and have been shown to provide reasonably accurate lattice energies for a benchmark of small molecular crystals.49 We present the first use of semi-empirical QM methods within a full CSP workflow. We aim to verify whether DFTB3-D3 can be used as an intermediate re-ranking step in CSP. There are at least four ways in which it could provide useful insights:
(1) Producing more accurate molecular and crystalline geometries and packings than the original crystal structures generated in the search, which could be used as starting points for better lattice energy evaluations.
(2) Obtaining a more accurate energy ranking than the one obtained after the crystal structure search, in order to limit the number of structures that need to be taken to the final optimisation stage.
(3) Producing fewer minima that are artefacts of the constraints imposed by the space group and conformational variables, by optimising all crystal structures with P1 symmetry.
(4) Computing the phonon modes of plausible crystal structures to estimate thermodynamic free energies in the harmonic approximation.
We test the suitability of DFTB3-D3 for these purposes on structures generated in a set of searches25 performed with the Ψmol method. Elatt, the energy needed to completely separate all the molecules in the static crystal structure to isolated molecules in their most stable conformation,10 can be described as:
| Elatt = Uinter + ΔEintra |
ΔEintra is the energy penalty for modifying the conformation of the molecule from its isolated gas-phase minimum, calculated with a good quality molecular electronic structure method, and Uinter is the intermolecular energy, which sums all the interactions between the molecules in the crystal.10 The search method variant used in this work treated the most flexible torsion angles as search variables, and the other conformational degrees of freedom correspond to the isolated molecule ab initio values, an approach that is comparable to using multiple rigid conformations.43 Five flexible molecules for which full CSP studies had already been performed (Fig. 1) are used to test the application of DFTB3-D3 methods: molecule XX was the first “pharmaceutical-like” molecule used in a Blind Test,50 XXIII and XXVI are from the sixth blind test,1 GSK269984B was used in an early pharmaceutical study,23 while the CSP study of mebendazole was performed for an on-going collaboration.51 These CSP methods were successful in predicting all the observed polymorphs with one molecule in the asymmetric unit cell (i.e. Z′ = 1), and so provide a stringent test for the suitability of an intermediate DFTB3-D3 step to extend CSP methods to even larger, more flexible molecules.
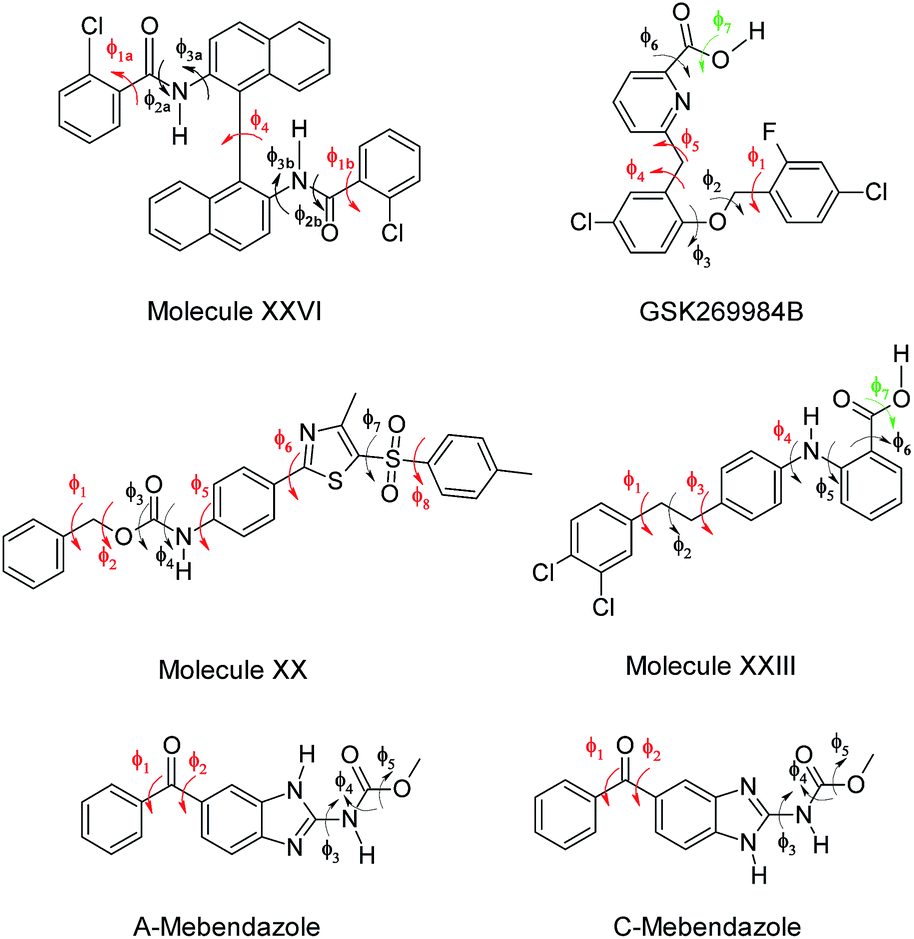 | ||
| Fig. 1 Chemical diagrams of the six molecules used in this work. The torsion angles in red were treated as explicitly flexible in the crystal structure searches, those in black were constrained to a set of values defined by the CSD Conformer Generator, and those in green were constrained to a set of values after an ab initio gas-phase scan. | ||
Methods
Crystal structure generation
The method for generating the crystal structures used statistical information retrieved from the Cambridge Structural Database (CSD) to guide the determination and the analysis of the conformational search space.25 For each molecule in Fig. 1, the CSD distributions of each rotatable bond were retrieved from the CSD knowledge base conformational libraries;52 the effect of each of these rotamers on the overall shape of each molecule was subsequently determined via ultra-fast shape recognition.53 This information was used to divide the torsion angles into two groups: those that were treated as explicitly flexible in the searches, and those that were constrained to a set of values, determined by the CSD Conformer Generator.54 Molecular electronic structure calculations at the PBE0 6-31G(d,p) level of theory using Gaussian09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 55 were used to limit the set of potential conformations to be considered and to calculate grids of ΔEintra values for the explicitly flexible torsion angles. These calculations were also used to determine the fixed atomic charges used by CrystalPredictor 1.832 to estimate Uinter in combination with the empirically fitted FIT repulsion–dispersion intermolecular potential.56 The number of crystal structures generated varied from ∼350000 for the C tautomer of mebendazole to ∼2200000 for molecule XXVI. The outputs from this search method and from the original CSP studies are compared in a previous publication.25 Thus the molecular geometries prior to refinement were mostly determined by PBE0 6-31G(d,p) isolated molecule optimisations, with the torsion angles shown in red in Fig. 1 at values specifically produced in the search.
55 were used to limit the set of potential conformations to be considered and to calculate grids of ΔEintra values for the explicitly flexible torsion angles. These calculations were also used to determine the fixed atomic charges used by CrystalPredictor 1.832 to estimate Uinter in combination with the empirically fitted FIT repulsion–dispersion intermolecular potential.56 The number of crystal structures generated varied from ∼350000 for the C tautomer of mebendazole to ∼2200000 for molecule XXVI. The outputs from this search method and from the original CSP studies are compared in a previous publication.25 Thus the molecular geometries prior to refinement were mostly determined by PBE0 6-31G(d,p) isolated molecule optimisations, with the torsion angles shown in red in Fig. 1 at values specifically produced in the search.
Density functional tight-binding method
As an established semi-empirical method, we used a third order density functional tight-binding Hamiltonian with self-consistent charge redistribution as implemented in dftb+.57–59 We used the 3OB Slater–Koster files,60,61 and an additional damping of all hydrogen-containing pair-potentials, and we corrected for missing London dispersion interactions with the atom-pairwise D3 scheme in the rational damping variant.62,63 The Brillouin zone was then sampled with a Γ-centred mesh with a density of 0.025 Å−1. The original DFTB3-D3 parametrisation focused on energetic properties only.49 However, it has been shown that the geometries of large molecular complexes and molecular crystals are not reproduced sufficiently accurately.64,65 A re-parametrisation of the DFTB3-MBD model has been shown to improve the geometrical properties substantially,66 so we followed a similar strategy and optimised the D3 damping parameter to minimise the errors in centre-of-mass distances on a set of small molecular dimers (S66).67,68 This results in a DFTB3-D3 parameter set (S6 = 1.0, S8 = 0.0, a1 = 0.841, a2 = 3.834 Bohr) with 8 kJ mol−1 mean absolute error on the S66 binding energies and 1.3% error in the centre-of-mass distances, compared to 3.8 kJ mol−1 and 3.2%, respectively with the original parametrisation. This re-parametrisation also improves the mass density errors of a set of small molecular crystals (X23)69,70 from 11.6% to 4.2%. Additional atom pair-wise corrections to DFTB3-D3, such as geometrical hydrogen-bonding corrections71,72 and halogen-bonding corrections,73 were not utilised.Structure relaxations have been conducted with a quasi-Newton optimisation in internal redundant coordinates as implemented in the CRYSTAL14 programme.74 As DFTB3-D3 is used as an intermediate optimisation method, it is important to test the relaxation thresholds. This was tested on the POLY59 benchmark set75 and an optimum trade-off between the accuracy in relative energy ranking and computational cost was obtained with thresholds of 0.12 a.u. and 0.003 a.u. for the root-mean-square (RMS) displacement and RMS gradient.
Phonon calculations were used as a final step to estimate the free energies for the experimental crystal structures and a few diverse low energy structures (given in ESI Table 10†). Phonon calculations using symmetric finite displacements required geometry optimisations with one order of magnitude tighter thresholds, corresponding to the default settings in CRYSTAL14. The Brillouin zone sampling for the phonon calculations was initially formed by the common practice of constructing supercells with minimum cell lengths of 10 Å.70 As we are interested in relative polymorph energies, we additionally ensured that a similar number of atoms are covered in each supercell to enhance possible error compensations. The Helmholtz free energy, A, can be defined as:
| A = Elatt + Fvib |
DFTB3-D3 intermediate optimisation of all the CrystalPredictor generated structures
All the unique generated crystal structures within 40 kJ mol−1 of the global minimum in CrystalPredictor energy were optimised with DFTB3-D3. In order to perform the DFTB3-D3 optimisations, the symmetry of each crystal structure had to be reduced to the P1 space group. 9215 crystal structures were optimised for molecule XXVI, 16744 for GSK269984B, 28249 for molecule XXIII, 26650 for molecule XX, 4165 for the A-tautomer of mebendazole and 4284 for its C-tautomer. The symmetries of all the crystal structures that were successfully optimised were reintroduced with Platon,76 and then they were clustered to remove duplicates using the Crystal Packing Similarity tool within the CSD Python API;77 details on the clustering procedure can be found in ESI Section 1.1.†
The energy ranking calculated via DFTB3-D3 proved to be very different, as shown in Fig. 2 for XXVI, with the calculated Elatt values having a larger energy spread than after the search stage. However, the poor energies of the structures corresponding to the experimental forms showed that, contrary to the original hope, DFTB3-D3 had not produced a more accurate energy ranking. Hence, all the unique optimised crystal structures within a large window of 50 kJ mol−1 of the global minimum in DFTB3-D3 energy were taken forward. Clustering to remove duplicates and applying the energy cut-off each reduced the number of structures by a molecule-dependent proportion, which is broken down in ESI Table 1.† This resulted in 3346 crystal structures for molecule XXVI, 5328 for GSK269984B, 13490 for molecule XXIII, 19146 for molecule XX, 3078 for the A tautomer of mebendazole and 3352 for its C tautomer, requiring optimisation with a more accurate method.
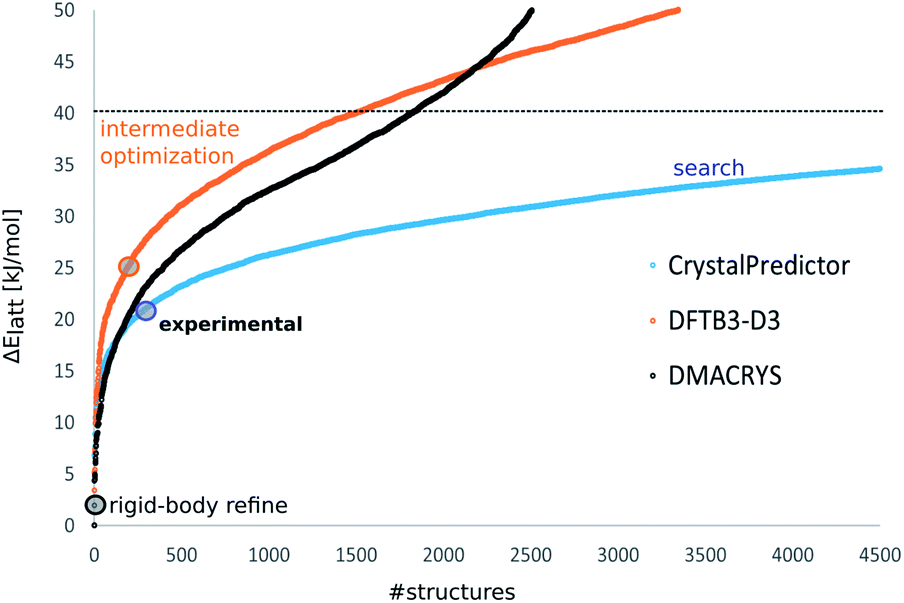 | ||
| Fig. 2 The energy distributions of the computer-generated crystal structures of molecule XXVI showing how the ∼9000 structures below 40 kJ mol−1 generated by the CrystalPredictor search (blue) are optimised by DFTB3-D3 (orange) to provide only ∼3000 structures below 50 kJ mol−1, and their corresponding energies calculated by DMACRYS using the ΨPBE0+FITmol model (black). The coloured circles indicate the energies of the experimental structures relative to the global minimum in Elatt at that stage. Similar plots for the other molecules are shown in ESI Fig. 1–5.† | ||
Re-ranking using improved molecular wave-functions
The lattice energies with DFTB3-D3 were clearly inadequate, hence a further lattice energy evaluation with a more accurate description of the wave-function was required. We assumed that the DFTB3-D3 optimisation had improved the molecular geometries, and this was exploited by reverting to the Ψmol approach used to generate the crystal structures, but using distributed multipoles instead of atomic charges, and a more accurate evaluation of ΔEintra. For all the DFTB3-D3 optimised crystal structures, the molecular wave-function of each distinct molecular conformation was calculated at the PBE0 6-31G(d,p) level of theory using Gaussian09, and distributed multipoles up to hexadecapoles were derived from the charge density using GDMA 2.2.78 Finally the intermolecular interactions were optimised with DMACRYS.79Uinter was modelled using an electrostatic component calculated from distributed multipoles combined with a repulsion–dispersion component calculated with the empirically-fitted exp-6 FIT potential. The crystal structures optimised with this specific method, denoted ΨPBE0+FITmol, were ranked in terms of Elatt, with ΔEintra estimated as the difference between the energy of each crystalline conformer and the PBE0 6-31G(d,p) energy of the DFTB3-D3 optimised gas-phase minimum calculated with Gaussian09. This was used to avoid the absolute values of Elatt being affected by a ΔEintra component calculated relative to a value optimised with a different wave-function. Although the energies of the DFTB3-D3 optimised gas-phase minima are 12–25 kJ mol−1 higher than their PBE0 6-31G(d,p) optimised counterparts, this systematic error does not change the energy differences between polymorphs, which are the main assessment criteria of CSP studies. For each molecule, all the crystal structures within 50 kJ mol−1 were clustered to remove duplicates.Rigid-molecule harmonic phonon calculations were carried out on the same structures as for the DFTB3-D3 phonons with DMACRYS using supercells generated following the methodology of Nyman and Day.80 This computationally cheap approach effectively assumes that the molecular modes do not affect ΔFvib, in contrast to the DFTB3-D3 calculations that allow the molecular and lattice modes to couple. The rigid-molecule ΨPBE0+FITmol model assumes that the molecular modes make an identical contribution to Fvib for all structures. This is a necessary assumption since the pure molecular modes could only be calculated at a local isolated-molecule minimum, and many conformations are far from such minima (although they would not necessarily be defined as conformational polymorphs).81
Assessment of the results
Crystal structures were compared with the experimental crystal structures and sets of the most significant computer-generated PPMs from the previous CSP studies identified in a previous work25 using the Crystal Packing Similarity tool in the CSD Python API, trying to match 15-molecule clusters with 20% distance and 20° angle tolerances. In a few cases, the tolerances had to be slightly increased to find matches with some of the computer-generated structures.The generated crystal structures that upon optimisation ended up matching the experimental crystal structures and the significant computer-generated PPMs25 were optimised with CrystalOptimizer82 at the PBE0 6-31G(d,p) level of theory, as a function of a set of torsion and bond angles (shown in ESI Fig. 6†) that were selected according to rules defined by Nyman and Day26 based on chemical intuition derived from small model molecules. These calculations were used as a stringent test to verify how the geometries and energies obtained using DFTB3-D3 and ΨPBE0+FITmol compare with those obtained through optimising the most flexible conformational degrees of freedom using a more accurate and computationally expensive Ψmol model.
Results
Intra- and intermolecular geometries
The quality of reproduction of each experimental Z′ = 1 crystal structure and the molecular conformation within it, before and after optimisations with DTFB3-D3 and ΨPBE0+FITmol is shown in Table 1, and contrasted with the reproductions achieved with the more computationally demanding CrystalOptimizer.| Experimental crystal structure (CSD refcode) | Reproduction of the experimental conformers (RMSD1) | Reproduction of the experimental crystal lattices (RMSD15) | |||||
|---|---|---|---|---|---|---|---|
| Search/Å | DFTB3-D3/Å | CrystalOptimizer/Å | Search/Å | DFTB3-D3/Å | ΨPBE0+FITmol/Å | CrystalOptimizer/Å | |
| XXVI (XAFQIH) | 0.2 | 0.133 | 0.123 | 0.533 | 0.425 | 0.376 | 0.294 |
| GSK269984B (BIFHOP) | 0.093 | 0.071 | 0.094 | 0.219 | 0.329 | 0.212 | 0.21 |
| XXIII a (XAFPAY) | 0.236 | 0.216 | 0.212 | 0.689 | 0.68 | 0.663 | 0.443 |
| XXIII b (XAFPAY01) | 0.278 | 0.18 | 0.159 | 0.626 | 0.528 | 0.415 | 0.373 |
| XXIII d (XAFPAY03) | 0.31 | 0.278 | 0.237 | 0.511 | 0.544 | 0.583 | 0.53 |
| XX (OBEQIX) | 0.227 | 0.137 | 0.101 | 0.455 | 0.469 | 0.39 | 0.218 |
| Mebendazole A (TUXPEJ) | 0.201 | 0.176 | 0.143 | 0.465 | 0.408 | 0.402 | 0.321 |
| Mebendazole C (YULGIW) | 0.096 | 0.067 | 0.066 | 0.825 | 0.763 | 0.313 | 0.28 |
| Average | 0.205 | 0.157 | 0.142 | 0.540 | 0.518 | 0.419 | 0.334 |
| Standard deviation | 0.073 | 0.067 | 0.055 | 0.169 | 0.135 | 0.134 | 0.103 |
DFTB3-D3 optimisations are effective at improving all the crystalline molecular conformations from the search generated structure, with a 23% improvement in the average RMSD1. The improvement in the overall crystal packing is more variable, with some becoming slightly worse and some slightly better with a mere 4% improvement in the average RMSD15. Keeping the molecule rigid at the DFTB3-D3 geometry but using the distributed multipoles and intramolecular energy from a superior electronic structure method improves the intermolecular packing, leading to an overall improvement of the average RMSD15 of 22% after the ΨPBE0+FITmol optimisations. Both the molecular and crystalline geometries are of a slightly worse quality than those obtained after the CrystalOptimizer optimisations, which improve the search structure by an average RMSD1 of 31% and an average RMSD15 of 38%. However, DFTB3-D3 refinements could be carried out at a much lower computational cost for these types of molecules, and yet successfully reproduced the structures.
Lattice energy rankings
The energy rankings of the crystal structures matching the experimental ones and their stabilities relative to the most stable one at that stage are shown in Table 2, and compared to those obtained in the original studies.| Experimental crystal structure | Ranking after search | ΔElatt after search/kJ mol−1 | Ranking after DFTB3-D3 | ΔElatt after DFTB3-D3/kJ mol−1 | Ranking after ΨPBE0+FITmol | ΔElatt after ΨPBE0+FITmol/kJ mol−1 | Ranking in previous CSP study | ΔElatt in previous CSP study/kJ mol−1 |
|---|---|---|---|---|---|---|---|---|
| a The crystal structures of the two tautomers of mebendazole are ranked together. | ||||||||
| XXVI (XAFQIH) | 262 | 20.5 | 208 | 25.44 | 2 | 1.96 | 2 | 0.49 |
| GSK269984B (BIFHOP) | 54 | 6.63 | 1803 | 37.2 | 1 | 0 | 1 | 0 |
| XXIII a (XAFPAY) | 4311 | 20.57 | 3670 | 33.8 | 232 | 12.31 | 280 | 13.52 |
| XXIII B (XAFPAY01) | 55 | 7.72 | 805 | 23.49 | 3 | 0.82 | 1 | 0 |
| XXIII D (XAFPAY03) | 1318 | 16.28 | 3748 | 33.99 | 114 | 10.25 | 85 | 9.21 |
| XX (OBEQIX) | 10 | 3.86 | 10 | 2.94 | 1 | 0 | 1 | 0 |
| Mebendazole A (TUXPEJ)a | 2 | 1.37 | 14 | 4.32 | 1 | 0 | 1 | 0 |
| Mebendazole C (YULGIW)a | 92 | 9.64 | 101 | 8.96 | 15 | 5.78 | 4 | 2.55 |
DFTB3-D3 energies are inadequate for CSP and in most cases the experimental crystal structures are ranked worse than after the search. On the other hand, the final optimisation of just Uinter (i.e. keeping the DFTB3-D3-optimised molecular geometries rigid) with the ΨPBE0+FITmol model leads to a large improvement in the relative energies, with the crystal structures matching their experimental counterparts having rankings similar to those in the original CSP studies, which had been performed at a much larger computational cost. In three cases (molecule XX, GSK269984B, and mebendazole form A) the experimental crystal structure is still the global minimum. Experimental form B of molecule XXIII, form C of mebendazole, and XXVI are all within a few kJ mol−1 of the global minimum, while forms A and D of molecule XXIII are higher in energy, although still within a sensible energy window considering the issues with predicting the stability of the polymorphs of XXIII with any method as shown in Fig. 5.1
A question that arises is whether the energy landscapes, such as the two examples in Fig. 3, differ significantly in the range of putative polymorphs produced. Any comparison with the original studies is not straightforward because of the developments in the algorithms, variations in settings and the potential energy surfaces being used. A selection of the lowest energy PPMs and a few conformationally diverse structures from the original CSP studies had been selected for testing the workflow for generating the crystal structures25 and we find that the vast majority of these structures remain (see ESI Tables 3–7†) on the crystal energy landscapes (Fig. 3 and ESI Fig. 7–9†). Out of the 180 targeted PPMs for the five molecules, 21 were not found in the set of ΨPBE0+FITmol optimised crystal structures. However, in eight of those cases the PPMs were already missing after the search, and for a further nine the recent search structure was a very poor match.25 Only four PPMs that had been considered as ‘certainly found’ in the search25 were lost in this study, and either these were towards the high energy end of the sample, or similar types of packing are present in the crystal energy landscape. Some PPMs were found, but only above 20 kJ mol−1 from the global minimum. This can reflect the huge sensitivity of Elatt to quite subtle changes in conformation, particularly when hydrogen bonds can be inter- or intramolecular as in GSK269984B, and a crystal structure containing a molecular conformation optimised at the DFTB3-D3 level may converge to a less favourable local minimum when the intermolecular interactions are optimised with the ΨPBE0+FITmol method. The huge dependence of Elatt on subtle changes is further highlighted by the differences between the energies obtained in previous CSP studies and those obtained via re-optimisation of the key crystal structures with CrystalOptimizer, as shown in ESI Tables 3–7.† These optimisations also seem to lead to different geometries compared to the ΨPBE0+FITmol optimised crystal structures, highlighting how different routes can lead to different outcomes.
 | ||
| Fig. 3 Crystal energy landscapes of molecules (left) XX and (right) XXIII, with Elatt calculated by ΨPBE0+FITmol, plotted against the density of each crystal structure. The structures corresponding to the three experimental forms with Z′ = 1 only are given, with the relative energies of the Z′ = 2 polymorphs of XXIII being shown in Fig. 5. | ||
Phonon calculations
The calculation of phonons by DFTB3-D3 methods proved difficult to automate for reasons that would apply equally to significantly more expensive periodic electronic structure methods. For certain specific crystal structures, it was necessary to perform optimisations to an even tighter convergence on the energies and gradients in order to remove small imaginary frequencies (<100 i cm−1). The recipe of only needing supercells to have a minimum length of 10 Å proved insufficient to converge the Brillouin sampling so that the difference in Fvib between structures could be converged below 1 kJ mol−1, mainly because of the differences in entropy. Thus the contrasting calculations in Fvib (Fig. 4) contain significant numerical uncertainties, which do not cancel between different structures of the same molecule. Overall, the DFTB3-D3 phonons do not cause significant re-ranking; the most notable exception is for molecule XXVI, where the experimental crystal structure becomes lower in free energy than the global minimum. | ||
| Fig. 4 The relative vibrational contributions to the Helmholtz free energy ΔFvib, which can be added to Elatt to calculate A. For each molecule, ΔFvib is calculated relative to the structure which is the Elatt global minimum within the ΨPBE0+FITmol method, for which ΔFvib = 0. The experimental structures are in green. The rigid-body modes are pure lattice modes, calculated from the Ψmol method. Full details about the crystal structures and their energies at the different stages can be found in ESI Table 10.† | ||
It is clear that the inclusion of the coupling of the molecular modes with the lattice modes in the DFTB3-D3 calculations leads to a wider range of free energy differences than with rigid-body phonons, implying that there is a considerable effect from the variations of the modes in different conformations and their couplings. The rigid-body phonons, which used 4 or 5 linear supercells to converge the Brillouin zone sampling, may be subject to greater errors in ignoring the coupling of the modes for this type of molecule.
Computational cost
The savings in computational cost are quite large with this methodology, when compared with the original CSP studies, as detailed in ESI Tables 11–13.† The smallest saving (30%) was for the two tautomers of mebendazole, which is the least complex molecule in terms of size and conformational flexibility, and the greatest was for XXVI (90%). Some of this reduction in computational cost would be due to the use of different computer clusters, but the saving from replacing CrystalOptimizer and sometimes intermediate refinement methods with DFTB3-D3 structure refinement and ΨPBE0+FITmol seems to increase as a function of molecular size and flexibility. The rigid-molecule phonons were much cheaper to calculate than the DFTB3-D3 phonons.Discussion
This study pioneers the assessment of the DFTB3-D3 method for CSP studies of pharmaceuticals. In principle, the free energy calculated using a dispersion corrected PBE0 functional for each crystal structure (i.e. Ψcrys using the same wavefunction as in the ΨPBE0+FITmol method) should be more accurate than using the PBE functional that is commonly used for the final lattice energy evaluation in CSP studies,1 with both being more accurate than DFTB3-D3 calculations. However, the enormous computational cost makes this impractical for use in accurate CSP studies of large flexible molecules with current computer resources. Full DFTB3-D3 optimisations are significantly better than fixed cell optimisations of selected torsion angles with a transferable force-field, a previously used intermediate step in CSP for flexible molecules,43,44,50 where the energies were discarded. In practice, theoretical accuracy, which is very dependent on the molecule and range of crystal structures, has to be compromised by the huge number of structures that need to be considered, and the need to make such studies affordable for larger pharmaceutical studies. Thus the time saving produced by an intermediate DFTB3-D3 step is an important consideration for extending CSP to be used more widely in pharmaceutical development. DFTB3-D3 scales as Nln(N), in contrast to N3 for periodic DFT-D methods,58 where N is the number of atoms in the unit cell. The cost of the Ψmol approach is more variable, as by using databases of previous calculations and methods of interpolation82 the cost of this approach decreases as more structures are optimised. However, the Ψmol approach scales badly with the number of conformational degrees of freedom that are allowed to vary in response to the packing forces of specific crystals. The other conformational variables cannot be fixed, but have to be simultaneously ab initio optimised as intramolecular energy barriers can be drastically lowered by small changes in other parts of the molecule.83 Pharmaceutical molecules, where packing forces acting on large aromatic rings at different ends of the molecule strain the conformation of linking groups, and inter- and intramolecular dispersion need to be balanced, are very demanding of the quality of the modelling of the potential energy surface.
The numerical results shown in Table 2 suggest that DFTB3-D3 is of similar accuracy to PBE0 partial optimisation using CrystalOptimizer for reproducing the experimental conformers, as judged by RMSD1 values. Holding the DFTB3-D3 conformations as rigid during the final optimisation with the ΨPBE0+FITmol model gives a slightly worse reproduction of the crystal structures in terms of RMSD15, but at a reduced cost. Comparisons with the experimental conformations (see ESI Table 2†) show that in most cases the DFTB3-D3 reproduction is good, probably within the amplitude of the vibrational motions of the terminal groups. The only exception is XXIII form D, where the optimisations have found a conformer close to the search generated structure, but visually different from the experimental one. The differences between crystal structures, as judged by RMSD15, powder pattern similarity, or any other measure, raise the issue of clustering: how can one eliminate duplicates while being certain not to eliminate potential polymorphs? Many polymorphs are very similar in their packing.84 Determining which structures correspond to distinct free energy minima at crystallisation temperatures requires realistic and expensive modelling of molecular motions within the crystals and would only ever be applied to the most promising PPMs resulting from a CSP study. The question of whether structures could remain distinct during crystallisation and growth is closely linked to the possibility of disorder within crystal structures, and indeed CSP can help rationalise disordered pharmaceuticals.8,11 For example, molecules that can pack in hydrogen-bonded layers that can stack in different ways with similar energies could form polytypic packings or have stacking faults.20 Closely related structures can manifest as static or dynamic disorder depending on the energy barriers.17 Hence, decisions about the extent of the search and the clustering criteria can have a major effect on the number of structures that need to be considered and their interpretation. It appears that using an electronic structure level optimisation (DFTB3-D3) has not significantly reduced the number of minima (only by 1–4%, see ESI Table 1†) for the molecules considered in this study, hinting that few structures are artefacts of the Ψmol approach.
The disappointment is that the relative energies generated by DFTB3-D3 are too poor (see ESI Section 2.3.1†) to allow a confident, drastic reduction in the number of structures to be investigated with more accurate and expensive methods. This is probably due to the limitations of this methodology, such as the approximate Hamiltonian used in the molecular overlap regime where atoms are in van der Waals contact, and approximating the long-range electrostatic potential by atomic monopoles, although they are iterated to self-consistency to allow some polarisation of the molecules by the field of the other molecules.59 There is also an underestimation of the Pauli repulsion that can lead to an overestimate of the density of molecular crystals;45 the unbalanced description of Pauli repulsion in the parametrization of DFTB seems to originate from the underlying basis confinement.60 Some observed deficiencies in hydrogen bond and halogen bond geometries and energies have led to the proposal of additional corrections,71,73 but these do not seem to improve the results for large molecular assemblies.46 The long range electrostatic interactions are described by atomic charges in both DFTB3-D3 and in the crystal generation stage, but from atomic multipoles in the final energy evaluation, and the quality of the electrostatic model is very critical in crystal structure prediction, particularly for polar hydrogen bonded molecules;85 this explains why the final energy ranking appears to be much more accurate than the initial and intermediate ones. Several other differences between the various methodologies employed exist in the ways in which intra- and intermolecular interactions are treated and how they are combined to optimise the crystal structures and calculate Elatt. All these differences play a part in determining the quality of the geometries and of the energy ranking, and it appears that despite the clear advantages of periodic electronic methods a high-quality crystal energy landscape cannot be calculated with a simplified wave-function.
DFTB3-D3 and the Ψmol approach are less applicable in the presence of strong induction forces, suggesting they should not be used for salts or zwitterions, though there have been successful studies which make allowance for these deficiencies.86,87 The affordable density functionals such as PBE also have known problems with these types of systems.88–90 Furthermore, it is very challenging to have computational methods that are equally reliable for intramolecular dispersion effects91 and intermolecular dispersion extending through the range of separations sampled in crystal structures, but this balance underlies the observed preference for flexible molecules to pack in extended crystal structures.92 The large dispersion contributions make the energies very density dependent, as well as being sensitive to conformational distortion and the stronger more directional interactions such as hydrogen bonding.
The phonon calculations within the Ψcrys approach, which are only affordable using DFTB3-D3, are conceptually different from the rigid-body Ψmol approach, in including effects for molecules having many molecular modes that are of comparable frequency to the lattice modes.41 The use of DFTB3-D3 accounts for differences in the molecular modes within different crystalline geometries. This leads to an even larger spread in Fvib values than with the rigid-body model, which already shows a range of ΔFvib that is comparable with polymorph energy differences,4 and was estimated to reorder the calculated stability of about 9% of over 500 observed pairs of polymorphs.80 This raises a question as to whether thermal effects are likely to be more significant in determining the relative thermodynamic stability of pharmaceutical polymorphs than for more rigid molecules, particularly when there is a larger range of densities amongst the PPMs. DFTB3-D3 calculates all phonon modes within the specific crystal in a reasonable timeframe, and these can be added to Elatt calculated with ΨPBE0+FITmol or a better periodic DFT-D method.70,93,94 The disadvantages of a poorer potential energy surface need to be balanced with the problems of converging energies with the size of the supercell to equivalent accuracy for different polymorphs demonstrated in this study.95 Even if it were possible to calculate completely reliable harmonic modes, which the current results in Fig. 5 show has yet to be achieved, thermal expansion is likely to be important for both absolute free energies65 and the differences in thermal expansion between polymorphs, which will affect the relative free energies.41,96 These estimates assume that the (quasi)-harmonic model is equally adequate for all polymorphs, which is questionable for larger molecules where methyl groups may be rotating and phenyl rings may have different large amplitude motions, depending on the packing. Hence, the potential energy surface should be explored more explicitly, for example by molecular dynamics simulations,97,98 as none of the methodologies currently available can fully describe all the possible thermal effects that can affect crystal structures and their relative free energy ranking.
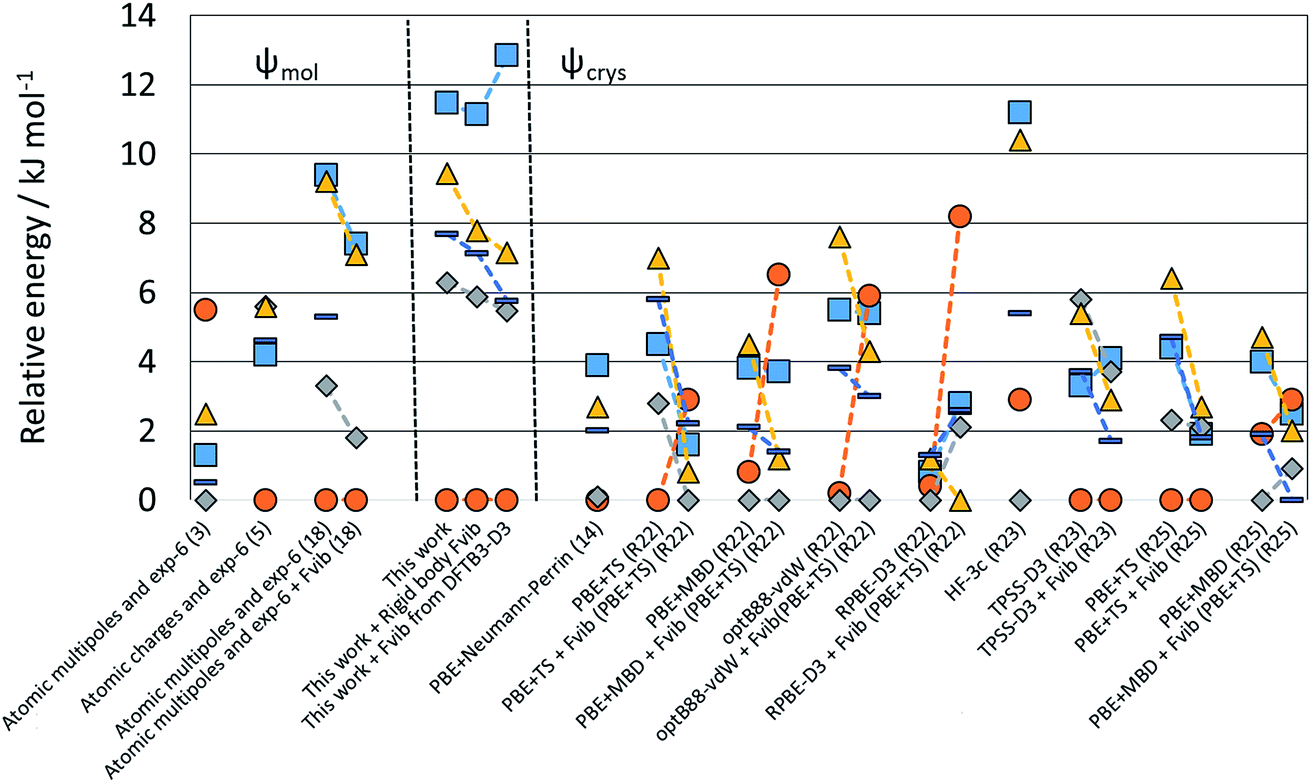 | ||
| Fig. 5 Relative energies of the polymorphs of XXIII calculated in this study compared with those reported by the participants of the 6th Blind Test.1 Note that values linked by dashed lines denote the changes from adding free energy estimates, with the group identifier in parentheses, and R denoting groups which only re-ranked the crystal structures. | ||
Many of the challenges in CSP studies are illustrated by the contrast between the crystal energy landscapes of XX and XXIII in Fig. 3, which from the chemical diagrams look like similar extended pharmaceutical molecules. The crystal energy landscape of molecule XX represents the simplest outcome of a CSP study, with a clear prediction of one crystal structure being more stable than any of its competitors. This structural refinement has led to a larger energy gap than previously found with two different Ψmol approaches,50 using CrystalOptimizer (FCC) and a force-field (RCM) for modifying the molecular conformation in response to the packing forces. Both had the experimentally-known crystal structure as the global minimum in Elatt but with a margin of less than 1 kJ mol−1 from the second most favourable PPM. Comparing the quality of this landscape with those of the previous studies is impossible as only three structures among the lowest ten found with the RCM method were among the top ten when optimised with the FCC method. ESI Table 5† shows that seven of the ten lowest-energy FCC crystal structures are among the ten lowest energy ΨPBE0+FITmol crystal structures, while for RCM this is only true for the crystal structure that matches the experimental form (see ESI Table 8†). The sensitivity of energies to the Elatt model is further shown by a generally very effective Ψcrys methodology developed by Neumann et al.21,24,99–101 having the experimental crystal structure ranked 7th,27 although that search included some Z′ = 2 crystal structures. Some of these Z′ = 2 structures also appear to be highly competitive when optimised with DFTB3-D3 and ΨPBE0+FITmol, as shown in ESI Table 9.† Nevertheless in our study the structure matching the only known experimental form is calculated to be the most stable by approximately 3 kJ mol−1, and there are relatively few alternatives to be considered as possible metastable polymorphs.
In contrast, the crystal energy landscape of XXIII has successfully found the three Z′ = 1 polymorphs, but amongst a plethora of competitive Z′ = 1 structures. Two further Z′ = 2 polymorphs are known from the industrial polymorph screening. Only form B is among the lowest energy crystal structures, while forms A and D are much higher in energy amongst many other structures of similar energy and density. Comparing these results with the sensitivity of the known polymorphs to different energy models, Fig. 5 suggests that the energy differences would have been reduced if the final energy optimisation had been performed using a good periodic DFT-D method, like TPSS-D3,102 PBE-MBD98 or B86bPBE-XDM.94,103 However, this contrasts with experimental stabilities from slurrying data, where form A is the most stable at 257 K and form D at 293 K.1 Overall, the sensitivity of the energies of the known forms of XXIII highlights the challenges of modelling the thermodynamics of the organic solid state. The contrast between the crystal energy landscapes of XX and XXIII emphasises the need for more CSP studies to be performed in collaboration with experimentalists in industry and academia.
Conclusions
Periodic DFTB3-D3 is suitable for optimising the crystal structures of flexible molecules, but does not provide a sufficiently accurate energy ranking. Experimental conformers and crystal structures are reproduced by DFTB3-D3 with average RMSD1 and RMSD15 values of 0.16 Å and 0.52 Å, respectively. DFTB3-D3 calculations suggest that the phonon modes may cause a more significant re-ranking of the energies of polymorphs of flexible pharmaceuticals than for smaller, more rigid molecules. The demands of performing CSP for flexible pharmaceuticals, and hence the most cost-efficient strategy, are very dependent on the molecule and the number of competitive crystal structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the CCDC for organising the Blind Tests of CSP and for partial financial support of L. I. along with the M3S Centre for Doctoral Training (EPSRC GRANT EP/G036675/1). We also thank Profs Pantelides and Adjiman (Imperial College) for the use of CrystalPredictor and CrystalOptimizer. J. G. B acknowledges support by the Alexander von Humboldt foundation within the Feodor Lynen program. The CSP computational software is developed under EP/K039229/1.References
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell, R. Car, D. H. Case, R. Chadha, J. C. Cole, K. Cosburn, H. M. Cuppen, F. Curtis, G. M. Day, R. A. DiStasio Jr, A. Dzyabchenko, B. P. van Eijck, D. M. Elking, J. A. van den Ende, J. C. Facelli, M. B. Ferraro, L. Fusti-Molnar, C.-A. Gatsiou, T. S. Gee, R. de Gelder, L. M. Ghiringhelli, H. Goto, S. Grimme, R. Guo, D. W. M. Hofmann, J. Hoja, R. K. Hylton, L. Iuzzolino, W. Jankiewicz, D. T. de Jong, J. Kendrick, N. J. J. de Klerk, H.-Y. Ko, L. N. Kuleshova, X. Li, S. Lohani, F. J. J. Leusen, A. M. Lund, J. Lv, Y. Ma, N. Marom, A. E. Masunov, P. McCabe, D. P. McMahon, H. Meekes, M. P. Metz, A. J. Misquitta, S. Mohamed, B. Monserrat, R. J. Needs, M. A. Neumann, J. Nyman, S. Obata, H. Oberhofer, A. R. Oganov, A. M. Orendt, G. I. Pagola, C. C. Pantelides, C. J. Pickard, R. Podeszwa, L. S. Price, S. L. Price, A. Pulido, M. G. Read, K. Reuter, E. Schneider, C. Schober, G. P. Shields, P. Singh, I. J. Sugden, K. Szalewicz, C. R. Taylor, A. Tkatchenko, M. E. Tuckerman, F. Vacarro, M. Vasileiadis, A. Vazquez-Mayagoitia, L. Vogt, Y. Wang, R. E. Watson, G. A. de Wijs, J. Z. Yang, Q. Zhu and C. R. Groom, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 439–459 CrossRef PubMed.
- S. Curtarolo, G. L. W. Hart, M. B. Nardelli, N. Mingo, S. Sanvito and O. Levy, Nat. Mater., 2013, 12, 191 CrossRef PubMed.
- N. Marzari, Nat. Mater., 2016, 15, 381 CrossRef PubMed.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- J. Bernstein, Polymorphism in Molecular Crystals, Clarendon Press, Oxford, 2002 Search PubMed.
- Y. Abramov, Org. Process Res. Dev., 2013, 17, 472–485 CrossRef.
- A. Y. Lee, D. Erdemir and A. S. Myerson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef PubMed.
- S. L. Price and S. M. Reutzel-Edens, Drug Discovery Today, 2016, 21, 912–923 CrossRef PubMed.
- A. Pulido, L. Chen, T. Kaczorowski, D. Holden, M. Little, S. Chong, B. Slater, D. McMahon, B. Bonillo, C. Stackhouse, A. Stephenson, C. Kane, R. Clowes, T. Hasell, A. Cooper and G. Day, Nature, 2017, 543, 657–664 CrossRef PubMed.
- S. L. Price, Chem. Soc. Rev., 2014, 43, 2098–2111 RSC.
- S. L. Price, D. E. Braun and S. M. Reutzel-Edens, Chem. Commun., 2016, 52, 7065–7077 RSC.
- J. T. A. Jones, T. Hasell, X. F. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef PubMed.
- E. O. Pyzer-Knapp, H. P. G. Thompson, F. Schiffmann, K. E. Jelfs, S. Y. Chong, M. A. Little, A. I. Cooper and G. M. Day, Chem. Sci., 2014, 5, 2235–2245 RSC.
- C. J. Pickard and R. J. Needs, J. Phys.: Condens. Matter, 2011, 23 CrossRef PubMed.
- R. T. Strong, C. J. Pickard, V. Milman, G. Thimm and B. Winkler, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 045101 CrossRef.
- M. Martinez-Canales, C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2012, 108, 045704 CrossRef PubMed.
- D. E. Braun, J. A. McMahon, L. H. Koztecki, S. L. Price and S. M. Reutzel-Edens, Cryst. Growth Des., 2014, 14, 2056–2072 CrossRef.
- R. M. Bhardwaj, L. S. Price, S. L. Price, S. M. Reutzel-Edens, G. J. Miller, I. D. H. Oswald, B. Johnston and A. J. Florence, Cryst. Growth Des., 2013, 13, 1602–1617 CrossRef.
- M. Baias, J. N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, J. Am. Chem. Soc., 2013, 135, 17501–17507 CrossRef PubMed.
- L. S. Price, J. A. McMahon, S. R. Lingireddy, S. F. Lau, B. A. Diseroad, S. L. Price and S. M. Reutzel-Edens, J. Mol. Struct., 2014, 1078, 26–42 CrossRef.
- M. A. Neumann, J. V. de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef PubMed.
- M. Vasileiadis, C. C. Pantelides and C. S. Adjiman, Chem. Eng. Sci., 2015, 121, 60–76 CrossRef.
- S. Z. Ismail, C. L. Anderton, R. C. Copley, L. S. Price and S. L. Price, Cryst. Growth Des., 2013, 13, 2396–2406 CrossRef.
- J. Kendrick, G. A. Stephenson, M. A. Neumann and F. J. J. Leusen, Cryst. Growth Des., 2013, 13, 581–589 CrossRef.
- L. Iuzzolino, A. M. Reilly, P. McCabe and S. L. Price, J. Chem. Theory Comput., 2017, 13, 5163–5171 CrossRef PubMed.
- J. Nyman and G. Day, Phys. Chem. Chem. Phys., 2016, 18, 31132–31143 RSC.
- D. A. Bardwell, C. S. Adjiman, Y. A. Arnautova, E. Bartashevich, S. X. M. Boerrigter, D. E. Braun, A. J. Cruz-Cabeza, G. M. Day, R. G. Della Valle, G. R. Desiraju, B. P. van Eijck, J. C. Facelli, M. B. Ferraro, D. Grillo, M. Habgood, D. W. M. Hofmann, F. Hofmann, K. V. J. Jose, P. G. Karamertzanis, A. V. Kazantsev, J. Kendrick, L. N. Kuleshova, F. J. J. Leusen, A. V. Maleev, A. J. Misquitta, S. Mohamed, R. J. Needs, M. A. Neumann, D. Nikylov, A. M. Orendt, R. Pal, C. C. Pantelides, C. J. Pickard, L. S. Price, S. L. Price, H. A. Scheraga, J. van de Streek, T. S. Thakur, S. Tiwari, E. Venuti and I. K. Zhitkov, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 535–551 CrossRef PubMed.
- G. M. Day, T. G. Cooper, A. J. Cruz-Cabeza, K. E. Hejczyk, H. L. Ammon, S. X. M. Boerrigter, J. Tan, R. G. Della Valle, E. Venuti, J. Jose, S. R. Gadre, G. R. Desiraju, T. S. Thakur, B. P. van Eijck, J. C. Facelli, V. E. Bazterra, M. B. Ferraro, D. W. M. Hofmann, M. Neumann, F. J. J. Leusen, J. Kendrick, S. L. Price, A. J. Misquitta, P. G. Karamertzanis, G. W. A. Welch, H. A. Scheraga, Y. A. Arnautova, M. U. Schmidt, J. van de Streek, A. Wolf and B. Schweizer, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 107–125 CrossRef PubMed.
- C. C. Pantelides, C. S. Adjiman and A. V. Kazantsev, Top. Curr. Chem., 2014, 345, 25–58 CrossRef PubMed.
- M. A. Neumann, J. Phys. Chem. B, 2008, 112, 9810–9829 CrossRef PubMed.
- M. Habgood, I. J. Sugdan, A. V. Kazantsev, C. S. Adjiman and C. Pantelides, J. Chem. Theory Comput., 2015, 11, 1957–1969 CrossRef PubMed.
- P. G. Karamertzanis and C. C. Pantelides, Mol. Phys., 2007, 105, 273–291 CrossRef.
- S. Kim, A. M. Orendt, M. B. Ferraro and J. C. Facelli, J. Comput. Chem., 2009, 30, 1973–1985 CrossRef PubMed.
- R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989 Search PubMed.
- W. Kohn, Rev. Mod. Phys., 1999, 71, 1253–1266 CrossRef.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef PubMed.
- L. Kronik and A. Tkatchenko, Acc. Chem. Res., 2014, 47, 3208–3216 CrossRef PubMed.
- J. Hoja, A. Reilly and A. Tkatchenko, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1294 Search PubMed.
- G. J. O. Beran, Chem. Rev., 2016, 116, 5567–5613 CrossRef PubMed.
- Y. N. Heit, K. D. Nanda and G. J. O. Beran, Chem. Sci., 2016, 7, 246–255 RSC.
- H. K. Buchholz, R. K. Hylton, J. G. Brandenburg, A. Seidel-Morgenstern, H. Lorenz, M. Stein and S. L. Price, Cryst. Growth Des., 2017, 17, 4676–4686 CrossRef.
- S. L. Price and J. G. Brandenburg, in Non-Covalent Interactions in Quantum Chemistry and Physics, Elsevier, 2017, pp. 333–363 Search PubMed.
- G. M. Day, W. D. S. Motherwell and W. Jones, Phys. Chem. Chem. Phys., 2007, 9, 1693–1704 RSC.
- G. M. Day and T. G. Cooper, CrystEngComm, 2010, 12, 2443–2453 RSC.
- A. S. Christensen, T. Kubař, Q. Cui and M. Elstner, Chem. Rev., 2016, 116, 5301–5337 CrossRef PubMed.
- J. G. Brandenburg, M. Hochheim, T. Bredow and S. Grimme, J. Phys. Chem. Lett., 2014, 5, 4275–4284 CrossRef PubMed.
- A. V. Akimov and O. V. Prezhdo, Chem. Rev., 2015, 115, 5797–5890 CrossRef PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef PubMed.
- J. G. Brandenburg and S. Grimme, J. Phys. Chem. Lett., 2014, 5, 1785–1789 CrossRef PubMed.
- A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman, C. C. Pantelides, S. L. Price, P. T. A. Galek, G. M. Day and A. J. Cruz-Cabeza, Int. J. Pharm., 2011, 418, 168–178 CrossRef PubMed.
- M. K. Corpinot, L. Iuzzolino, S. L. Price and D.-K. Bučar, unpublished work.
- R. Taylor, J. Cole, O. Korb and P. McCabe, J. Chem. Inf. Model., 2014, 54, 2500–2514 CrossRef PubMed.
- P. J. Ballester and W. G. Richards, J. Comput. Chem., 2007, 28, 1711–1723 CrossRef PubMed.
- J. C. Cole, O. Korb, P. McCabe, M. G. Read and R. Taylor, J. Chem. Inf. Model., 2018, 58, 615–629 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- D. S. Coombes, S. L. Price, D. J. Willock and M. Leslie, J. Phys. Chem., 1996, 100, 7352–7360 CrossRef.
- G. Seifert and J.-O. Joswig, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 456–465 Search PubMed.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef PubMed.
- M. Elstner, J. Phys. Chem. A, 2007, 111, 5614–5621 CrossRef PubMed.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef PubMed.
- M. Kubillus, T. Kubař, M. Gaus, J. Řezáč and M. Elstner, J. Chem. Theory Comput., 2015, 11, 332–342 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- R. Sure, J. G. Brandenburg and S. Grimme, ChemistryOpen, 2016, 5, 94–109 CrossRef PubMed.
- J. G. Brandenburg, J. Potticary, H. A. Sparkes, S. L. Price and S. R. Hall, J. Phys. Chem. Lett., 2017, 8, 4319–4324 CrossRef PubMed.
- M. Mortazavi, J. G. Brandenburg, R. J. Maurer and A. Tkatchenko, J. Phys. Chem. Lett., 2018, 9, 399–405 CrossRef PubMed.
- J. Řezáč, K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2011, 7, 2427–2438 CrossRef PubMed.
- B. Brauer, M. K. Kesharwani, S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2016, 18, 20905–20925 RSC.
- A. Otero-de-la-Roza and E. R. Johnson, J. Chem. Phys., 2012, 137, 054103 CrossRef PubMed.
- A. M. Reilly and A. Tkatchenko, J. Chem. Phys., 2013, 139, 024705 CrossRef PubMed.
- M. Korth, M. Pitoňák, J. Řezáč and P. Hobza, J. Chem. Theory Comput., 2010, 6, 344–352 CrossRef PubMed.
- V. M. Miriyala and J. Rezac, J. Comput. Chem., 2017, 38, 688–697 CrossRef PubMed.
- J. Rezac and P. Hobza, Chem. Phys. Lett., 2011, 506, 286–289 CrossRef.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D’Arco, Y. Noël, M. Causà, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef.
- J. G. Brandenburg and S. Grimme, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 502–513 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef PubMed.
- A. J. Stone, J. Chem. Theory Comput., 2005, 1, 1128–1132 CrossRef PubMed.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478–8490 RSC.
- J. Nyman and G. M. Day, CrystEngComm, 2015, 17, 5154–5165 RSC.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef PubMed.
- A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman and C. C. Pantelides, in Molecular System Engineering, ed. C. S. Adjiman and A. Galindo, Wiley-VCH Verlag GmbH & Co., Weinheim, 2010, vol. 6, pp. 1–42 Search PubMed.
- O. G. Uzoh, P. T. A. Galek and S. L. Price, Phys. Chem. Chem. Phys., 2015, 17, 7936–7948 RSC.
- S. J. Coles, T. L. Threlfall and G. J. Tizzard, Cryst. Growth Des., 2014, 14, 1623–1628 CrossRef.
- G. M. Day, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1023–1033 CrossRef.
- D. E. Braun, S. R. Lingireddy, M. D. Beidelschies, R. Guo, P. Muller, S. L. Price and S. M. Reutzel-Edens, Cryst. Growth Des., 2017, 17, 5349–5365 CrossRef PubMed.
- D. E. Braun, M. Orlova and U. J. Griesser, Cryst. Growth Des., 2014, 14, 4895–4900 CrossRef PubMed.
- J. G. Brandenburg, T. Maas and S. Grimme, J. Chem. Phys., 2015, 142 Search PubMed.
- J. Klimes, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- L. Goerigk, H. Kruse and S. Grimme, ChemPhysChem, 2011, 12, 3421–3433 CrossRef PubMed.
- A. Fokin, T. Zhuk, S. Blomeyer, C. Perez, L. Chernish, A. Pashenko, J. Antony, Y. Vishnevskiy, R. Berger, S. Grimme, C. Logemann, M. Schnell, N. Mitzel and P. Schreiner, J. Am. Chem. Soc., 2017, 139, 16696–16707 CrossRef PubMed.
- H. P. G. Thompson and G. M. Day, Chem. Sci., 2014, 5, 3173–3182 RSC.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef PubMed.
- S. R. Whittleton, A. Otero-de-la-Roza and E. R. Johnson, J. Chem. Theory Comput., 2017, 13, 441–450 CrossRef PubMed.
- R. Guo, K. Refson and S. L. Price, personal communication.
- Y. Heit and G. Beran, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 514–529 CrossRef PubMed.
- M. Rossi, P. Gasparotto and M. Ceriotti, Phys. Rev. Lett., 2016, 117, 115702 CrossRef PubMed.
- A. Shtukenberg, Q. Zhu, D. Carter, L. Vogt, J. Hoja, E. Schneider, H. Song, B. Pokroy, I. Polishchuk, A. Tkatchenko, A. Oganov, A. Rohl, M. Tuckerman and B. Kahr, Chem. Sci., 2017, 8, 4926–4940 RSC.
- M. A. Neumann, F. J. J. Leusen and J. Kendrick, Angew. Chem., Int. Ed., 2008, 47, 2427–2430 CrossRef PubMed.
- A. Asmadi, M. A. Neumann, J. Kendrick, P. Girard, M. A. Perrin and F. J. J. Leusen, J. Phys. Chem. B, 2009, 113, 16303–16313 CrossRef PubMed.
- J. van de Streek and M. A. Neumann, CrystEngComm, 2011, 13, 7135–7142 RSC.
- J. G. Brandenburg, J. E. Bates, J. Sun and J. P. Perdew, Phys. Rev. B, 2016, 94, 115144 CrossRef.
- S. R. Whittleton, A. Otero-de-la-Roza and E. R. Johnson, J. Chem. Theory Comput., 2017, 13, 5332–5342 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full details of results for each molecule. See DOI: 10.1039/c8fd00010g |
| ‡ Current address: Institute for Physical Chemistry, University of Goettingen, Tammannstr. 6, 37077 Goettingen, Germany. |
| This journal is © The Royal Society of Chemistry 2018 |